
Exercise Training Enhances BDNF/TrkB Signaling Pathway and Inhibits Apoptosis in Diabetic Cerebral Cortex



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Diabetes Induction
2.3. Experimental Groups and Procedures
2.4. Hematoxylin and Eosin (H&E) Stain
2.5. TUNEL Assay
2.6. Western Blot Analysis
2.7. Statistical Analysis
3. Results
3.1. Neural Histopathology and TUNEL(+) Apoptotic Cells
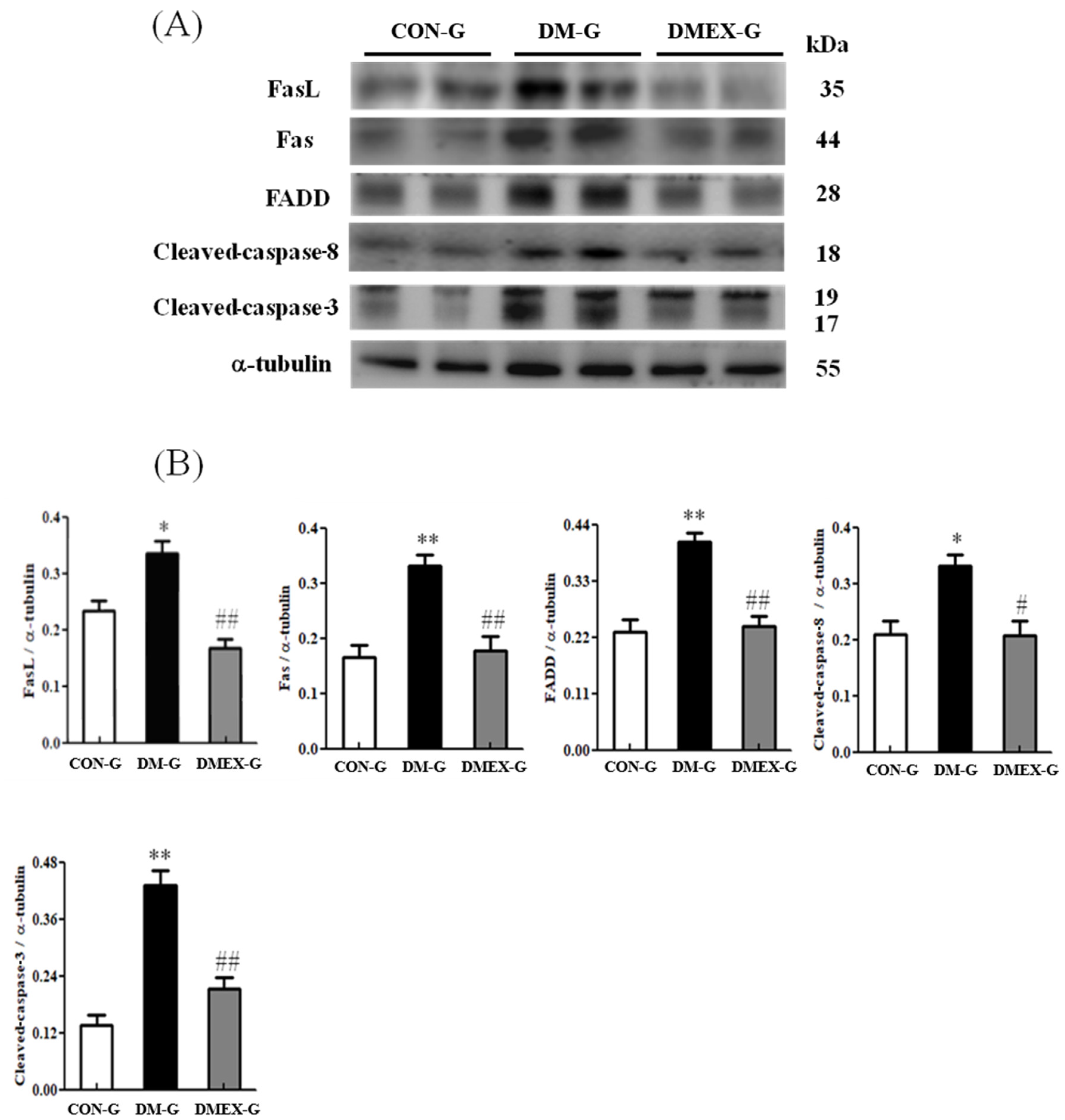
3.2. Neural Extrinsic (Fas/FasL-Mediated) Apoptotic Pathway
3.3. Neural Intrinsic (Mitochondria-Initiated) Apoptotic Pathway
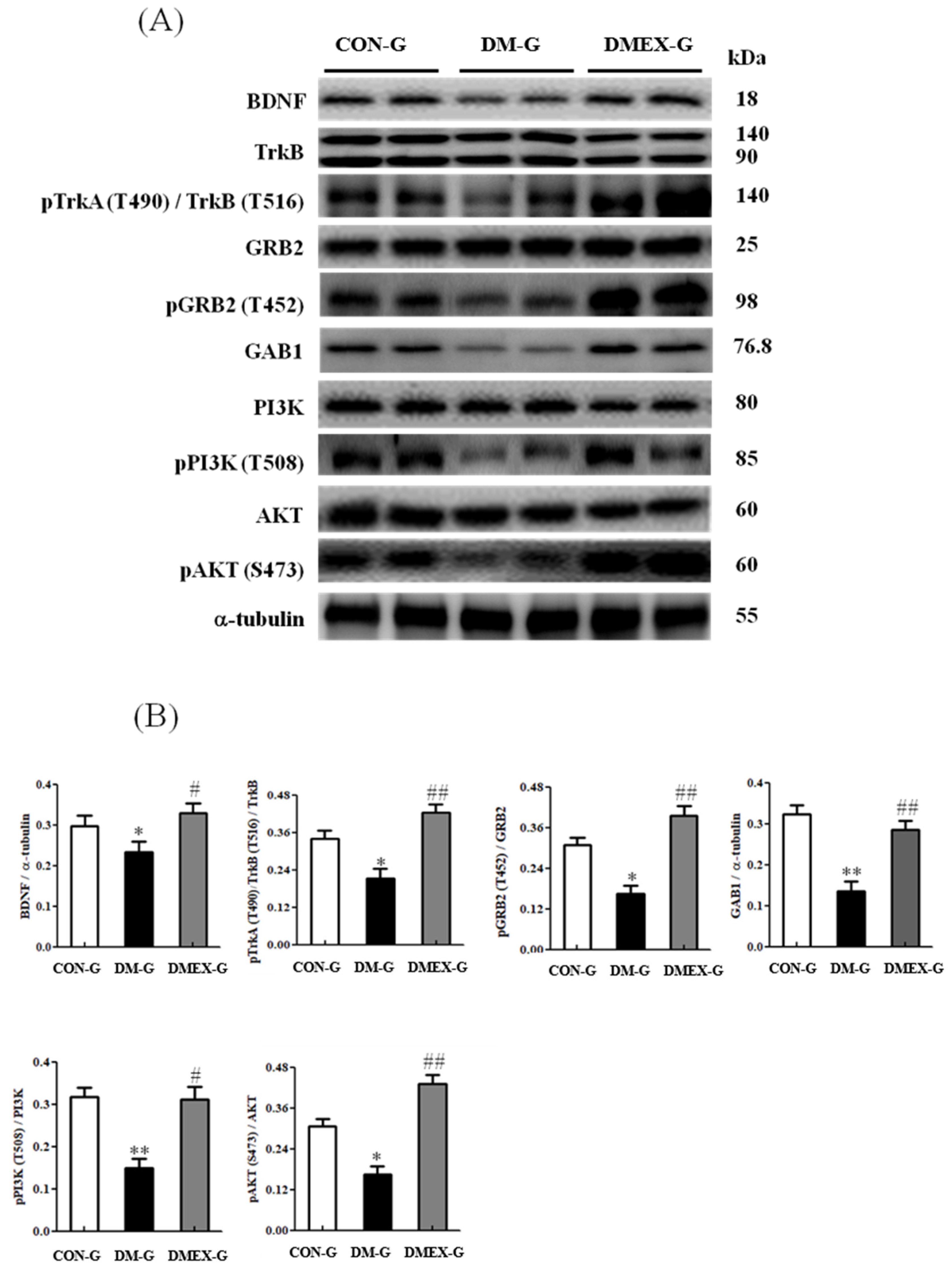
3.4. Neural BDNF/TrkB and PI3K/AKT Survival Pathway
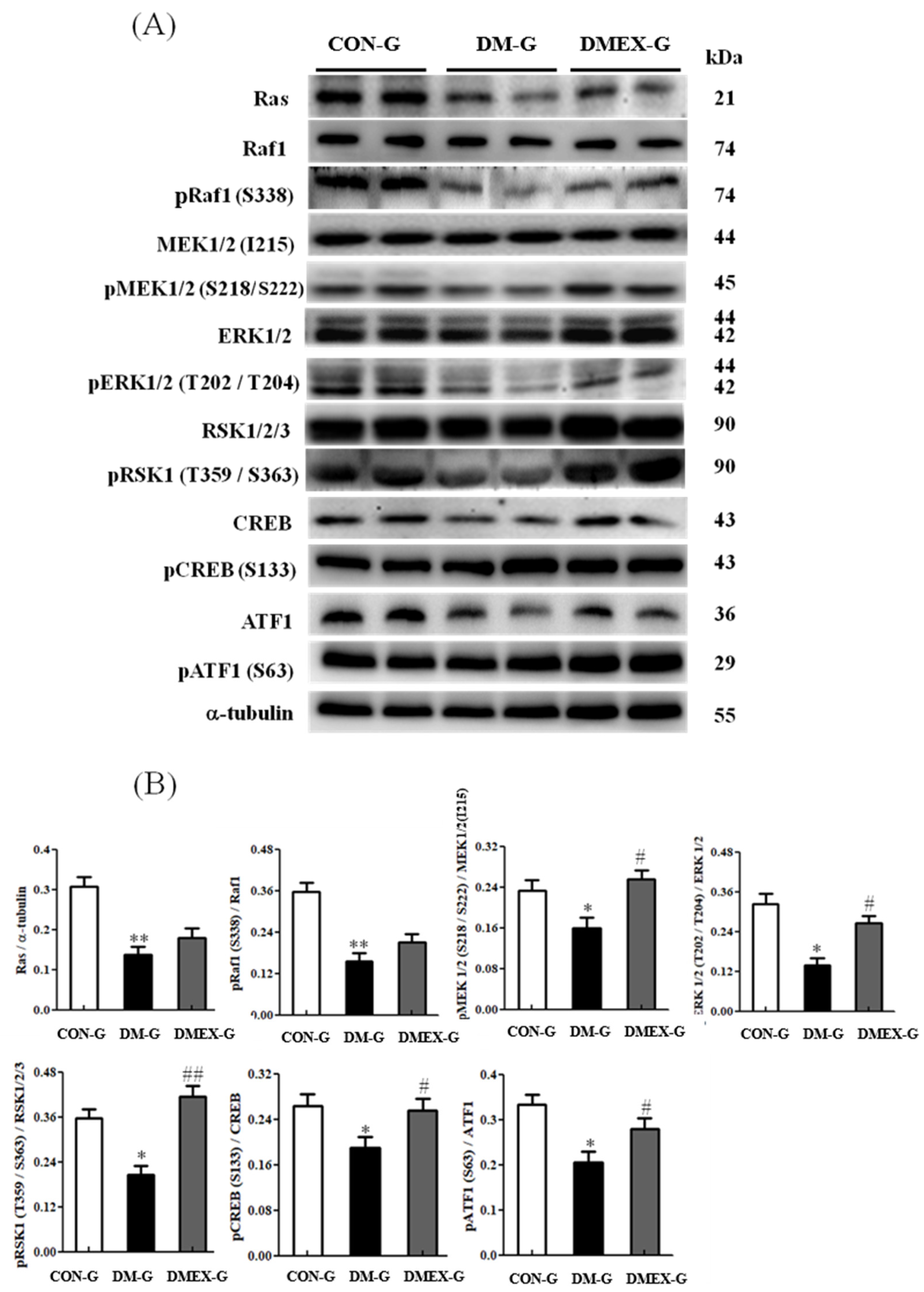
3.5. Neural Ras/MEK/MAPK/ERK Signaling Pathway
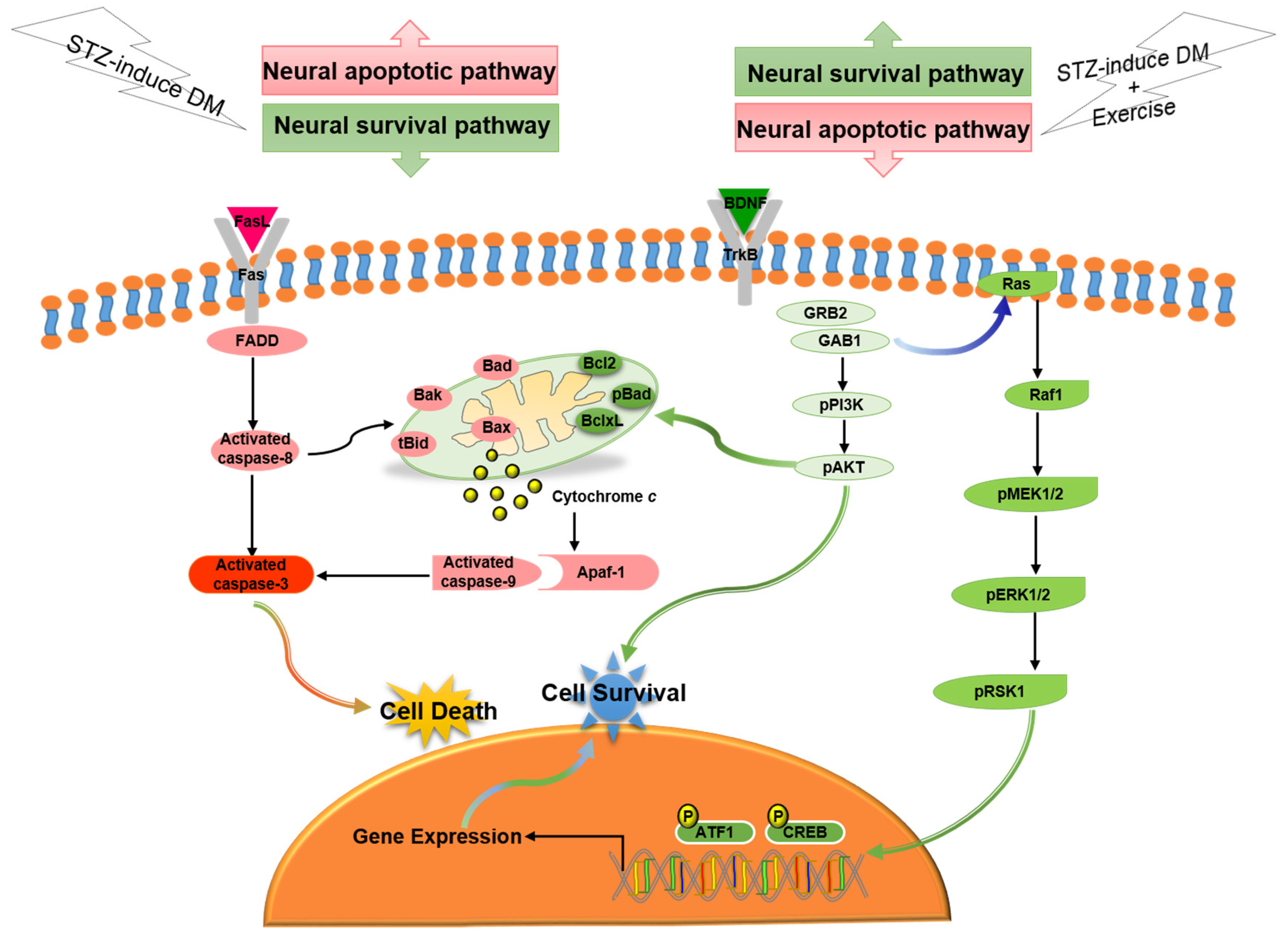
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zimmet, P.; Alberti, K.G.M.M.; Shaw, J. Global and societal implications of the diabetes epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Manschot, S.M.; Biessels, G.J.; Rutten, G.E.H.M.; Kessels, R.C.P.; Gispen, W.H.; Kappelle, L.J. Peripheral and central neurologic complications in type 2 diabetes mellitus: No association in individual patients. J. Neurol. Sci. 2008, 264, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Toth, C. Diabetes and neurodegeneration in the brain. Handb. Clin. Neurol. 2014, 126, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.G.; Sima, A.A. C-peptide and central nervous system complications in diabetes. Exp. Diabesity Res. 2004, 5, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Moheet, A.; Mangia, S.; Seaquist, E.R. Impact of diabetes on cognitive function and brain structure. Ann. N. Y. Acad. Sci. 2015, 1353, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.; Han, X.R.; Wang, Y.J.; Wang, S.; Shen, M.; Zhang, Z.F.; Fan, S.H.; Shan, Q.; Wang, L.; Li, M.Q.; et al. Down-regulated long non-coding RNA ANRIL restores the learning and memory abilities and rescues hippocampal pyramidal neurons from apoptosis in streptozotocin-induced diabetic rats via the NF-kappaB signaling pathway. J. Cell. Biochem. 2018, 119, 5821–5833. [Google Scholar] [CrossRef]
- Li, Z.G.; Zhang, W.; Sima, A.A. The role of impaired insulin/IGF action in primary diabetic encephalopathy. Brain Res. 2005, 1037, 12–24. [Google Scholar] [CrossRef]
- Meng, Y.; Wang, W.; Kang, J.; Wang, X.; Sun, L. Role of the PI3K/AKT signalling pathway in apoptotic cell death in the cerebral cortex of streptozotocin-induced diabetic rats. Exp. Ther. Med. 2017, 13, 2417–2422. [Google Scholar] [CrossRef] [Green Version]
- Bordier, L.; Doucet, J.; Boudet, J.; Bauduceau, B. Update on cognitive decline and dementia in elderly patients with diabetes. Diabetes Metab. 2014, 40, 331–337. [Google Scholar] [CrossRef]
- Ninomiya, T. Diabetes mellitus and dementia. Curr. Diabetes Rep. 2014, 14, 487. [Google Scholar] [CrossRef]
- Lima Giacobbo, B.; Doorduin, J.; Klein, H.C.; Dierckx, R.; Bromberg, E.; de Vries, E.F.J. Brain-Derived Neurotrophic Factor in Brain Disorders: Focus on Neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisonpierre, P.C.; Le Beau, M.M.; Espinosa, R., 3rd; Ip, N.Y.; Belluscio, L.; de la Monte, S.M.; Squinto, S.; Furth, M.E.; Yancopoulos, G.D. Human and rat brain-derived neurotrophic factor and neurotrophin-3: Gene structures, distributions, and chromosomal localizations. Genomics 1991, 10, 558–568. [Google Scholar] [CrossRef]
- Noble, E.E.; Billington, C.J.; Kotz, C.M.; Wang, C. The lighter side of BDNF. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1053–R1069. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. The biology of neurotrophins, signalling pathways, and functional peptide mimetics of neurotrophins and their receptors. CNS Neurol. Disord.-Drug Targets 2008, 7, 46–62. [Google Scholar] [CrossRef]
- Patapoutian, A.; Reichardt, L.F. Trk receptors: Mediators of neurotrophin action. Curr. Opin. Neurobiol. 2001, 11, 272–280. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, J.; Xu, M.; Wang, Y.; Zhang, M.; Zhou, Y. Resveratrol Improves Cognitive Impairment by Regulating Apoptosis and Synaptic Plasticity in Streptozotocin-Induced Diabetic Rats. Cell. Physiol. Biochem. 2016, 40, 1670–1677. [Google Scholar] [CrossRef]
- Nitta, A.; Murai, R.; Suzuki, N.; Ito, H.; Nomoto, H.; Katoh, G.; Furukawa, Y.; Furukawa, S. Diabetic neuropathies in brain are induced by deficiency of BDNF. Neurotoxicol. Teratol. 2002, 24, 695–701. [Google Scholar] [CrossRef]
- Liu, P.; Li, H.; Wang, Y.; Su, X.; Li, Y.; Yan, M.; Ma, L.; Che, H. Harmine Ameliorates Cognitive Impairment by Inhibiting NLRP3 Inflammasome Activation and Enhancing the BDNF/TrkB Signaling Pathway in STZ-Induced Diabetic Rats. Front. Pharmcol. 2020, 11, 535. [Google Scholar] [CrossRef]
- Chan, C.B.; Ahuja, P.; Ye, K. Developing Insulin and BDNF Mimetics for Diabetes Therapy. Curr. Top. Med. Chem. 2019, 19, 2188–2204. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.F.; Zhang, J.; Liu, X.Y.; Fang, H.; Tian, L.B.; Zhou, D.H.; Kosten, T.R.; Zhang, X.Y. Low BDNF is associated with cognitive deficits in patients with type 2 diabetes. Psychopharmacology 2013, 227, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, K.S.; Nielsen, A.R.; Krogh-Madsen, R.; Plomgaard, P.; Rasmussen, P.; Erikstrup, C.; Fischer, C.P.; Lindegaard, B.; Petersen, A.M.; Taudorf, S.; et al. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia 2007, 50, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Ferris, L.T.; Williams, J.S.; Shen, C.L. The effect of acute exercise on serum brain-derived neurotrophic factor levels and cognitive function. Med. Sci. Sports Exerc. 2007, 39, 728–734. [Google Scholar] [CrossRef]
- Seifert, T.; Brassard, P.; Wissenberg, M.; Rasmussen, P.; Nordby, P.; Stallknecht, B.; Adser, H.; Jakobsen, A.H.; Pilegaard, H.; Nielsen, H.B.; et al. Endurance training enhances BDNF release from the human brain. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2010, 298, R372–R377. [Google Scholar] [CrossRef] [Green Version]
- Vaynman, S.; Ying, Z.; Gomez-Pinilla, F. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur. J. Neurosci. 2004, 20, 2580–2590. [Google Scholar] [CrossRef]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp. Physiol. 2009, 94, 1062–1069. [Google Scholar] [CrossRef]
- Lang, X.; Zhao, N.; He, Q.; Li, X.; Sun, C.; Zhang, X. Treadmill exercise mitigates neuroinflammation and increases BDNF via activation of SIRT1 signaling in a mouse model of T2DM. Brain Res. Bull. 2020, 165, 30–39. [Google Scholar] [CrossRef]
- Żebrowska, A.; Hall, B.; Maszczyk, A.; Banas, R.; Urban, J. Brain-derived neurotrophic factor, insulin like growth factor-1 and inflammatory cytokine responses to continuous and intermittent exercise in patients with type 1 diabetes. Diabetes Res. Clin. Pract. 2018, 144, 126–136. [Google Scholar] [CrossRef]
- Tang, L.; Kang, Y.T.; Yin, B.; Sun, L.J.; Fan, X.S. Effects of weight-bearing ladder and aerobic treadmill exercise on learning and memory ability of diabetic rats and its mechanism. Chin. J. Appl. Physiol. 2017, 33, 436–440. [Google Scholar] [CrossRef]
- Lay, I.S.; Kuo, W.W.; Shibu, M.A.; Ho, T.J.; Cheng, S.M.; Day, C.H.; Ban, B.; Wang, S.; Li, Q.; Huang, C.Y. Exercise training restores IGFIR survival signaling in d-galactose induced-aging rats to suppress cardiac apoptosis. J. Adv. Res. 2021, 28, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.M.; Ho, T.J.; Yang, A.L.; Chen, I.J.; Kao, C.L.; Wu, F.N.; Lin, J.A.; Kuo, C.H.; Ou, H.C.; Huang, C.Y.; et al. Exercise training enhances cardiac IGFI-R/PI3K/Akt and Bcl-2 family associated pro-survival pathways in streptozotocin-induced diabetic rats. Int. J. Cardiol. 2013, 167, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.M.; Cheng, Y.J.; Wu, L.Y.; Kuo, C.H.; Lee, Y.S.; Wu, M.C.; Huang, C.Y.; Ting, H.; Lee, S.D. Activated apoptotic and anti-survival effects on rat hearts with fructose induced metabolic syndrome. Cell Biochem. Funct. 2014, 32, 133–141. [Google Scholar] [CrossRef] [PubMed]
- de Lemos, E.T.; Reis, F.; Baptista, S.; Pinto, R.; Sepodes, B.; Vala, H.; Rocha-Pereira, P.; da Silva, G.C.; Teixeira, N.; Silva, A.S.; et al. Exercise training decreases proinflammatory profile in Zucker diabetic (type 2) fatty rats. Nutrition 2009, 25, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Anti-inflammatory nature of exercise. Nutrition 2004, 20, 323–326. [Google Scholar] [CrossRef]
- UN, D. Molecular, biochemical, and physiological basis of beneficial actions of exercise. In Diet and Exercise in Cognitive Function and Neurological Diseases, 1st ed.; Farooqui, T., Farooqui, A.A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 183–204. [Google Scholar]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef]
- Liu, J.; Feng, L.; Ma, D.; Zhang, M.; Gu, J.; Wang, S.; Fu, Q.; Song, Y.; Lan, Z.; Qu, R.; et al. Neuroprotective effect of paeonol on cognition deficits of diabetic encephalopathy in streptozotocin-induced diabetic rat. Neurosci. Lett. 2013, 549, 63–68. [Google Scholar] [CrossRef]
- Cheng, S.M.; Ho, Y.J.; Yu, S.H.; Liu, Y.F.; Lin, Y.Y.; Huang, C.Y.; Ou, H.C.; Huang, H.L.; Lee, S.D. Anti-Apoptotic Effects of Diosgenin in D-Galactose-Induced Aging Brain. Am. J. Chin. Med. 2020, 48, 391–406. [Google Scholar] [CrossRef]
- Rozanska, O.; Uruska, A.; Zozulinska-Ziolkiewicz, D. Brain-Derived Neurotrophic Factor and Diabetes. Int. J. Mol. Sci. 2020, 21, 841. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, J.N.; Czernik, A.J.; Fienberg, A.A.; Greengard, P.; Sihra, T.S. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat. Neurosci. 2000, 3, 323–329. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.-M.; Lee, S.-D. Exercise Training Enhances BDNF/TrkB Signaling Pathway and Inhibits Apoptosis in Diabetic Cerebral Cortex. Int. J. Mol. Sci. 2022, 23, 6740. https://doi.org/10.3390/ijms23126740
Cheng S-M, Lee S-D. Exercise Training Enhances BDNF/TrkB Signaling Pathway and Inhibits Apoptosis in Diabetic Cerebral Cortex. International Journal of Molecular Sciences. 2022; 23(12):6740. https://doi.org/10.3390/ijms23126740
Chicago/Turabian StyleCheng, Shiu-Min, and Shin-Da Lee. 2022. "Exercise Training Enhances BDNF/TrkB Signaling Pathway and Inhibits Apoptosis in Diabetic Cerebral Cortex" International Journal of Molecular Sciences 23, no. 12: 6740. https://doi.org/10.3390/ijms23126740
APA StyleCheng, S.-M., & Lee, S.-D. (2022). Exercise Training Enhances BDNF/TrkB Signaling Pathway and Inhibits Apoptosis in Diabetic Cerebral Cortex. International Journal of Molecular Sciences, 23(12), 6740. https://doi.org/10.3390/ijms23126740