
Integrated Methylome and Transcriptome Analysis Provides Insights into the DNA Methylation Underlying the Mechanism of Cytoplasmic Male Sterility in Kenaf (Hibiscus cannabinus L.)

 and
and 
Abstract
:1. Introduction
2. Results
2.1. Hormones, Photosynthetic Products and ATP Contents
2.2. Genomic DNA Methylation Levels in Kenaf
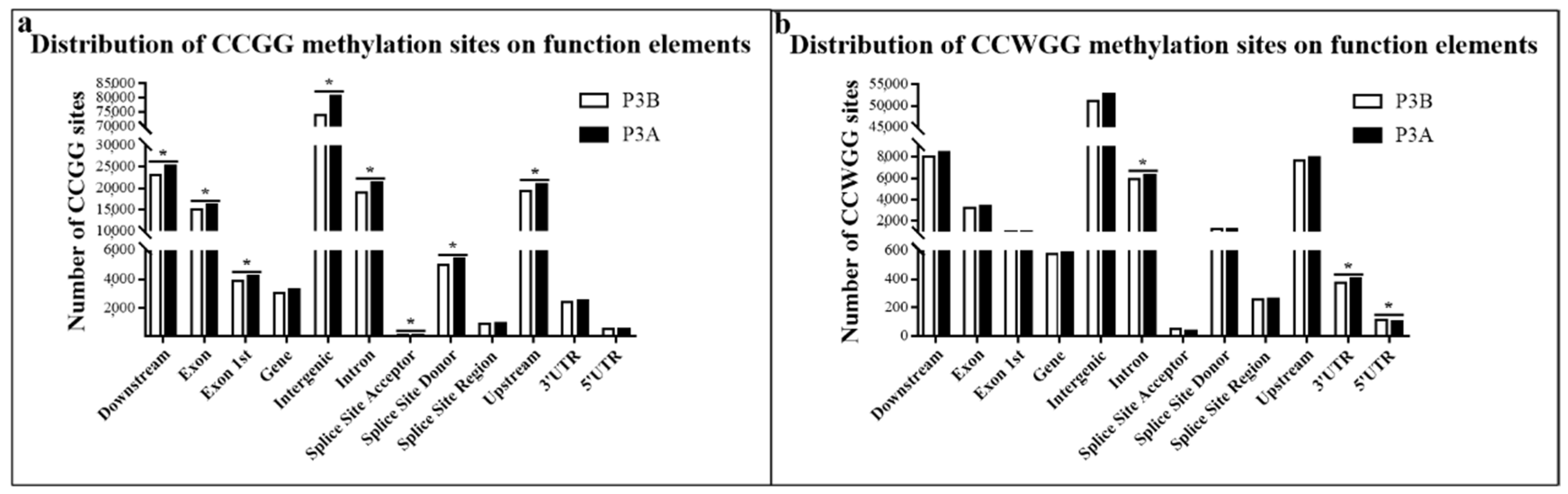
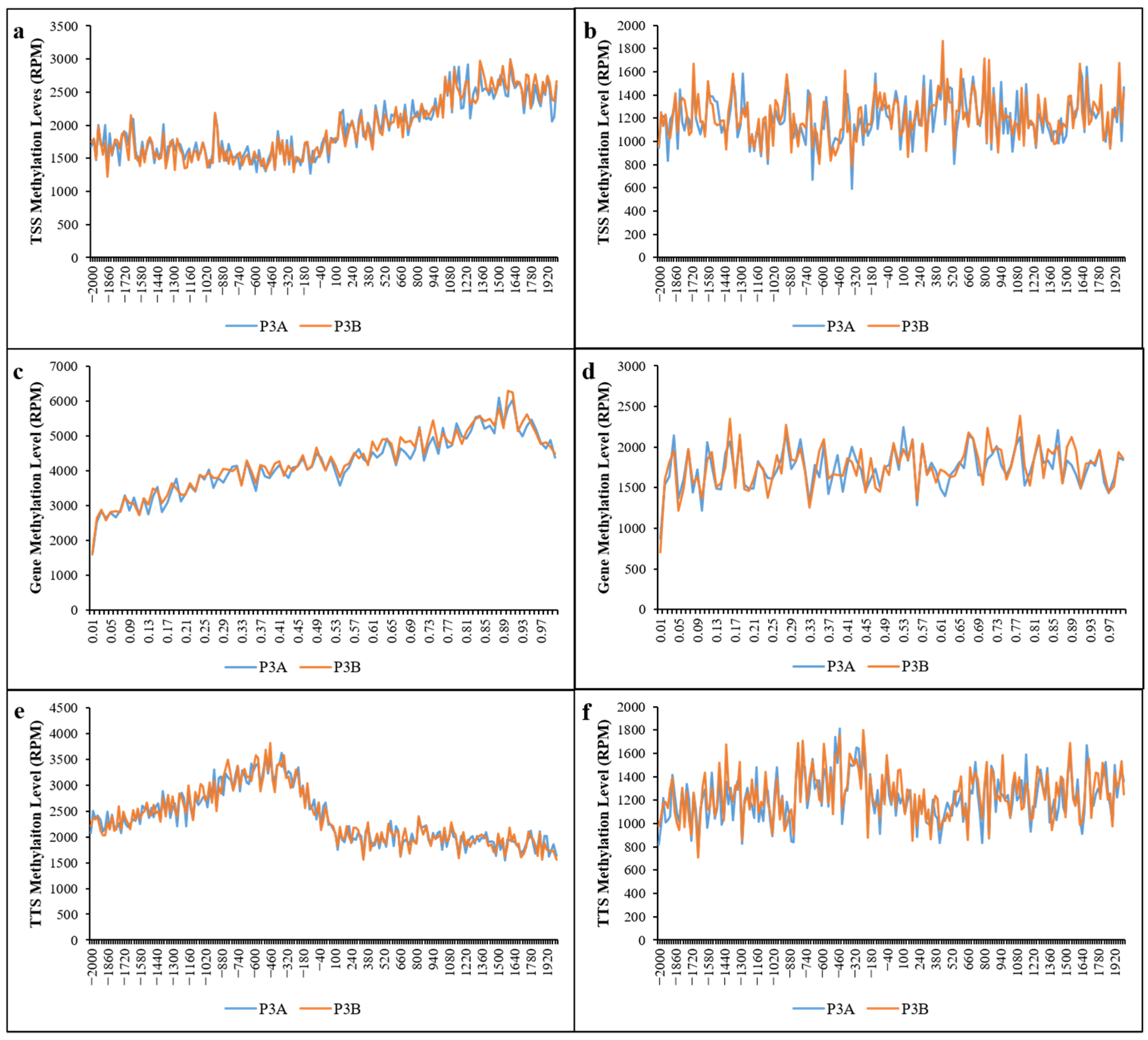
2.3. Distribution of Methylated Sites in Different Functional Elements and Gene Regions
2.4. Differentially Methylated Sites (DMSs) and Differentially Methylated Genes (DMGs) between P3A and P3B
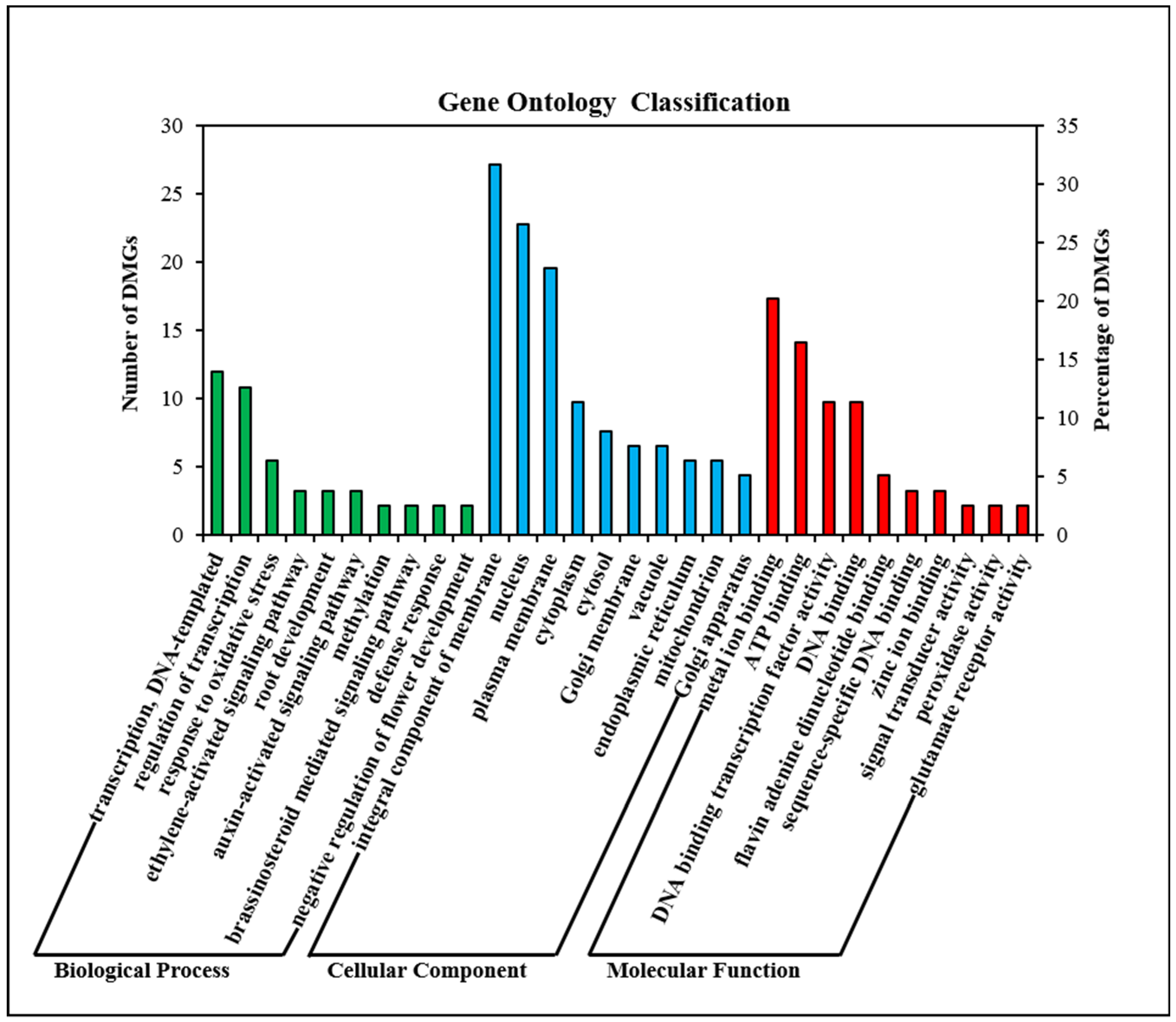
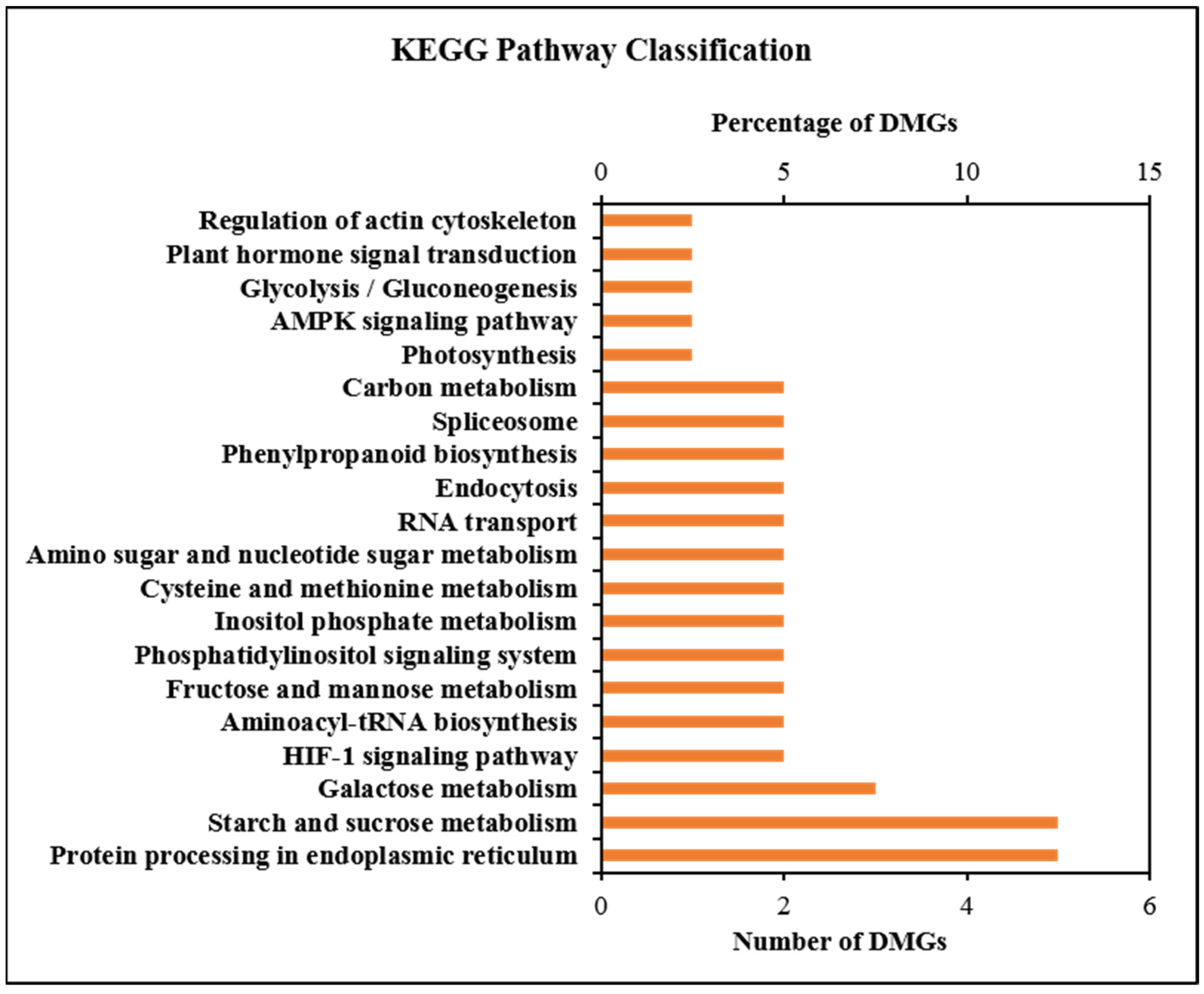
2.5. GO and KEGG Enrichment Analysis of DMGs
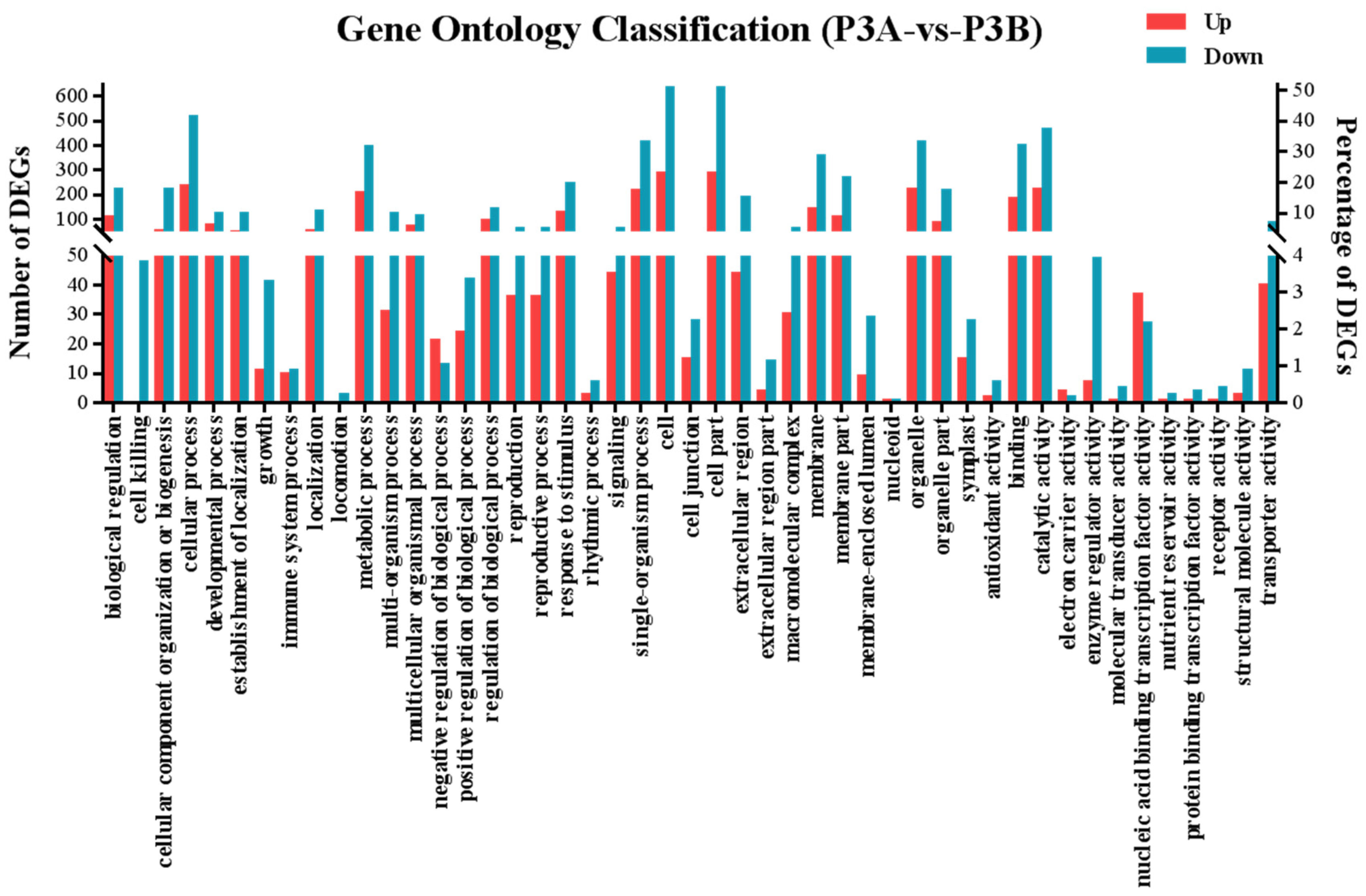
2.6. Differentially Expressed Gene (DEG) Analysis
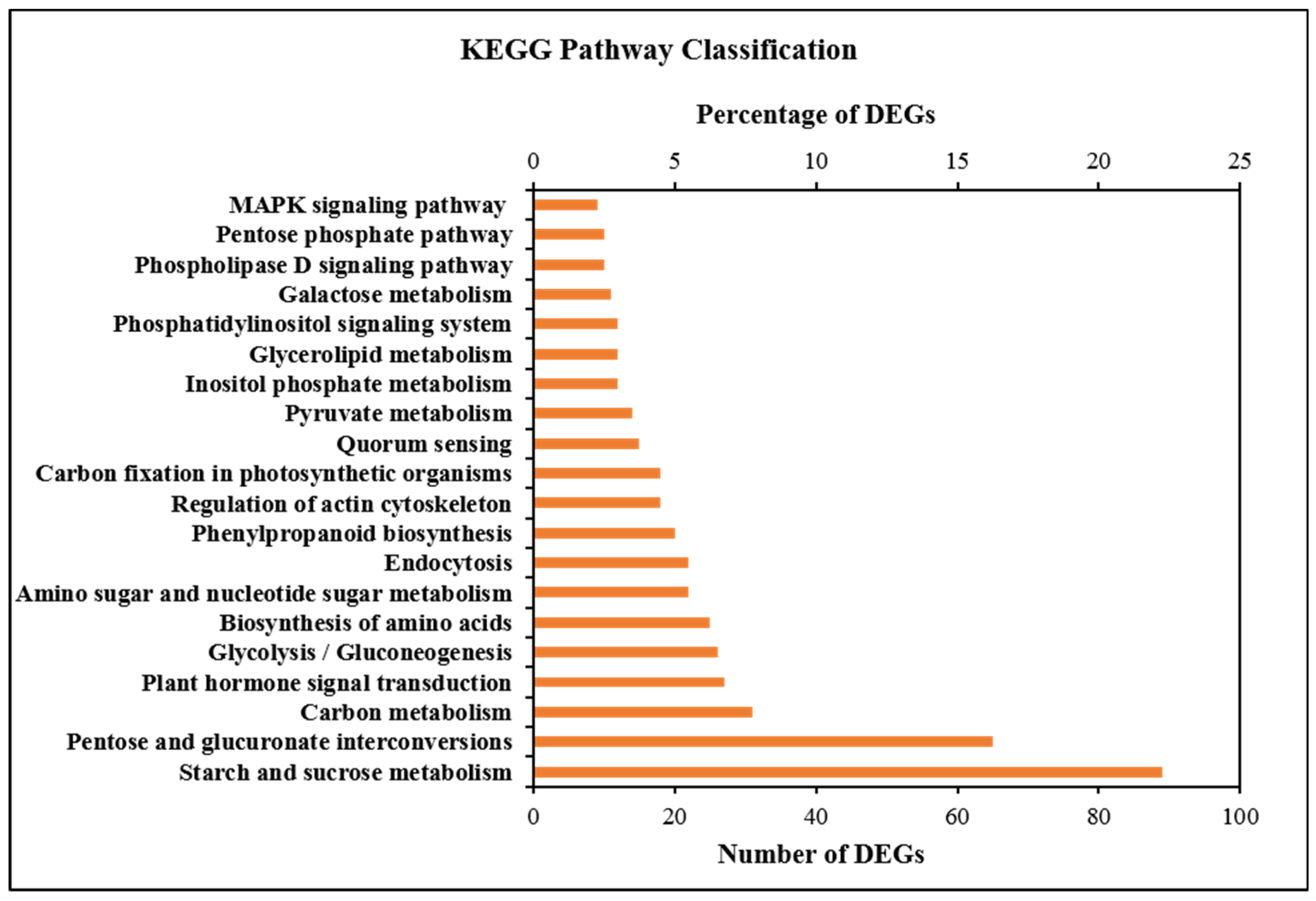
2.7. GO and KEGG Enrichment Analysis of DEGs
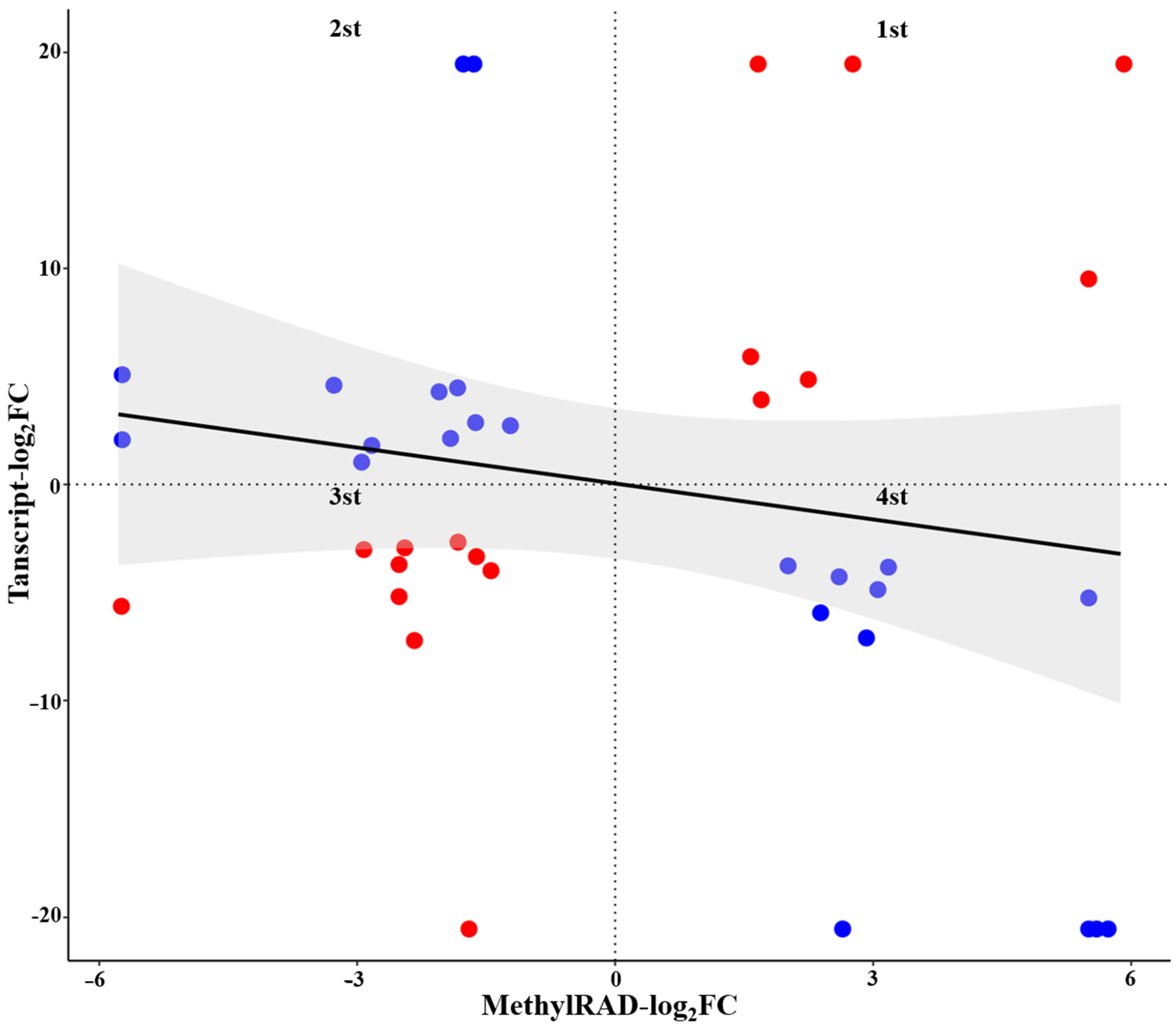
2.8. Integrated Analysis of DMGs and DEGs
2.9. qRT–PCR Verification
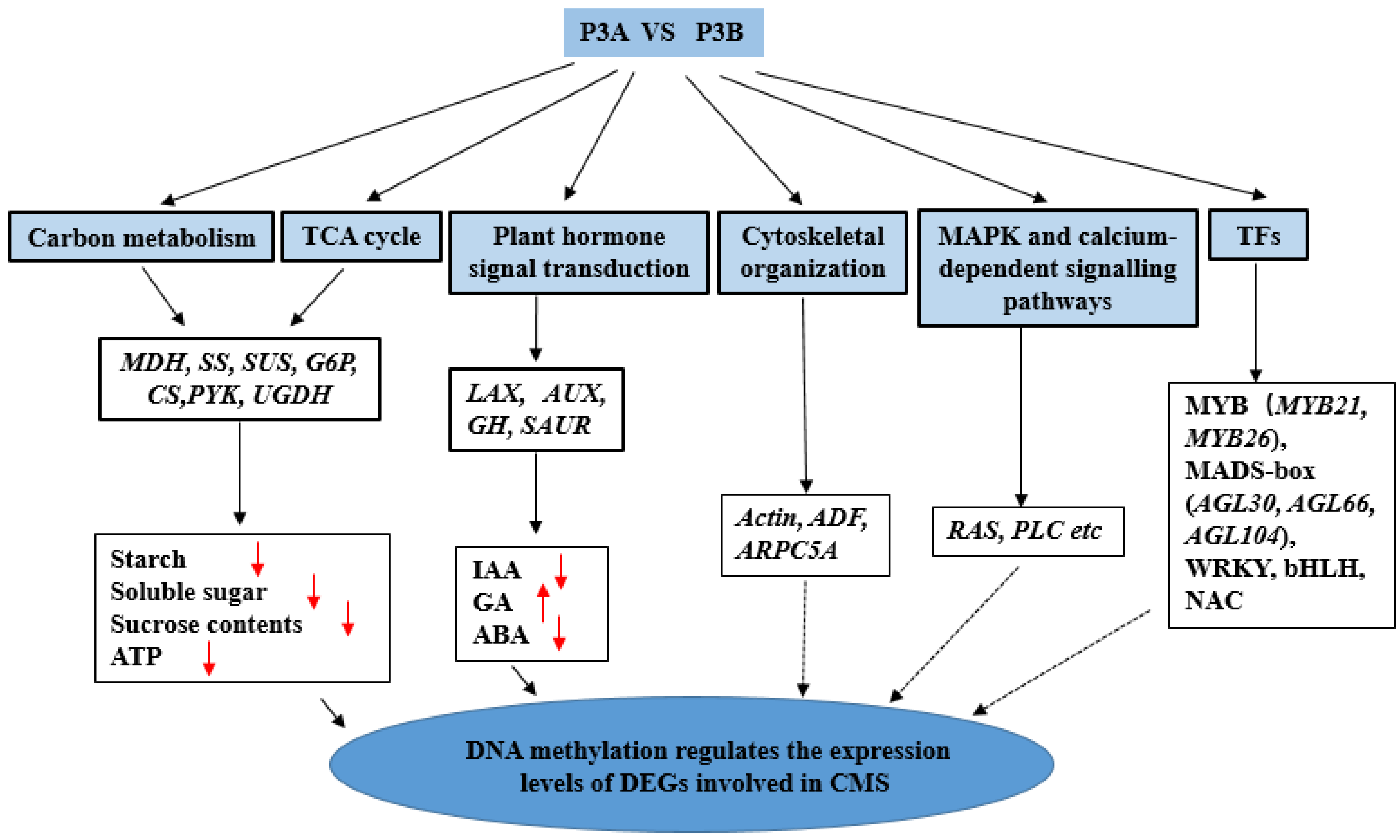
3. Discussion
3.1. DMGs and DEGs Related to Carbohydrate Metabolism Processes and the TCA Cycle
3.2. DMGs and DEGs Related to Plant Hormone Signal Transduction
3.3. DMGs and DEGs Related to Cytoskeletal Organization
3.4. DMGs and DEGs Related to Transcription Factors (TFs)
3.5. DMGs and DEGs Involved in MAPK and Calcium-Dependent Signalling Pathway
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Physiological Indexes
4.3. MethylRAD Library Construction, Sequencing, and Data Analysis
4.4. cDNA Library Preparation, Sequencing and Data Analysis
4.5. qRT–PCR Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chen, P.; Shi, Q.; Liang, Z.; Lu, H.; Li, R. Comparative profile analysis reveals differentially expressed microRNAs regulate anther and pollen development in kenaf cytoplasmic male sterility line. Genome 2019, 62, 455–466. [Google Scholar] [CrossRef]
- Chen, P.; Li, Z.; Luo, D.; Jia, R.; Lu, H.; Tang, M.; Hu, Y.; Yue, J.; Huang, Z. Comparative transcriptomic analysis reveals key genes and pathways in two different cadmium tolerance kenaf (Hibiscus cannabinus L.) cultivars. Chemosphere 2020, 263, 128211. [Google Scholar] [CrossRef]
- Kashif, M.H.; Wei, F.; Tang, D.; Tang, M.; Luo, D.; Lu, H.; Li, R.; Chen, P. iTRAQ-based comparative proteomic response analysis reveals regulatory pathways and divergent protein targets associated with salt-stress tolerance in kenaf (Hibiscus cannabinus L.). Ind. Crops. Prod. 2020, 153, 112566. [Google Scholar] [CrossRef]
- Zhou, R.; Zhang, X.; Zhang, J.; Gan, Z.; Wei, H. A breakthrough in kenaf cytoplasmic male sterile lines breeding and heterosis utilization. Sci. Agric. Sin. 2008, 41, 313–314. [Google Scholar]
- Chen, P.; Liao, J.; Huang, Z.; Li, R.; Zhao, Y.; Ran, S.; Zhou, R. Comparative proteomics study on anther mitochondria between cytoplasmic male sterility line and its maintainer in kenaf (Hibiscus cannabinus L.). Crop Sci. 2014, 54, 1103–1114. [Google Scholar] [CrossRef]
- Chen, P.; Wei, F.; Li, R.; Li, Z.Q.; Kashif, M.H.; Zhou, R.Y. Comparative acetylenic analysis reveals differentially acetylated proteins regulating anther and pollen development in kenaf cytoplasmic male sterility line. Physiol. Plant. 2018, 166, 960–978. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, R.; Zhou, R. Comparative phosphoproteomic analysis reveals differentially phosphorylated proteins regulate anther and pollen development in kenaf cytoplasmic male sterility line. Amino Acids 2018, 50, 841–862. [Google Scholar] [CrossRef]
- Eveland, A.L.; Jackson, D.P. Sugars, signaling, and plant development. J. Exp. Bot. 2012, 63, 3367–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, M.; Godt, D.E.; Guivarc’h, A.; Kahmann, U.; Chriqui, D.; Roitsch, T. Induction of male sterility in plants by metabolic engineering of the carbohydrate supply. Proc. Natl. Acad. Sci. USA 2001, 98, 6522–6527. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Tian, H.; Huang, Y.Q.; Hu, J.; Ji, Y.X.; Li, S.Q.; Feng, Y.Q.; Guo, L.; Zhu, Y.G. Alterations of mitochondrial protein assembly and jasmonic acid biosynthesis pathway in Honglian (HL)-type cytoplasmic male sterility rice. J. Biol. Chem. 2012, 287, 40051–40060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, S.N.; Dennis, E.S.; Dolferus, R. ABA regulates apoplastic sugar transport and is a potential signal for cold induced pollen sterility in rice. Plant Cell Physiol. 2007, 48, 1319–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liang, W.; Yang, X.; Luo, X.; Jiang, N.; Ma, H.; Zhang, D. Carbon starved anther encodes a MYB domain protein that regulates sugar partitioning required for rice pollen development. Plant Cell 2010, 22, 672–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, P.; Edqvist, J.; Farbos, I.; Glimelius, K. Male-sterile tobacco displays abnormal mitochondrial atp1 transcript accumulation and reduced floral atp/adp ratio. Plant Mol. Biol. 2000, 42, 531. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, R.T.; Knorpp, C.; Glimelius, K. Modified sucrose, starch, and ATP levels in two alloplasmic male-sterile lines of B. napus. J. Exp. Bot. 2005, 56, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Khan, A.; Li, B.; Zheng, J.; Dawar, F.U.; Li, M.; You, J.; Li, Z.; Zhou, R. Deviant DNA methylation play a key role in the pollen abortion of Gossypium barbadense L. cytoplasmic male sterility. Ind. Crops Prod. 2020, 154, 112622. [Google Scholar] [CrossRef]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.L.; Chen, H.; Henderson, L.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Hewezi, T. Editorial: Epigenetic regulation of plant development and stress responses. Plant Cell Rep. 2017, 37, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Mou, Z. Elongator and its epigenetic role in plant development and responses to abiotic and biotic stresses. Front. Plant Sci. 2015, 6, 296. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Yan, W.; He, J.; Li, Y.; Zhang, H.; Peng, H.; Wu, X. DNA methylation affected by male sterile cytoplasm in rice (Oryza sativa L.). Mol. Breed. 2013, 31, 719–727. [Google Scholar] [CrossRef]
- Chen, X.; Hu, J.; Zhang, H.; Ding, Y. DNA methylation changes in photoperiod-thermo-sensitive male sterile rice PA64S under two different conditions. Gene 2014, 537, 143–148. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, Y.; Lu, Y.; Wang, J.; Zhang, S.; Lan, H.; Rong, T. DNA methylation analysis of sterile and fertile CMS-C hybrids and their parents in maize. J. Plant Biochem. Biot. 2016, 25, 3–11. [Google Scholar] [CrossRef]
- Li, Y.; Ding, X.; Wang, X.; He, T.; Zhang, H.; Yang, L.; Wang, T.; Chen, L.; Gai, J.; Yang, S. Genome-wide comparative analysis of DNA methylation between soybean cytoplasmic male-sterile line NJCMS5A and its maintainer NJCMS5B. BMC Genom. 2017, 8, 596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Li, Y.; Li, J.; Zhang, H.; Ding, X.; He, T.; Gai, J.; Yang, S. Genome-wide analysis of DNA methylation to identify genes and pathways associated with male sterility in soybean. Mol. Breed. 2018, 38, 118. [Google Scholar] [CrossRef]
- Krizek, B.A. Auxin regulation of Arabidopsis flower development involves members of the AINTEGUMENTA-LIKE/PLETHORA (AIL/PLT) family. J. Exp. Bot. 2011, 62, 3311–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Zhao, Y. A role for auxin in flower development. J. Integr. Plant Biol. 2007, 49, 99–104. [Google Scholar] [CrossRef]
- Law, J.A.; Jcaobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Stitt, M.; Zeeman, S.C. Starch turnover: Pathways, regulation and role in growth. Curr. Opin. Plant Biol. 2012, 15, 282–292. [Google Scholar] [CrossRef]
- Lastdrager, J.; Hanson, J.; Smeekens, S. Sugar signals and the control of plant growth and development. J. Exp. Bot. 2014, 65, 799–807. [Google Scholar] [CrossRef]
- Bizaquerz, M.A.; Green, R.; Nillsson, O.; Sussman, M.R.; Weigel, D. Gibberellins promote flowering of arabidopsis by activating the LEAFY promoter. Plant Cell 1998, 10, 791–800. [Google Scholar]
- Zhang, J.; Zhang, C.; Cheng, Y.; Qi, L.; Wang, S.; Hou, X. Microtubule and male sterility in a gene-cytoplasmic male sterile line of non-heading Chinese cabbage. J. Sci. Food Agric. 2012, 92, 3046–3054. [Google Scholar] [CrossRef]
- Fu, S.; Zhang, Y.N.; Wang, Y.Y.; Zhu, X.Y.; Tian, H.Q. Defects in cytoskeletal microtubule deployment of microsporocytes contribute to fertility loss in genic male-sterile Chinese cabbage. Plant Reprod. 2013, 26, 55–61. [Google Scholar] [CrossRef]
- Xu, C.; Liu, Z.; Zhang, L.; Zhao, C.; Yuan, S.; Zhang, F. Organization of actin cytoskeleton during meiosis I in a wheat thermo-sensitive genic male sterile line. Protoplasma 2013, 250, 415–422. [Google Scholar] [CrossRef]
- Yan, L.; Liu, G.; Xiao, X. From pollen actin to crop male sterility. Sci. Bull. 2000, 45, 784–788. [Google Scholar] [CrossRef]
- Chen, C.Y.; Wong, E.I.; Vidali, L.; Estavillo, A.; Hepler, P.K.; Wu, H.M.; Cheung, A.Y. The regulation of actin organization by actin-depolymerizing factor in elongating pollen tubes. Plant Cell 2002, 14, 2175–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Nan, Q.; Qin, T.; Qian, D.; Mao, T.; Yuan, S.; Wu, X.; Niu, Y.; Bai, Q.; An, L.; et al. Higher-ordered actin structures remodeled by Arabidopsis ACTIN-DEPOLYMERIZING FACTOR5 are important for pollen germination and pollen tube growth. Mol. Plant 2017, 10, 1065–1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, X.F.; Tang, Y.; Li, J.G.; Zhao, M.L. RNA-Seq provides new insights into the molecular events involved in “Ball-Skin versus Bladder Effect” on fruit cracking in litchi. Int. J. Mol. Sci. 2021, 22, 454. [Google Scholar] [CrossRef] [PubMed]
- Niedojadlo, K.; Niedojadlo, J.; Bednarskakozakiewicz, E. Localization of poly(A) RNA in male gametes of Arabidopsis thaliana and Hyacinthus orientalis. Acta Biol. Cracov. Bot. 2016, 58, 88. [Google Scholar]
- Mandaokar, A.; Thines, B.; Shin, B.; Lange, B.M.; Choi, G.; Koo, Y.J.; Yoo, Y.J.; Choi, Y.D.; Choi, G.; Browse, J. Transcriptional regulators of stamen development in Arabidopsis identified by transcriptional profiling. Plant J. 2006, 46, 984–1008. [Google Scholar] [CrossRef]
- Song, S.; Qi, T.; Huang, H.; Ren, Q.; Wu, D.; Chang, C.; Peng, W.; Liu, Y.; Peng, J.; Xie, D. The jasmonate-ZIM domain proteins interact with the R2R3-MYB transcription factors MYB21 and MYB24 to affect jasmonate-regulated stamen development in Arabidopsis. Plant Cell 2011, 23, 1000–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Jiang, J.; Du, M.L.; Li, L.; Wang, X.L.; Li, X.B. A cotton gene encoding MYB-Like transcription factor is specifically expressed in pollen and is involved in regulation of late anther/pollen development. Plant Cell Physiol. 2013, 54, 893–906. [Google Scholar] [CrossRef]
- Yang, C.; Xu, Z.; Song, J.; Conner, K.; Barrena, G.V.; Wilson, Z.A. Arabidopsis MYB26/MALE STERILE35 regulates secondary thickening in the endothecium and is essential for anther dehiscence. Plant Cell 2007, 19, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honys, D.; Twell, D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 2004, 5, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, S. Control of male gametophyte development. Plant Cell 2004, 16, S142–S153. [Google Scholar] [CrossRef] [PubMed]
- Hord, C.L.H.; Sun, Y.J.; Pillitteri, L.J.; Torii, K.U.; Wang, H.; Zhang, S.; Ma, H. Regulation of Arabidopsis early anther development by the mitogen-activated protein kinases, MPK3 and MPK6, and the ERECTA and related receptor-like kinases. Mol. Plant 2008, 1, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Schulz, P.; Herde, M.; Romeis, T. Calcium-dependent protein kinases: Hubs in plant stress signaling and development. Plant Physiol. 2013, 163, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Tamguney, T.M.; Collisson, E.A.; Lin, L.J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef] [Green Version]
- Tasma, I.M.; Brendel, V.; Whitham, S.A.; Bhattacharyya, M.K. Expression and evolution of the phosphoinositide-specific phospholipase C gene family in Arabidopsis thaliana. Plant Physiol. Bioch. 2008, 46, 627–637. [Google Scholar] [CrossRef]
- Dowd, P.E.; Coursol, S.; Skirpan, A.L.; Gilroy, K.S. Petunia phospholipase C1 is involved in pollen tube growth. Plant Cell 2006, 18, 1438–1453. [Google Scholar] [CrossRef] [Green Version]
- Helling, D.; Possart, A.; Cottier, S.; Klahre, U.; Kost, B. Pollen tube tip growth depends on plasma membrane polarization mediated by tobacco PLC3 activity and endocytic membrane recycling. Plant Cell 2006, 18, 3519–3534. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liu, P.; Lv, J.; Li, Y.; Cheng, T.; Zhang, L.; Xia, Y.; Sun, H.; Hu, X.; Bao, Z. Serial sequencing of isolength RAD tags for cost-efficient genome-wide profiling of genetic and epigenetic variations. Nat. Protoc. 2016, 11, 2189–2200. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Tang, D.; Li, Z.; Kashif, M.H.; Khan, A.; Lu, H.; Jia, R.; Chen, P. Molecular cloning and subcellular localization of six HDACs and their roles in response to salt and drought stress in kenaf (Hibiscus cannabinus L.). Biol. Res. 2019, 52, 20. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Enzyme Reads | Mapping Reads | Mapping Reads Ratio | CCGG Sites | CCWGG Sites | ||
|---|---|---|---|---|---|---|---|---|
| Number | Depth | Number | Depth | |||||
| P3A | 18,283,466 | 5,264,564 | 4,716,458 | 89.59% | 120,323 | 3.51 | 63,240 | 4.05 |
| P3B | 17,513,339 | 5,733,725 | 5,246,070 | 91.50% | 131,529 | 4.03 | 65,376 | 4.17 |
| Diff_Type | DMS Number | DMG Number | ||
|---|---|---|---|---|
| CCGG Site | CCWGG Site | CCGG Site | CCWGG Site | |
| Diff | 1792 | 1014 | 521 | 129 |
| Up | 786 | 446 | 274 | 39 |
| Down | 1006 | 568 | 247 | 90 |
| Gene Name | Functional Annotation | Fold Change (P3A vs. P3B) | ||
|---|---|---|---|---|
| Methylation | Transcriptome | qRT–PCR | ||
| atp8 | Phospholipid-transporting atpase 8 | 2.18 | 0.49 | 0.47 |
| COX2 | Cytochrome c oxidase subunit II | 2.19 | 4.53 | 6.39 |
| DDM1 | ATP-dependent DNA helicase DDM1 | 0 | 0.49 | 0.86 |
| DME | Transcriptional activator DEMETER | 2.16 | 0.48 | 0.6 |
| DRM1 | DNA(cytosine-5)-methyltransferase DRM1 | 2.04 | 0.47 | 0.35 |
| DRM2 | DNA(cytosine-5)-methyltransferase DRM2 | 0.72 | 0.47 | 0.72 |
| FAAH | Fatty acid amide hydrolase | 2.81 | 0.09 | 0.12 |
| FRU | Acid beta-fructofuranosidase | 5.59 | 0.19 | 0.14 |
| GA2ox6 | Gibberellin 2-oxidase | 0.03 | 104.5 | 100 |
| GLU | Beta-glucosidase bogh3b-like | 3.34 | 0.18 | 0.12 |
| IAA32 | Auxin-responsive protein IAA32 | 4.49 | 3.32 | 1.52 |
| ILR1 | IAA-amino acid hydrolase ILR1 | 0.67 | 0.1 | 0.48 |
| LIM2 | LIM domain-containing protein WLIM2b | 5.88 | 0.04 | 0.03 |
| LSD | Lysine-specific demethylase | 2.02 | 0.49 | 0.33 |
| MADS2 | MADS-box transcription factor 2 | 6.38 | 0.45 | 0.49 |
| MADS23 | MADS-box transcription factor 23 | 0.63 | 0.45 | 0.25 |
| AGL29 | MADS-box gene, AGL29 | 0.81 | 0.37 | 0.2 |
| AGL30 | MADS-box gene, AGL30 | 0.84 | 0.19 | 0.13 |
| AGL61 | MADS-box gene, AGL61 | 0.79 | 0/15.6 | 0.03 |
| AGL62 | MADS-box gene, AGL62 | 6.78 | 0.42 | 0.17 |
| AGL104 | MADS-box gene, AGL104 | 0.54 | 0.29 | 0.27 |
| MET1 | DNA methylation 1 | 1.46 | 0.5 | 0.58 |
| MYB21 | Myb21 | 0/2.1 | 14.08 | 2.81 |
| MYB26 | Myb26 | 0/1 | 0.5 | 0.47 |
| PHY | Phytochrome B | 0.58 | 0.46 | 0.33 |
| PK | Pyruvate kinase, cytosolic isozyme | 1.53 | 0.12 | 0.16 |
| ROS1 | ROS1, Repressor of silencing 1 | 1.34 | 0.46 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Luo, D.; Tang, M.; Cao, S.; Pan, J.; Zhang, W.; Hu, Y.; Yue, J.; Huang, Z.; Li, R.; et al. Integrated Methylome and Transcriptome Analysis Provides Insights into the DNA Methylation Underlying the Mechanism of Cytoplasmic Male Sterility in Kenaf (Hibiscus cannabinus L.). Int. J. Mol. Sci. 2022, 23, 6864. https://doi.org/10.3390/ijms23126864
Li Z, Luo D, Tang M, Cao S, Pan J, Zhang W, Hu Y, Yue J, Huang Z, Li R, et al. Integrated Methylome and Transcriptome Analysis Provides Insights into the DNA Methylation Underlying the Mechanism of Cytoplasmic Male Sterility in Kenaf (Hibiscus cannabinus L.). International Journal of Molecular Sciences. 2022; 23(12):6864. https://doi.org/10.3390/ijms23126864
Chicago/Turabian StyleLi, Zengqiang, Dengjie Luo, Meiqiong Tang, Shan Cao, Jiao Pan, Wenxian Zhang, Yali Hu, Jiao Yue, Zhen Huang, Ru Li, and et al. 2022. "Integrated Methylome and Transcriptome Analysis Provides Insights into the DNA Methylation Underlying the Mechanism of Cytoplasmic Male Sterility in Kenaf (Hibiscus cannabinus L.)" International Journal of Molecular Sciences 23, no. 12: 6864. https://doi.org/10.3390/ijms23126864
APA StyleLi, Z., Luo, D., Tang, M., Cao, S., Pan, J., Zhang, W., Hu, Y., Yue, J., Huang, Z., Li, R., & Chen, P. (2022). Integrated Methylome and Transcriptome Analysis Provides Insights into the DNA Methylation Underlying the Mechanism of Cytoplasmic Male Sterility in Kenaf (Hibiscus cannabinus L.). International Journal of Molecular Sciences, 23(12), 6864. https://doi.org/10.3390/ijms23126864