
Rapid Identification of Pollen- and Anther-Specific Genes in Response to High-Temperature Stress Based on Transcriptome Profiling Analysis in Cotton

 , and
, and
Abstract
:1. Introduction
2. Results
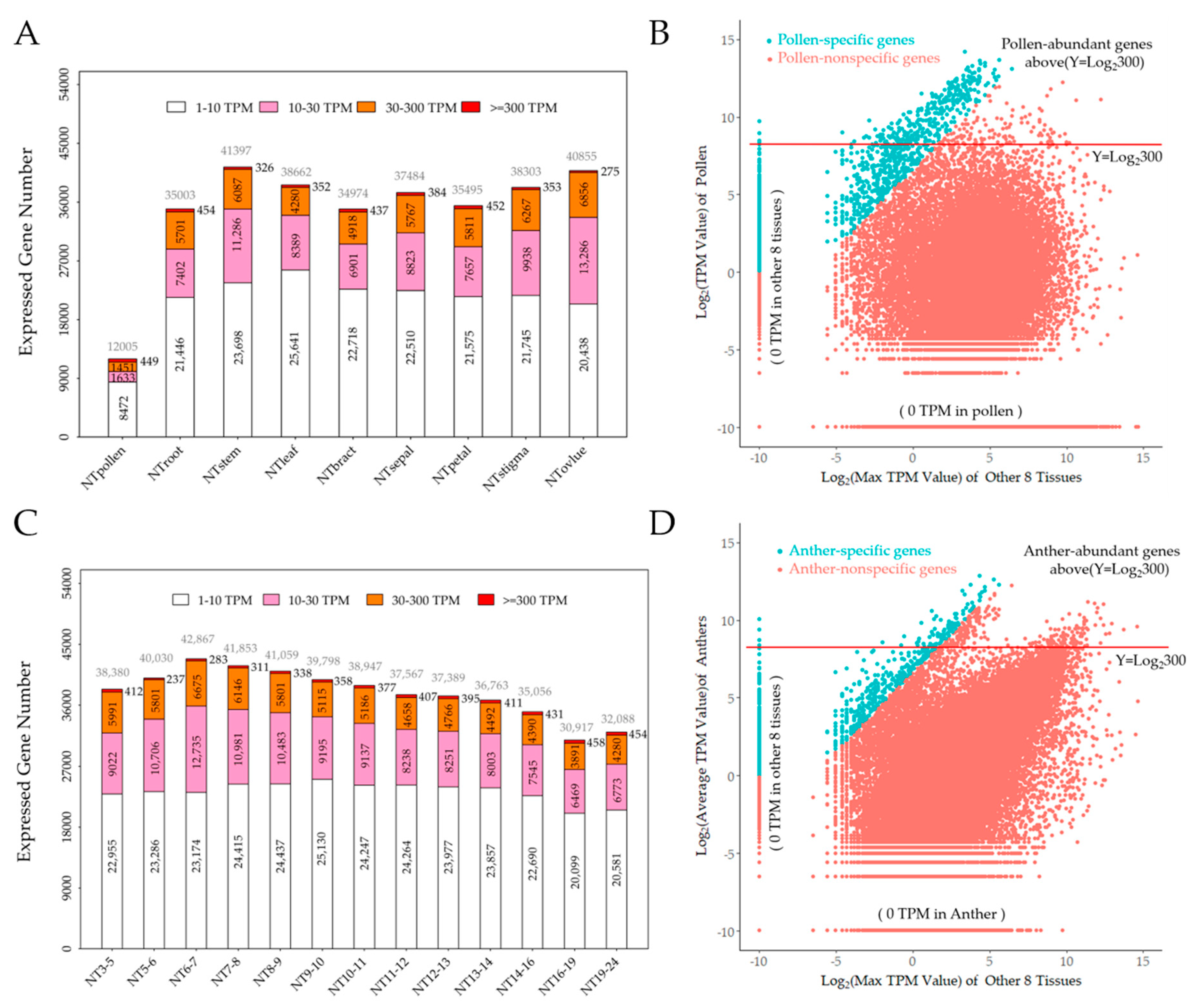
2.1. Cotton Pollen Possesses a Unique Transcriptome
2.2. Cotton Anther Express a Transformable Transcriptome
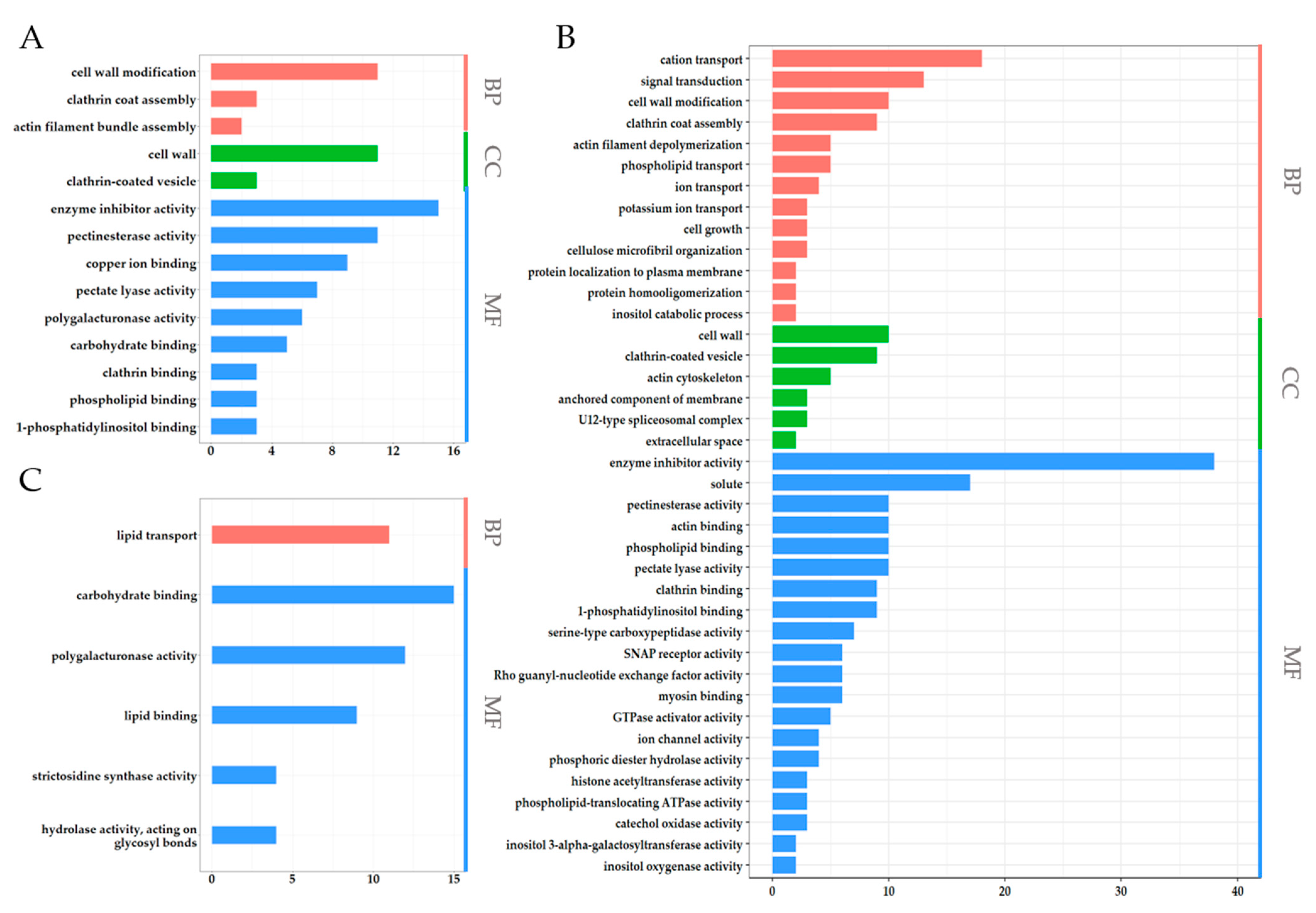
2.3. The Function of Anther- and Pollen-Specific Genes (APSGs) Are Predicted to Be More Like PSGs
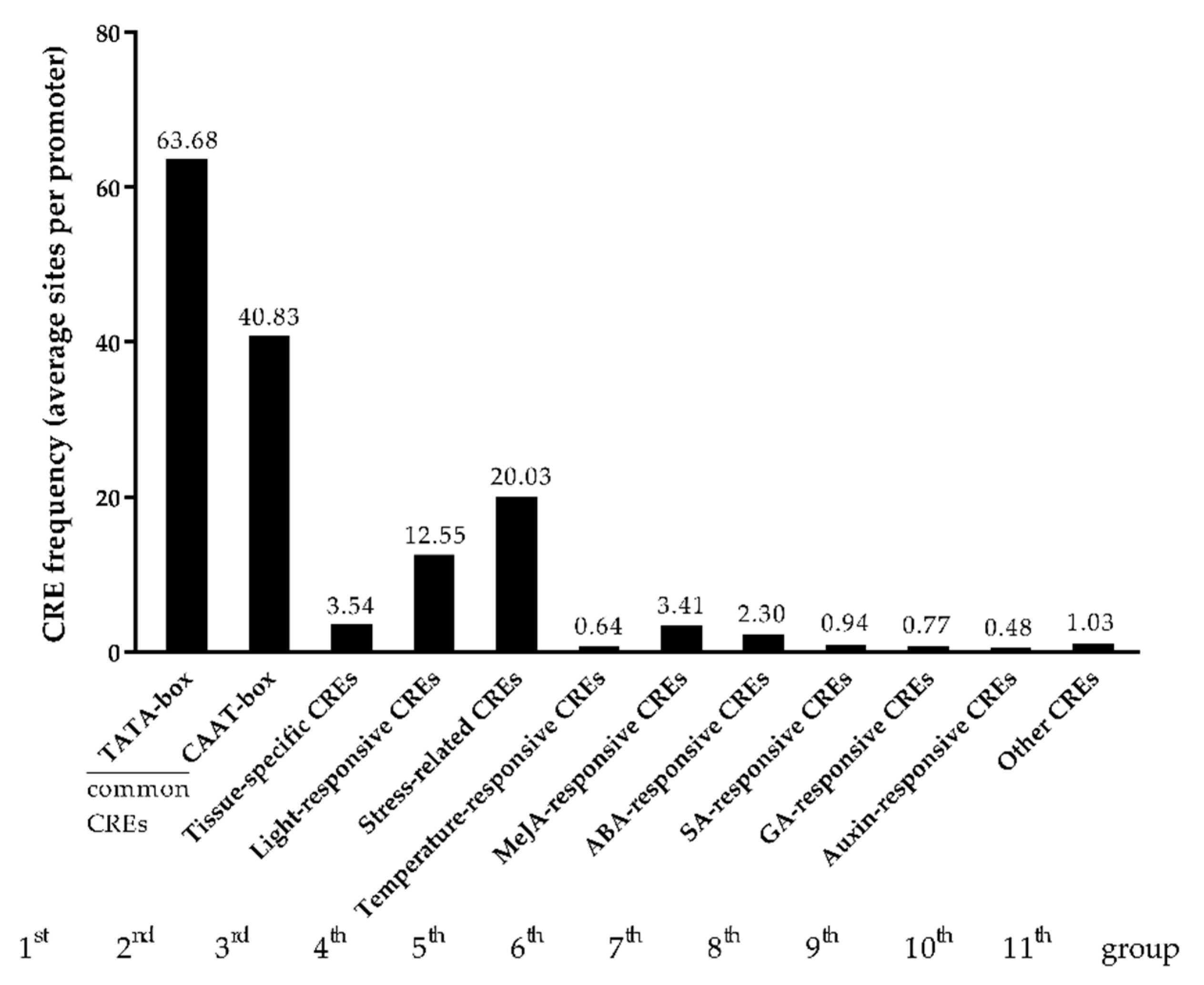
2.4. Stress-Related and Hormone-Responsive Cis-Elements Enrich in the Promoters of APSGs
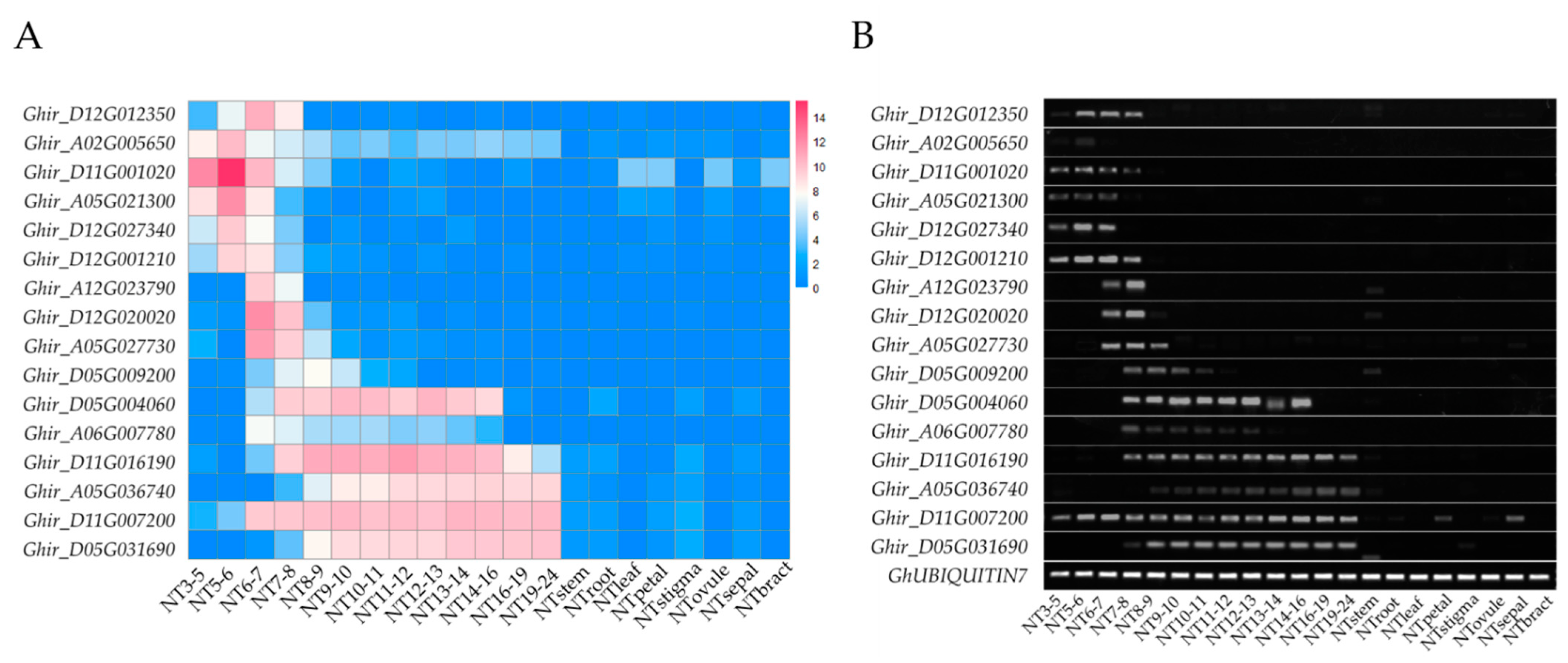
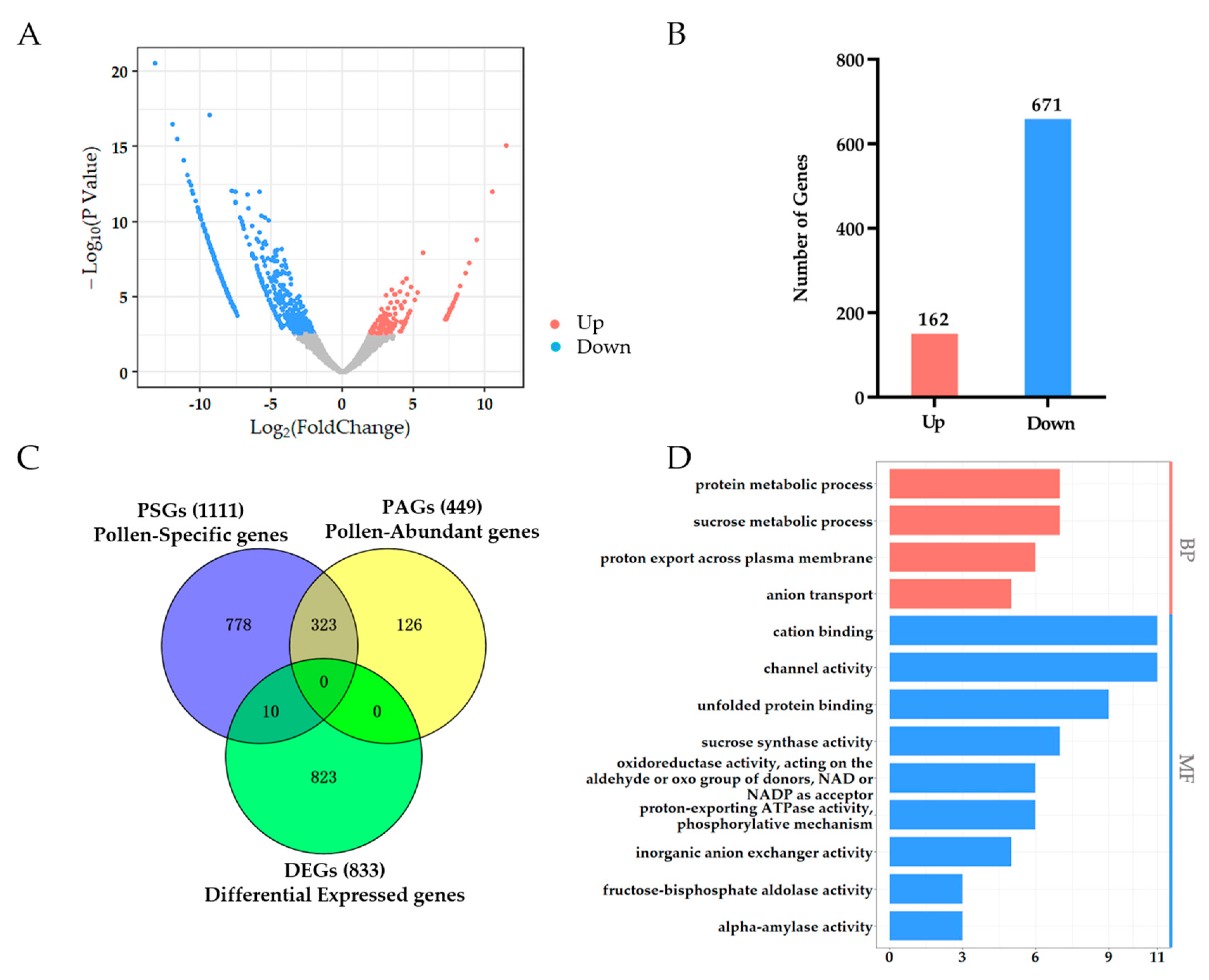
2.5. PSGs Mostly Do Not Respond to HT and Have Distinct Differences from Pollen Differentially Expressed Genes (DEGs)
3. Discussion
3.1. Transcriptome Diversity in Cotton Anthers and Pollen
3.2. The Late Stage of Cotton Anther Contains More Anther- and Pollen-Specific Genes
3.3. Few Pollen-Specific Genes (PSGs) Are Involved in Response to High-Temperature (HT) Stress
4. Materials and Methods
4.1. Plant Materials and HT Treatment
4.2. RNA-Seq, RT-PCR, and qRT-PCR
4.3. Identification of PSGs, ASGs, APSGs, and Pollen DEGs in the Cotton Genome Using RNA-Seq Datasets
4.4. GO Enrichment and Cis-Elements Analysis of PSGs, ASGs, and APSGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lobell, D.; Field, C.B. Global scale climate–crop yield relationships and the impacts of recent warming. Environ. Res. Lett. 2007, 2, 014002. [Google Scholar] [CrossRef]
- Shen, H.; Zhong, X.; Zhao, F.; Wang, Y.; Yan, B.; Li, Q.; Chen, G.; Mao, B.; Wang, J.; Li, Y.; et al. Overexpression of receptor-like kinase ERECTA improves thermotolerance in rice and tomato. Nat. Biotechnol. 2015, 33, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, S.I.; Rogelj, J.; Seferian, R.; Wartenburger, R.; Allen, M.R.; Cain, M.; Millar, R.J.; Ebi, K.L.; Ellis, N.; Hoegh-Guldberg, O.; et al. The many possible climates from the Paris Agreement’s aim of 1.5 degrees C warming. Nature 2018, 558, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Schauberger, B.; Archontoulis, S.; Arneth, A.; Balkovic, J.; Ciais, P.; Deryng, D.; Elliott, J.; Folberth, C.; Khabarov, N.; Müller, C.; et al. Consistent negative response of US crops to high temperatures in observations and crop models. Nat. Commun. 2017, 8, 13931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Liu, B.; Piao, S.; Wang, X.; Lobell, D.B.; Huang, Y.; Huang, M.T.; Yao, Y.T.; Bassu, S.; Ciais, P.; et al. Temperature increase reduces global yields of major crops in four independent estimates. Proc. Natl. Acad. Sci. USA 2017, 114, 9326–9331. [Google Scholar] [CrossRef] [Green Version]
- Pettigrew, W.T. The Effect of Higher Temperatures on Cotton Lint Yield Production and Fiber Quality. Crop Sci. 2008, 48, 278–285. [Google Scholar] [CrossRef] [Green Version]
- Min, L.; Li, Y.; Hu, Q.; Zhu, L.; Gao, W.; Wu, Y.; Ding, Y.; Liu, S.; Yang, X.; Zhang, X. Sugar and Auxin Signaling Pathways Respond to High-Temperature Stress during Anther Development as Revealed by Transcript Profiling Analysis in Cotton. Plant Physiol. 2014, 164, 1293–1308. [Google Scholar] [CrossRef] [Green Version]
- Zahid, K.R.; Ali, F.; Shah, F.; Younas, M.; Shah, T.; Shahwar, D.; Hassan, W.; Ahmad, Z.; Qi, C.; Lu, Y.; et al. Response and Tolerance Mechanism of Cotton Gossypium hirsutum L. to Elevated Temperature Stress: A Review. Front. Plant Sci. 2016, 7, 937. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Min, L.; Wang, J.; Li, Y.; Wu, Y.; Hu, Q.; Ding, Y.; Wang, M.; Liang, Y.; Gong, Z.; et al. A combination of genome-wide and transcriptome-wide association studies reveals genetic elements leading to male sterility during high temperature stress in cotton. New Phytol. 2021, 231, 165–181. [Google Scholar] [CrossRef]
- Ma, H. Molecular Genetic Analyses of Microsporogenesis and Microgametogenesis in Flowering Plants. Annu. Rev. Plant Biol. 2005, 56, 393–434. [Google Scholar] [CrossRef]
- Goldberg, R.B.; Beals, T.P.; Sanders, P.M. Anther Development—Basic Principles and Practical Applications. Plant Cell 1993, 5, 1217–1229. [Google Scholar] [PubMed] [Green Version]
- Begcy, K.; Nosenko, T.; Zhou, L.Z.; Fragner, L.; Weckwerth, W.; Dresselhaus, T. Male Sterility in Maize after Transient Heat Stress during the Tetrad Stage of Pollen Development. Plant Physiol. 2019, 181, 683–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Takeda, H.; Tsukaguchi, T.; Egawa, Y. Ultrastructural study on degeneration of tapetum in anther of snap bean ( Phaseolus vulgaris L.) under heat stress. Sex. Plant Reprod. 2001, 13, 293–299. [Google Scholar] [CrossRef]
- Zheng, X.; He, L.; Liu, Y.; Mao, Y.; Wang, C.; Zhao, B.; Li, Y.; He, H.; Guo, S.; Zhang, L.; et al. A study of male fertility control in Medicago truncatulauncovers an evolutionarily conserved recruitment of two tapetal bHLH subfamilies in plant sexual reproduction. New Phytol. 2020, 228, 1115–1133. [Google Scholar] [CrossRef]
- Li, N.; Zhang, D.S.; Liu, H.S.; Yin, C.S.; Li, X.X.; Liang, W.Q.; Yuan, Z.; Xu, B.; Chu, H.W.; Wang, J.; et al. The RiceTapetum Degeneration RetardationGene Is Required for Tapetum Degradation and Anther Development. Plant Cell 2006, 18, 2999–3014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Peet, M.M.; Thomas, J.F. Determining critical pre- and post-anthesis periods and physiological processes in Lycopersicon esculentum Mill. exposed to moderately elevated temperatures. J. Exp. Bot. 2002, 53, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; Tsuchiya, T.; Hamada, K.; Kawamura, S.; Yano, K.; Ohshima, M.; Higashitani, A.; Watanabe, M.; Kawagishi-Kobayashi, M. High Temperatures Cause Male Sterility in Rice Plants with Transcriptional Alterations during Pollen Development. Plant Cell Physiol. 2009, 50, 1911–1922. [Google Scholar] [CrossRef]
- Min, L.; Zhu, L.; Tu, L.; Deng, F.; Yuan, D.; Zhang, X. Cotton GhCKI disrupts normal male reproduction by delaying tapetum programmed cell death via inactivating starch synthase. Plant J. 2013, 75, 823–835. [Google Scholar] [CrossRef]
- Ma, Y.; Min, L.; Wang, M.; Wang, C.; Zhao, Y.; Li, Y.; Fang, Q.; Wu, Y.; Xie, S.; Ding, Y.; et al. Disrupted Genome Methylation in Response to High Temperature Has Distinct Affects on Microspore Abortion and Anther Indehiscence. Plant Cell 2018, 30, 1387–1403. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wei, H.; Song, M.; Pang, C.; Liu, J.; Wang, L.; Zhang, J.; Fan, S.; Yu, S. Transcriptome Profiling Analysis Reveals That Flavonoid and Ascorbate-Glutathione Cycle Are Important during Anther Development in Upland Cotton. PLoS ONE 2012, 7, e49244. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Min, L.; Wu, Z.; Yang, L.; Zhu, L.; Yang, X.; Yuan, D.; Guo, X.; Zhang, X.T. Defective pollen wall contributes to male sterility in the male sterile line 1355A of cotton. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Lou, Y.; Shi, Q.S.; Zhang, S.; Zhou, W.; Yang, J.; Zhang, C.; Yao, X.Z.; Xu, T.; Liu, J.L.; et al. Slowing development restores the fertility of thermo-sensitive male-sterile plant lines. Nat. Plants 2020, 6, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Guo, T.; Li, X.M.; Zhang, Y.M.; Yang, Y.B.; Ye, W.W.; Dong, N.Q.; Shi, C.L.; Kan, Y.; Xiang, Y.H.; et al. Translational Regulation of Plant Response to High Temperature by a Dual-Function tRNA(His) Guanylyltransferase in Rice. Mol. Plant 2019, 12, 1123–1142. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.; Zhang, H.; Hammond, R.; Huang, K.; Meyers, B.C.; Walbot, V. Dicer-like 5 deficiency confers temperature-sensitive male sterility in maize. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, L.; Ou, S.; Wang, R.; Wang, Y.; Chu, C.; Yao, S. Natural variations of SLG1 confer high-temperature tolerance in indica rice. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Abbas, M.; Hernández-García, J.; Blanco-Touriñán, N.; Aliaga, N.; Minguet, E.G.; Alabadí, D.; Blázquez, M.A. Reduction of indole-3-acetic acid methyltransferase activity compensates for high-temperature male sterility in Arabidopsis. Plant Biotechnol. J. 2017, 16, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Sakata, T.; Oshino, T.; Miura, S.; Tomabechi, M.; Tsunaga, Y.; Higashitani, N.; Miyazawa, Y.; Takahashi, H.; Watanabe, M.; Higashitani, A. Auxins reverse plant male sterility caused by high temperatures. Proc. Natl. Acad. Sci. USA 2010, 107, 8569–8574. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.H.; Min, L.; Ma, Y.; Wu, Y.; Ding, Y.; Li, Y.; Xie, S.; Ullah, A.; Shaban, M.; Manghwar, H.; et al. High day and night temperatures distinctively disrupt fatty acid and jasmonic acid metabolism, inducing male sterility in cotton. J. Exp. Bot. 2020, 71, 6128–6141. [Google Scholar] [CrossRef]
- Li, X.M.; Chao, D.Y.; Wu, Y.; Huang, X.; Chen, K.; Cui, L.G.; Su, L.; Ye, W.W.; Chen, H.; Chen, H.C.; et al. Natural alleles of a proteasome α2 subunit gene contribute to thermotolerance and adaptation of African rice. Nat. Genet. 2015, 47, 827–833. [Google Scholar] [CrossRef]
- Wittkopp, P.J.; Kalay, G. Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2011, 13, 59–69. [Google Scholar] [CrossRef]
- Bate, N.; Twell, D. Functional architecture of a late pollen promoter: Pollen-specific transcription is developmentally regulated by multiple stage-specific and co-dependent activator elements. Plant Mol. Biol. 1998, 37, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Gan, E.S.; Huang, J.; Ito, T. Functional Roles of Histone Modification, Chromatin Remodeling and MicroRNAs in Arabidopsis Flower Development. Int. Rev. Cell Mol. Biol. 2013, 305, 115–161. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Sun, B.; Looi, L.S.; Xu, Y.; Gan, E.S.; Huang, J.; Ito, T. Co-ordination of Flower Development through Epigenetic Regulation in Two Model Species: Rice and Arabidopsis. Plant Cell Physiol. 2015, 56, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oo, M.M.; Bae, H.K.; Nguyen, T.D.; Moon, S.; Oh, S.A.; Kim, J.H.; Soh, M.S.; Song, J.T.; Jung, K.H.; Park, S.K. Evaluation of rice promoters conferring pollen-specific expression in a heterologous system, Arabidopsis. Plant Reprod. 2014, 27, 47–58. [Google Scholar] [CrossRef]
- Moon, S.; Oo, M.M.; Kim, B.; Koh, H.J.; Oh, S.A.; Yi, G.; An, G.; Park, S.K.; Jung, K.H. Genome-wide analyses of late pollen-preferred genes conserved in various rice cultivars and functional identification of a gene involved in the key processes of late pollen development. Rice 2018, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yan, W.; Peng, X.; Chen, Z.; Xu, C.; Wu, J.; Deng, X.W.; Tang, X. Identification of late-stage pollen-specific promoters for construction of pollen-inactivation system in rice. J. Integr. Plant Biol. 2020, 62, 1246–1263. [Google Scholar] [CrossRef] [Green Version]
- Akasaka, M.; Taniguchi, Y.; Oshima, M.; Abe, K.; Tabei, Y.; Tanaka, J. Development of transgenic male-sterile rice by using anther-specific promoters identified by comprehensive screening of the gene expression profile database ‘RiceXPro’. Breed. Sci. 2018, 68, 420–431. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Shi, Y.; Li, Y.; Guo, Y.; Borrego, E.; Wei, Z.; Ren, H.; Ma, Z.; Yan, Y. A Rapid Pipeline for Pollen- and Anther-Specific Gene Discovery Based on Transcriptome Profiling Analysis of Maize Tissues. Int. J. Mol. Sci. 2021, 22, 6877. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, P.M.; Bui, A.Q.; Weterings, K.; McIntire, K.N.; Hsu, Y.C.; Lee, P.Y.; Truong, M.T.; Beals, T.P.; Goldberg, R.B. Anther developmental defects in Arabidopsis thaliana male-sterile mutants. Sex. Plant Reprod. 1999, 11, 297–322. [Google Scholar] [CrossRef]
- Liu, K.; Yu, Y.; Dong, A.; Shen, W. SET DOMAIN GROUP701 encodes a H3K4-methytransferase and regulates multiple key processes of rice plant development. New Phytol. 2017, 215, 609–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinon, V.; Yao, X.; Dong, A.; Shen, W.H. SDG2-Mediated H3K4me3 Is Crucial for Chromatin Condensation and Mitotic Division during Male Gametogenesis in Arabidopsis. Plant Physiol. 2017, 174, 1205–1215. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Skibbe, D.S.; Fernandes, J.; Walbot, V. Male reproductive development: Gene expression profiling of maize anther and pollen ontogeny. Genome Biol. 2008, 9, R181. [Google Scholar] [CrossRef] [Green Version]
- Deveshwar, P.; Bovill, W.D.; Sharma, R.; A Able, J.; Kapoor, S. Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice. BMC Plant Biol. 2011, 11, 78. [Google Scholar] [CrossRef] [Green Version]
- Honys, D.; Twell, D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 2004, 5, R85. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.; Boavida, L.; Carneiro, J.; Haury, M.; Feijó, J.A. Transcriptional Profiling of Arabidopsis Tissues Reveals the Unique Characteristics of the Pollen Transcriptome. Plant Physiol. 2003, 133, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; Tsuchiya, T.; Saito, H.; Matsubara, H.; Hakozaki, H.; Masuko, H.; Kamada, M.; Higashitani, A.; Takahashi, H.; Fukuda, H.; et al. Identification and molecular characterization of novel anther-specific genes in Oryza sativa L. by using cDNA microarray. Genes Genet. Syst. 2004, 79, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.D.; Moon, S.; Nguyen, V.N.T.; Gho, Y.; Chandran, A.K.N.; Soh, M.S.; Song, J.T.; An, G.; Oh, S.A.; Park, S.K.; et al. Genome-wide identification and analysis of rice genes preferentially expressed in pollen at an early developmental stage. Plant Mol. Biol. 2016, 92, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.S.; Chung, Y.Y.; Lee, S.; Yi, G.H.; Oh, B.G.; An, G. Isolation and characterization of an anther-specific gene, RA8, from rice (Oryza sativa L.). Plant Mol. Biol. 1999, 39, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Weterings, K.; Schrauwen, J.; Wullems, G.; Twell, D. Functional dissection of the promoter of the pollen-specific gene NTP303 reveals a novel pollen-specific, and conserved cis-regulatory element. Plant J. 1995, 8, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Turcich, M.P.; Hamilton, D.A.; Mascarenhas, J.P. Isolation and characterization of pollen-specific maize genes with sequence homology to ragweed allergens and pectate lyases. Plant Mol. Biol. 1993, 23, 1061–1065. [Google Scholar] [CrossRef]
- Wakeley, P.; Rogers, H.; Rozycka, M.; Greenland, A.J.; Hussey, P. A maize pectin methylesterase-like gene, ZmC5, specifically expressed in pollen. Plant Mol. Biol. 1998, 37, 187–192. [Google Scholar] [CrossRef]
- Kelliher, T.; Starr, D.; Richbourg, L.; Chintamanani, S.; Delzer, B.; Nuccio, M.L.; Green, J.; Chen, Z.; McCuiston, J.; Wang, W.; et al. MATRILINEAL, a sperm-specific phospholipase, triggers maize haploid induction. Nature 2017, 542, 105–109. [Google Scholar] [CrossRef]
- Wang, H.; Fan, M.; Wang, G.; Zhang, C.; Shi, L.; Wei, Z.; Ma, W.; Chang, J.; Huang, S.; Lin, F. Isolation and characterization of a novel pollen-specific promoter in maize (Zea mays L.). Genome 2017, 60, 485–495. [Google Scholar] [CrossRef]
- Twell, D.; Klein, T.M.; Fromm, M.E.; McCormick, S. Transient Expression of Chimeric Genes Delivered into Pollen by Microprojectile Bombardment. Plant Physiol. 1989, 91, 1270–1274. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, J.L.; E Ploense, S.; Snustad, D.P.; Silflow, C.D. Preferential expression of an alpha-tubulin gene of Arabidopsis in pollen. Plant Cell 1992, 4, 557–571. [Google Scholar] [CrossRef] [Green Version]
- Albani, D.; Robert, L.S.; Donaldson, P.A.; Altosaar, I.; Arnison, P.G.; Fabijanski, S.F. Characterization of a pollen-specific gene family from Brassica napus which is activated during early microspore development. Plant Mol. Biol. 1990, 15, 605–622. [Google Scholar] [CrossRef]
- Li, Y.; Min, L.; Zhang, L.; Hu, Q.; Wu, Y.; Li, J.; Xie, S.; Ma, Y.; Zhang, X.; Zhu, L. Promoters of Arabidopsis Casein kinase I-like 2 and 7 confer specific high-temperature response in anther. Plant Mol. Biol. 2018, 98, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissues a | Number of EGs b | Average Transcription of EGs (TPM) c | Average Transcription of Top 300 EGs (TPM) d | Average Transcription of Top 1000 EGs (TPM) e | Percentage of Top 300 EGs in Transcriptome f | Percentage of Top 1000 EGs in Transcriptome g |
|---|---|---|---|---|---|---|
| NTpollen | 12,005 | 82.8 | 2489.1 | 893.2 | 75.1% | 89.9% |
| NT3-5 | 38,380 | 25.9 | 779.4 | 396.8 | 23.5% | 39.9% |
| NT5-6 | 40,030 | 24.9 | 1000.2 | 421.9 | 30.1% | 42.3% |
| NT6-7 | 42,867 | 23.2 | 702.5 | 336.3 | 21.2% | 33.8% |
| NT7-8 | 41,853 | 23.8 | 789.6 | 373.7 | 23.8% | 37.5% |
| NT8-9 | 41,059 | 24.2 | 907.3 | 409.5 | 27.4% | 41.2% |
| NT9-10 | 39,798 | 25 | 1139.2 | 479 | 34.3% | 48.1% |
| NT10-11 | 38,947 | 25.5 | 1081.7 | 470 | 32.7% | 47.3% |
| NT11-12 | 37,567 | 26.5 | 1253.3 | 522.2 | 37.8% | 52.5% |
| NT12-13 | 37,389 | 26.6 | 1236.4 | 516.8 | 37.3% | 52.0% |
| NT13-14 | 36,763 | 27.1 | 1319.6 | 541.6 | 39.7% | 54.4% |
| NT14-16 | 35,056 | 28.4 | 1343.8 | 556.3 | 40.5% | 55.9% |
| NT16-19 | 30,917 | 32.2 | 1506.1 | 613.3 | 45.4% | 61.6% |
| NT19-24 | 32,088 | 31 | 1370.7 | 574.4 | 41.3% | 57.7% |
| NTroot | 35,003 | 28.4 | 856.7 | 431.1 | 25.9% | 43.4% |
| NTstem | 41,397 | 24.1 | 814.8 | 383.6 | 24.5% | 38.4% |
| NTleaf | 38,662 | 25.7 | 1416.4 | 551.6 | 42.8% | 55.5% |
| NTbract | 34,974 | 28.4 | 1138.7 | 508.3 | 34.4% | 51.2% |
| NTsepal | 37,484 | 26.6 | 866.6 | 418.2 | 26.1% | 41.9% |
| NTpetal | 35,495 | 28 | 822.8 | 419.5 | 24.8% | 42.2% |
| NTstigma | 38,303 | 26 | 760.6 | 378.7 | 22.9% | 38.0% |
| NTovlue | 40,855 | 24.4 | 634.1 | 322.9 | 19.1% | 32.4% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Zhou, L.; Li, Y.; Ma, H.; Li, Y.; Ma, Y.; Lv, R.; Yang, J.; Wang, W.; Alifu, A.; et al. Rapid Identification of Pollen- and Anther-Specific Genes in Response to High-Temperature Stress Based on Transcriptome Profiling Analysis in Cotton. Int. J. Mol. Sci. 2022, 23, 3378. https://doi.org/10.3390/ijms23063378
Zhang R, Zhou L, Li Y, Ma H, Li Y, Ma Y, Lv R, Yang J, Wang W, Alifu A, et al. Rapid Identification of Pollen- and Anther-Specific Genes in Response to High-Temperature Stress Based on Transcriptome Profiling Analysis in Cotton. International Journal of Molecular Sciences. 2022; 23(6):3378. https://doi.org/10.3390/ijms23063378
Chicago/Turabian StyleZhang, Rui, Lili Zhou, Yanlong Li, Huanhuan Ma, Yawei Li, Yizan Ma, Rongjie Lv, Jing Yang, Weiran Wang, Aierxi Alifu, and et al. 2022. "Rapid Identification of Pollen- and Anther-Specific Genes in Response to High-Temperature Stress Based on Transcriptome Profiling Analysis in Cotton" International Journal of Molecular Sciences 23, no. 6: 3378. https://doi.org/10.3390/ijms23063378
APA StyleZhang, R., Zhou, L., Li, Y., Ma, H., Li, Y., Ma, Y., Lv, R., Yang, J., Wang, W., Alifu, A., Zhang, X., Kong, J., & Min, L. (2022). Rapid Identification of Pollen- and Anther-Specific Genes in Response to High-Temperature Stress Based on Transcriptome Profiling Analysis in Cotton. International Journal of Molecular Sciences, 23(6), 3378. https://doi.org/10.3390/ijms23063378