
Inverse and Concordant Mucosal Pathway Gene Expressions in Inflamed and Non-Inflamed Ulcerative Colitis Patients: Potential Relevance to Aetiology and Pathogenesis


Abstract
:1. Introduction
2. Material and Methods
2.1. Study Samples
2.2. Sample Preparation and RNA-Sequencing
2.3. Statistical Analysis and Bioinformatics
2.4. Analysis of Differential Expression Using RNA-Seq Data
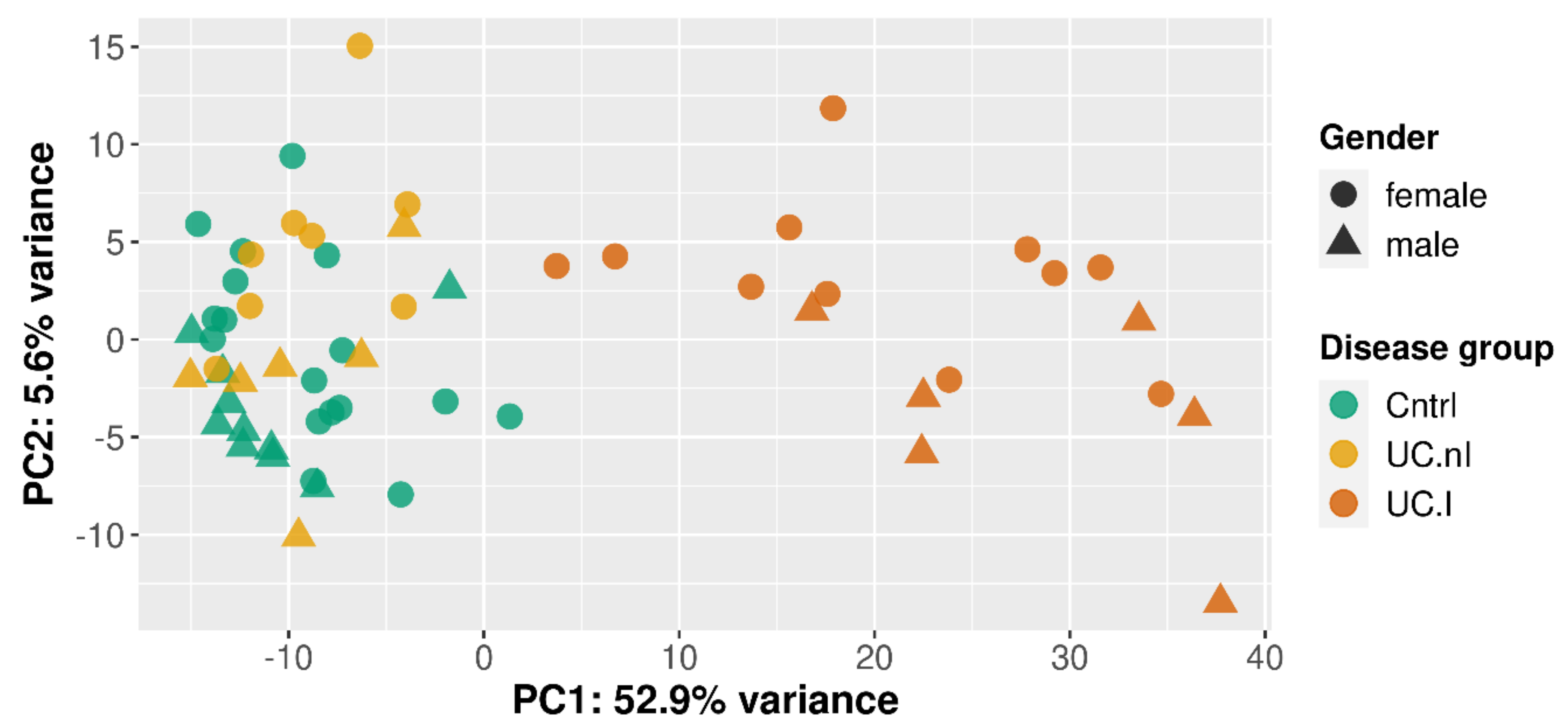
2.5. Principal Component Analysis
2.6. Enrichment Analysis
2.7. Text Analysis of UniProt Functional Information
3. Results
3.1. Unsupervised Clustering Based on Gene Expression
3.2. Differentially Expressed Genes
3.2.1. Gender Based Differential Gene Expression and Inflammation
3.2.2. Inflamed UC Mucosa vs. Controls
3.2.3. Non-Inflamed UC Mucosa vs. Controls
3.3. Gene Set Analysis
3.3.1. Gender and Inflammation DE Genes
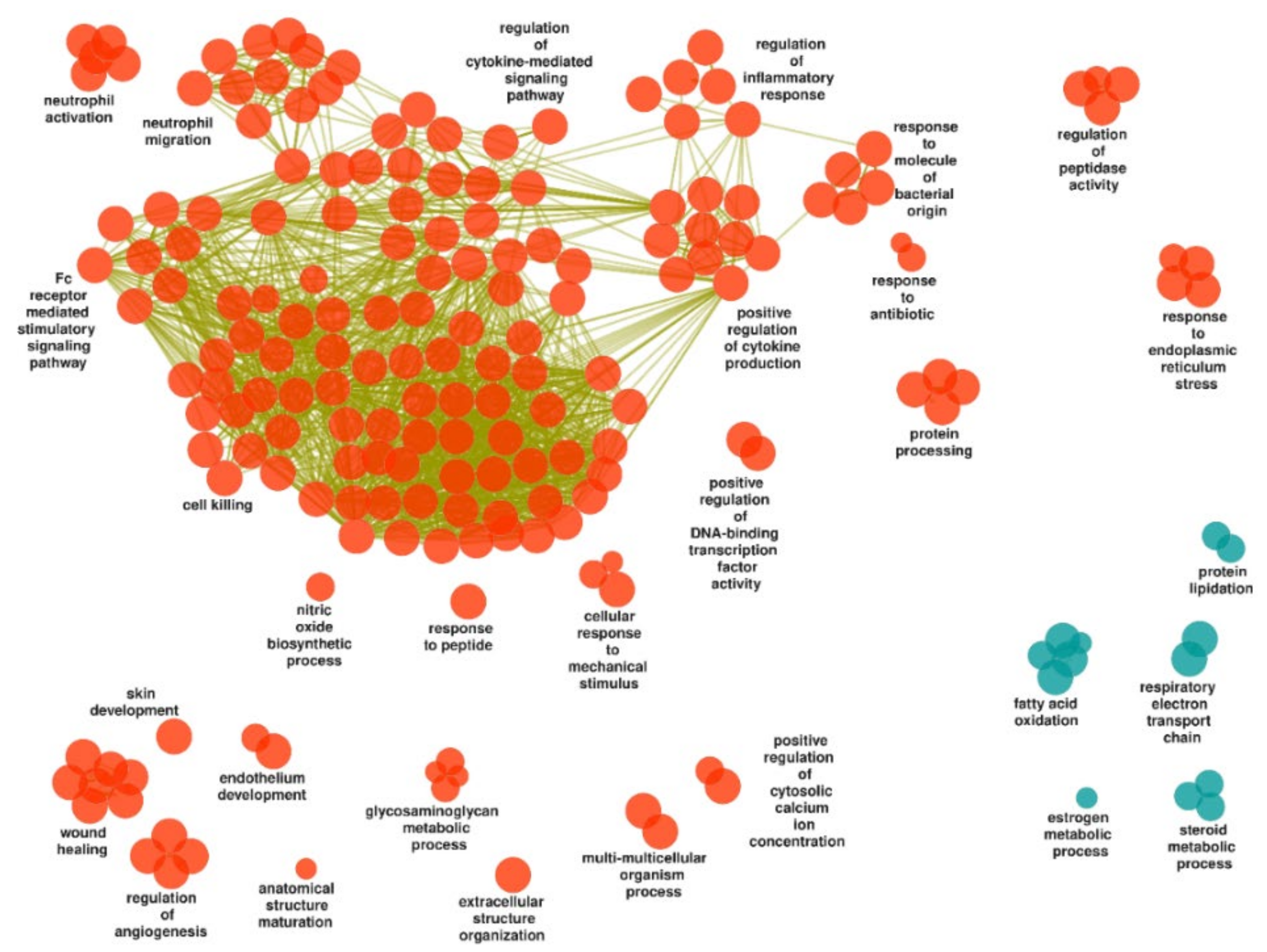
3.3.2. Inflamed UC Mucosa vs. Controls
Upregulated Gene Sets
Downregulated Gene Sets
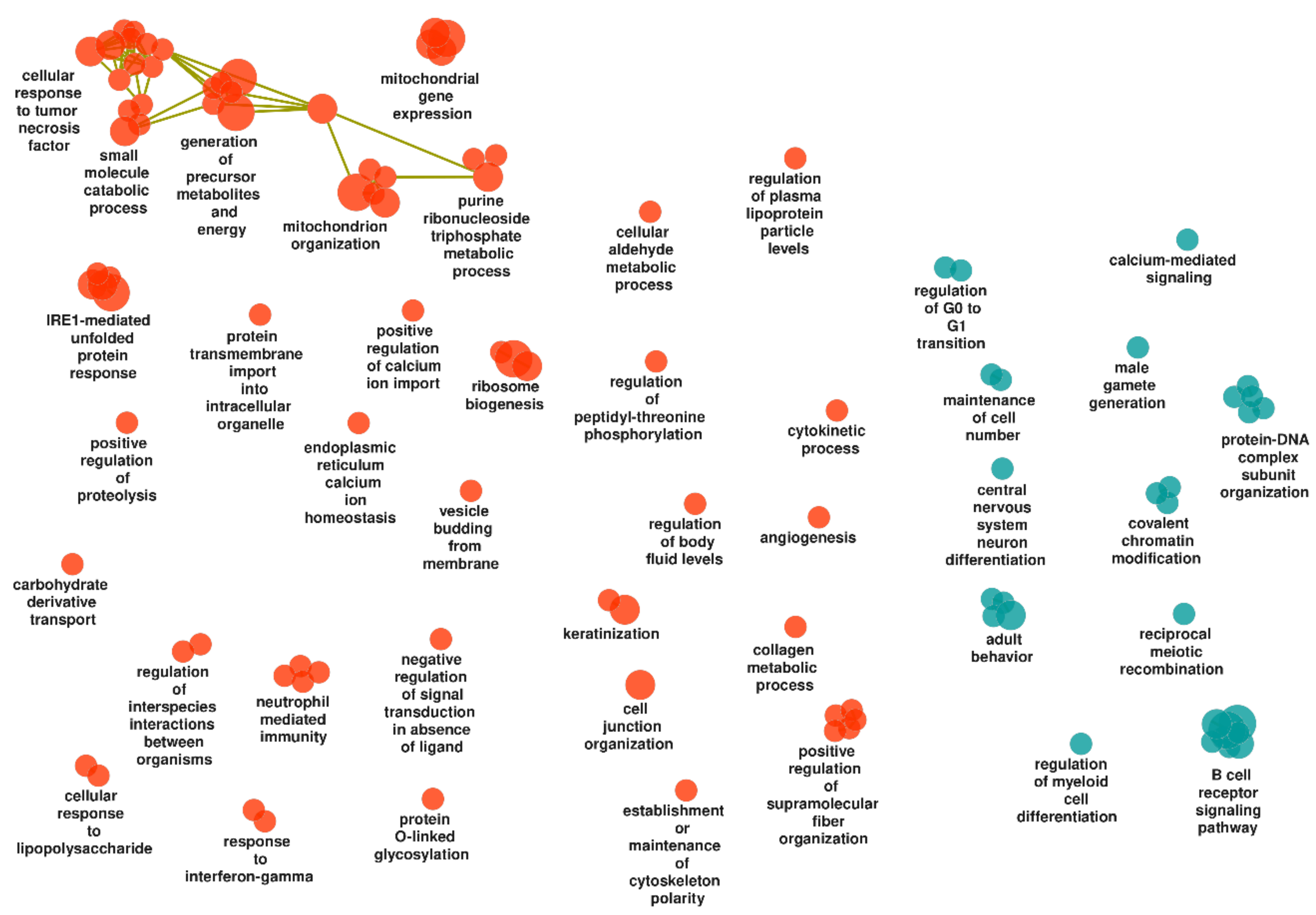
3.3.3. Non-Inflamed UC Mucosa vs. Controls
Upregulated Gene Sets
Downregulated Gene Sets
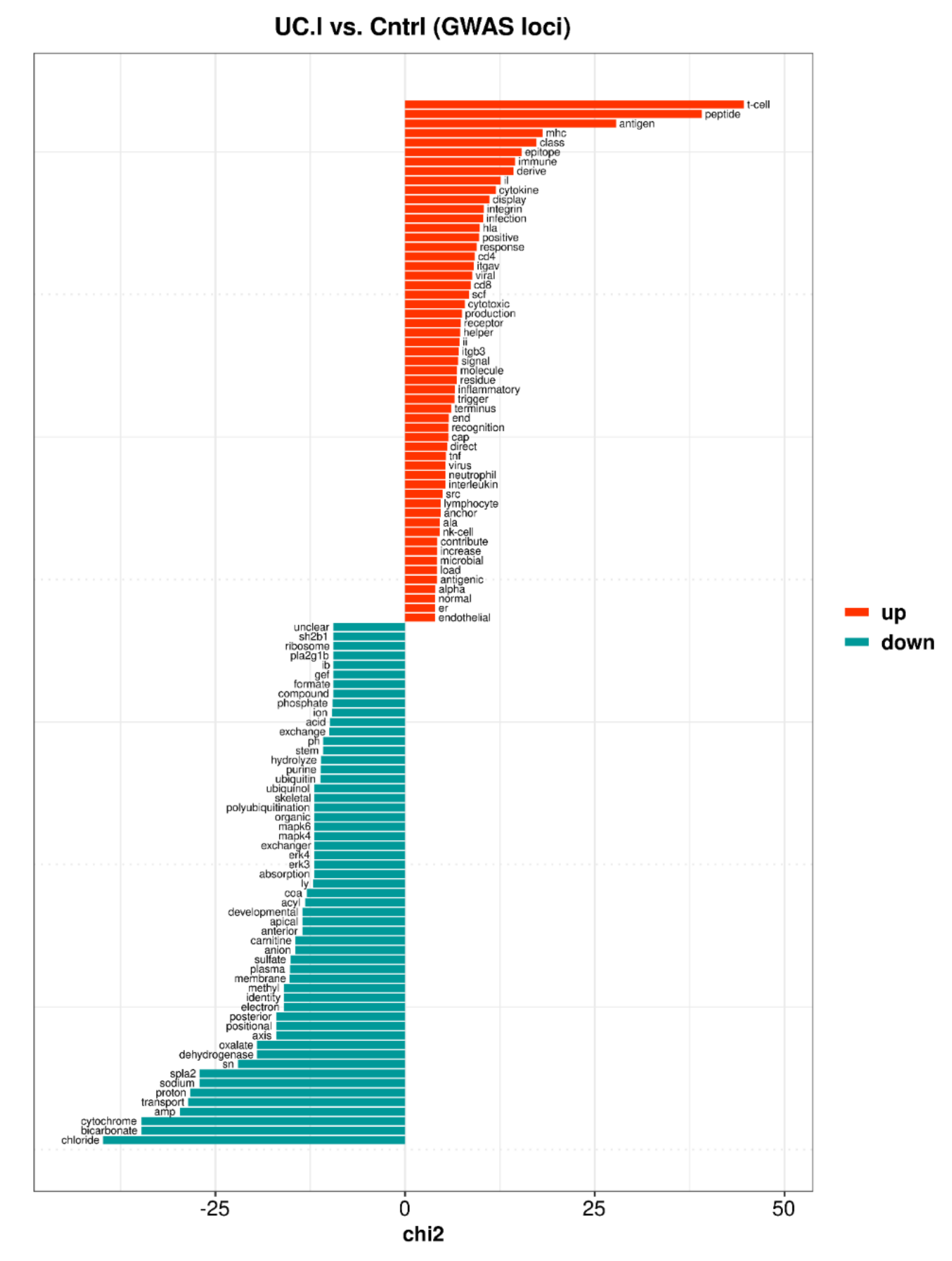
3.4. Core Enriched Genes and IBD Susceptibility Loci
3.4.1. Inflamed UC Mucosa vs. Controls
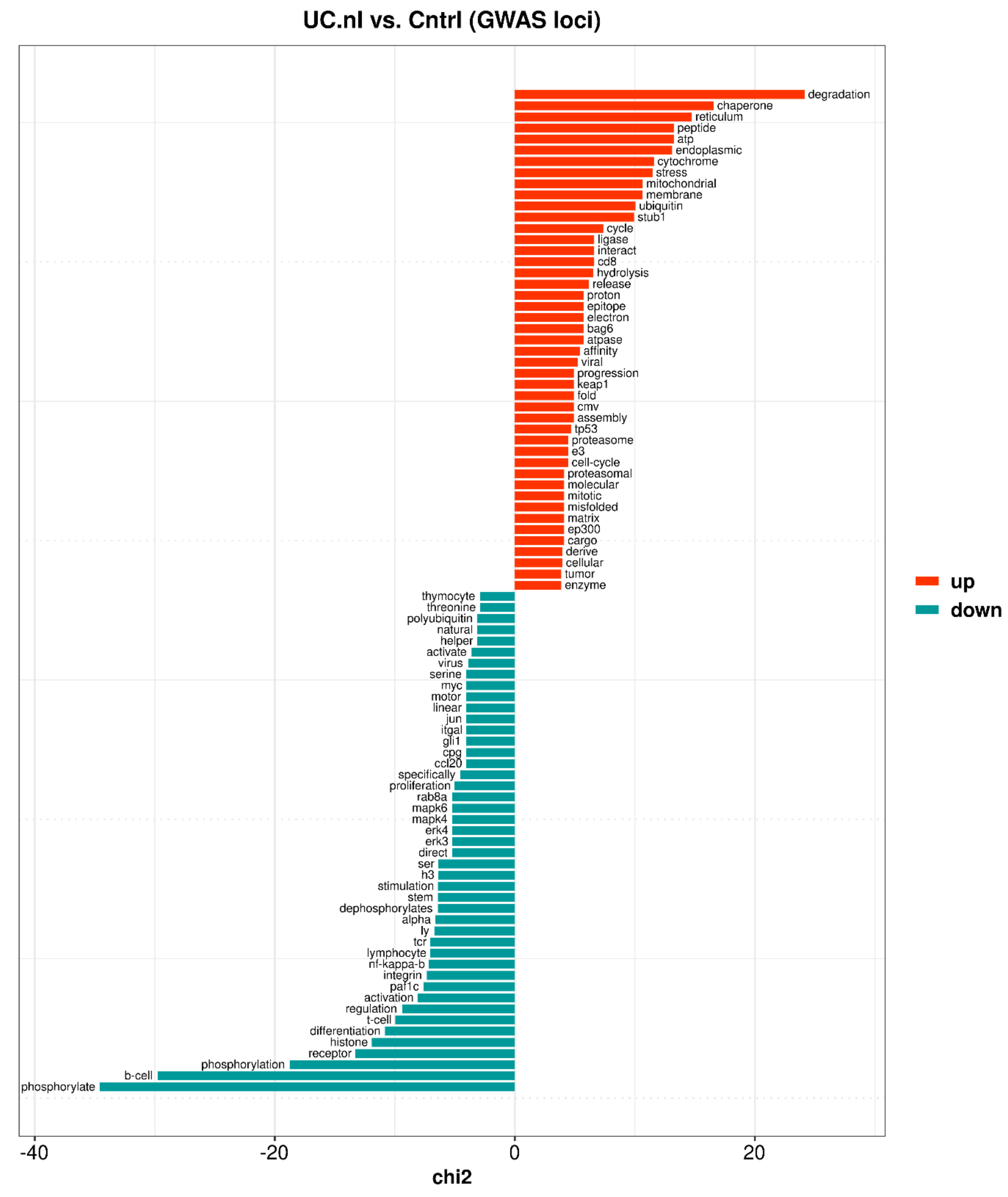
3.4.2. Non-Inflamed UC Mucosa vs. Controls
4. Discussion
4.1. Mitochondrial Dysfunction
4.2. Endoplasmic Reticulum Stress
4.3. Steroid Hormone Metabolism
4.4. Angiogenesis
4.5. Intestinal Permeability
4.6. Innate and Adaptive Immunity
4.7. Differential Expression and IBD Susceptibility Loci
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BP | biological process |
| Cntrl | controls |
| DE | differentially expressed |
| ER | endoplasmic reticulum |
| FDR | false discovery rate |
| GO | gene ontology |
| GWAS | genome-wide association study |
| IBD | inflammatory bowel disease |
| log2FC | log2-fold-change |
| PCA | principal components analysis |
| UC | ulcerative colitis |
| UC.I | inflamed UC mucosa |
| UC.nI | non-inflamed UC mucosa |
References
- De Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- De Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberman, Y.; Karns, R.; Dexheimer, P.J.; Schirmer, M.; Somekh, J.; Jurickova, I.; Braun, T.; Novak, E.; Bauman, L.; Collins, M.H.; et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, O.; Creanza, T.M.; Bossa, F.; Palumbo, O.; Maglietta, R.; Ancona, N.; Corritore, G.; Latiano, T.; Martino, G.; Biscaglia, G.; et al. Genome-wide Pathway Analysis Using Gene Expression Data of Colonic Mucosa in Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1260–1268. [Google Scholar] [CrossRef] [Green Version]
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019, 567, 49–55. [Google Scholar] [CrossRef]
- Juzenas, S.; Hubenthal, M.; Lindqvist, C.M.; Kruse, R.; Steiert, T.A.; Degenhardt, F.; Schulte, D.; Nikolaus, S.; Zeissig, S.; Bergemalm, D.; et al. Detailed transcriptional landscape of peripheral blood points to increased neutrophil activation in treatment-naive inflammatory bowel disease. J. Crohn's Colitis 2022. [Google Scholar] [CrossRef]
- Ostrowski, J.; Dabrowska, M.; Lazowska, I.; Paziewska, A.; Balabas, A.; Kluska, A.; Kulecka, M.; Karczmarski, J.; Ambrozkiewicz, F.; Piatkowska, M.; et al. Redefining the Practical Utility of Blood Transcriptome Biomarkers in Inflammatory Bowel Diseases. J. Crohn's Colitis 2019, 13, 626–633. [Google Scholar] [CrossRef]
- Park, S.K.; Kim, S.; Lee, G.Y.; Kim, S.Y.; Kim, W.; Lee, C.W.; Park, J.L.; Choi, C.H.; Kang, S.B.; Kim, T.O.; et al. Development of a Machine Learning Model to Distinguish between Ulcerative Colitis and Crohn’s Disease Using RNA Sequencing Data. Diagnostics 2021, 11, 2365. [Google Scholar] [CrossRef]
- Biasci, D.; Lee, J.C.; Noor, N.M.; Pombal, D.R.; Hou, M.; Lewis, N.; Ahmad, T.; Hart, A.; Parkes, M.; McKinney, E.F.; et al. A blood-based prognostic biomarker in IBD. Gut 2019, 68, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Soderman, J.; Berglind, L.; Almer, S. Gene Expression-Genotype Analysis Implicates GSDMA, GSDMB, and LRRC3C as Contributors to Inflammatory Bowel Disease Susceptibility. BioMed Res. Int. 2015, 2015, 834805. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- McGovern, D.P.; Gardet, A.; Torkvist, L.; Goyette, P.; Essers, J.; Taylor, K.D.; Neale, B.M.; Ong, R.T.; Lagace, C.; Li, C.; et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010, 42, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Burisch, J. Impact of Genes and the Environment on the Pathogenesis and Disease Course of Inflammatory Bowel Disease. Dig. Dis. Sci. 2019, 64, 1759–1769. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020, 578, 527–539. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Kolios, G. Animal models of inflammatory bowel disease: How useful are they really? Curr. Opin. Gastroenterol. 2016, 32, 251–257. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W.E. The colonic epithelium in ulcerative colitis: An energy-deficiency disease? Lancet 1980, 2, 712–715. [Google Scholar] [CrossRef]
- Novak, E.A.; Mollen, K.P. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell. Dev. Biol. 2015, 3, 62. [Google Scholar] [CrossRef] [Green Version]
- Van Haaften-Visser, D.Y.; Harakalova, M.; Mocholi, E.; van Montfrans, J.M.; Elkadri, A.; Rieter, E.; Fiedler, K.; van Hasselt, P.M.; Triffaux, E.M.M.; van Haelst, M.M.; et al. Ankyrin repeat and zinc-finger domain-containing 1 mutations are associated with infantile-onset inflammatory bowel disease. J. Biol. Chem. 2017, 292, 7904–7920. [Google Scholar] [CrossRef] [Green Version]
- Santhanam, S.; Rajamanickam, S.; Motamarry, A.; Ramakrishna, B.S.; Amirtharaj, J.G.; Ramachandran, A.; Pulimood, A.; Venkatraman, A. Mitochondrial electron transport chain complex dysfunction in the colonic mucosa in ulcerative colitis. Inflamm. Bowel Dis. 2012, 18, 2158–2168. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J.A.; Ranea-Robles, P. Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165720. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef]
- Powell, N.; Pantazi, E.; Pavlidis, P.; Tsakmaki, A.; Li, K.; Yang, F.; Parker, A.; Pin, C.; Cozzetto, D.; Minns, D.; et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut 2020, 69, 578–590. [Google Scholar] [CrossRef] [Green Version]
- Mohapatra, G.; Eisenberg-Lerner, A.; Merbl, Y. Gatekeepers of the Gut: The Roles of Proteasomes at the Gastrointestinal Barrier. Biomolecules 2021, 11, 989. [Google Scholar] [CrossRef]
- Moncan, M.; Mnich, K.; Blomme, A.; Almanza, A.; Samali, A.; Gorman, A.M. Regulation of lipid metabolism by the unfolded protein response. J. Cell Mol. Med. 2021, 25, 1359–1370. [Google Scholar] [CrossRef]
- Rohrl, C.; Eigner, K.; Winter, K.; Korbelius, M.; Obrowsky, S.; Kratky, D.; Kovacs, W.J.; Stangl, H. Endoplasmic reticulum stress impairs cholesterol efflux and synthesis in hepatic cells. J. Lipid Res. 2014, 55, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elustondo, P.; Martin, L.A.; Karten, B. Mitochondrial cholesterol import. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Schmidt, C.; Brunner, T. Extra-Adrenal Glucocorticoid Synthesis in the Intestinal Mucosa: Between Immune Homeostasis and Immune Escape. Front. Immunol. 2019, 10, 1438. [Google Scholar] [CrossRef] [PubMed]
- Stegk, J.P.; Ebert, B.; Martin, H.J.; Maser, E. Expression profiles of human 11beta-hydroxysteroid dehydrogenases type 1 and type 2 in inflammatory bowel diseases. Mol. Cell. Endocrinol. 2009, 301, 104–108. [Google Scholar] [CrossRef]
- Coste, A.; Dubuquoy, L.; Barnouin, R.; Annicotte, J.S.; Magnier, B.; Notti, M.; Corazza, N.; Antal, M.C.; Metzger, D.; Desreumaux, P.; et al. LRH-1-mediated glucocorticoid synthesis in enterocytes protects against inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2007, 104, 13098–13103. [Google Scholar] [CrossRef] [Green Version]
- Bouguen, G.; Langlois, A.; Djouina, M.; Branche, J.; Koriche, D.; Dewaeles, E.; Mongy, A.; Auwerx, J.; Colombel, J.F.; Desreumaux, P.; et al. Intestinal steroidogenesis controls PPARgamma expression in the colon and is impaired during ulcerative colitis. Gut 2015, 64, 901–910. [Google Scholar] [CrossRef]
- Bayrer, J.R.; Wang, H.; Nattiv, R.; Suzawa, M.; Escusa, H.S.; Fletterick, R.J.; Klein, O.D.; Moore, D.D.; Ingraham, H.A. LRH-1 mitigates intestinal inflammatory disease by maintaining epithelial homeostasis and cell survival. Nat. Commun. 2018, 9, 4055. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; de la Motte, C.; Graziani, C.; West, G.; Phillips, M.H.; Pola, R.; Rutella, S.; Willis, J.; Gasbarrini, A.; et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology 2006, 130, 2060–2073. [Google Scholar] [CrossRef] [Green Version]
- Scaldaferri, F.; Vetrano, S.; Sans, M.; Arena, V.; Straface, G.; Stigliano, E.; Repici, A.; Sturm, A.; Malesci, A.; Panes, J.; et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology 2009, 136, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Chidlow, J.H., Jr.; Greer, J.J.; Anthoni, C.; Bernatchez, P.; Fernandez-Hernando, C.; Bruce, M.; Abdelbaqi, M.; Shukla, D.; Granger, D.N.; Sessa, W.C.; et al. Endothelial caveolin-1 regulates pathologic angiogenesis in a mouse model of colitis. Gastroenterology 2009, 136, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Heidemann, J.; Ogawa, H.; Dwinell, M.B.; Rafiee, P.; Maaser, C.; Gockel, H.R.; Otterson, M.F.; Ota, D.M.; Lugering, N.; Domschke, W.; et al. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J. Biol. Chem. 2003, 278, 8508–8515. [Google Scholar] [CrossRef] [Green Version]
- Salcedo, R.; Young, H.A.; Ponce, M.L.; Ward, J.M.; Kleinman, H.K.; Murphy, W.J.; Oppenheim, J.J. Eotaxin (CCL11) induces in vivo angiogenic responses by human CCR3+ endothelial cells. J. Immunol. 2001, 166, 7571–7578. [Google Scholar] [CrossRef] [Green Version]
- Polosukhina, D.; Singh, K.; Asim, M.; Barry, D.P.; Allaman, M.M.; Hardbower, D.M.; Piazuelo, M.B.; Washington, M.K.; Gobert, A.P.; Wilson, K.T.; et al. CCL11 exacerbates colitis and inflammation-associated colon tumorigenesis. Oncogene 2021, 40, 6540–6546. [Google Scholar] [CrossRef]
- Cross, R.K.; Wilson, K.T. Nitric oxide in inflammatory bowel disease. Inflamm. Bowel Dis. 2003, 9, 179–189. [Google Scholar] [CrossRef]
- Okaniwa, N.; Sasaki, M.; Mizushima, T.; Ogasawara, N.; Funaki, Y.; Joh, T.; Kasugai, K. eNOS plays an important role in the regulation of colonic inflammation: A novel therapeutic target and a predictive marker for the prognosis of ulcerative colitis. Free Radic. Res. 2015, 49, 35–44. [Google Scholar] [CrossRef]
- Ahmad, R.; Sorrell, M.F.; Batra, S.K.; Dhawan, P.; Singh, A.B. Gut permeability and mucosal inflammation: Bad, good or context dependent. Mucosal Immunol. 2017, 10, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Leong, R.W.; Wasinger, V.C.; Ip, M.; Yang, M.; Phan, T.G. Impaired Intestinal Permeability Contributes to Ongoing Bowel Symptoms in Patients With Inflammatory Bowel Disease and Mucosal Healing. Gastroenterology 2017, 153, 723–731. [Google Scholar] [CrossRef]
- Soderman, J.; Noren, E.; Christiansson, M.; Bragde, H.; Thiebaut, R.; Hugot, J.P.; Tysk, C.; O’Morain, C.A.; Gassull, M.; Finkel, Y.; et al. Analysis of single nucleotide polymorphisms in the region of CLDN2-MORC4 in relation to inflammatory bowel disease. World J. Gastroenterol. WJG 2013, 19, 4935–4943. [Google Scholar] [CrossRef] [Green Version]
- Vancamelbeke, M.; Vanuytsel, T.; Farre, R.; Verstockt, S.; Ferrante, M.; Van Assche, G.; Rutgeerts, P.; Schuit, F.; Vermeire, S.; Arijs, I.; et al. Genetic and Transcriptomic Bases of Intestinal Epithelial Barrier Dysfunction in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2017, 23, 1718–1729. [Google Scholar] [CrossRef]
- Vatn, S.S.; Lindstrom, J.C.; Moen, A.E.F.; Brackmann, S.; Tannaes, T.M.; Olbjorn, C.; Bergemalm, D.; Keita, A.V.; Gomollon, F.; Detlie, T.E.; et al. Mucosal Gene Transcript Signatures in Treatment Naive Inflammatory Bowel Disease: A Comparative Analysis of Disease to Symptomatic and Healthy Controls in the European IBD-Character Cohort. Clin. Exp. Gastroenterol. 2022, 15, 5–25. [Google Scholar] [CrossRef]
- Momozawa, Y.; Dmitrieva, J.; Theatre, E.; Deffontaine, V.; Rahmouni, S.; Charloteaux, B.; Crins, F.; Docampo, E.; Elansary, M.; Gori, A.S.; et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat. Commun. 2018, 9, 2427. [Google Scholar] [CrossRef]
- Bergemalm, D.; Andersson, E.; Hultdin, J.; Eriksson, C.; Rush, S.T.; Kalla, R.; Adams, A.T.; Keita, A.V.; D’Amato, M.; Gomollon, F.; et al. Systemic Inflammation in Preclinical Ulcerative Colitis. Gastroenterology 2021, 161, 1526–1539. [Google Scholar] [CrossRef]
- Czarnewski, P.; Parigi, S.M.; Sorini, C.; Diaz, O.E.; Das, S.; Gagliani, N.; Villablanca, E.J. Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification. Nat. Commun. 2019, 10, 2892. [Google Scholar] [CrossRef]
- Holgersen, K.; Kutlu, B.; Fox, B.; Serikawa, K.; Lord, J.; Hansen, A.K.; Holm, T.L. High-resolution gene expression profiling using RNA sequencing in patients with inflammatory bowel disease and in mouse models of colitis. J. Crohn's Colitis 2015, 9, 492–506. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UC.I n = 17 | UC.nI n = 14 | Cntrl n = 27 | |
|---|---|---|---|
| Gender (male/female) | 6/11 | 6/8 | 10/17 |
| Age (years) b | 35 (18–68) | 43.5 (23–65) | 39 (19–81) |
| Smoker (yes/previous/no/no data) | 3/3/9/2 | 0/1/12/1 | 0/2/19/6 |
| Concomitant drug treatmentc | |||
| AS | 2 | 7 | 0 |
| AS, CS | 4 | 0 | 0 |
| AP, TP | 0 | 2 | 0 |
| AS, CS, TP | 2 | 1 | 0 |
| ATA, CS, TP | 1 | 0 | 0 |
| CS | 1 | 1 | 0 |
| CS, TP | 1 | 0 | 0 |
| TP | 0 | 1 | 0 |
| None | 6 | 2 | 27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Söderman, J.; Berglind, L.; Almer, S. Inverse and Concordant Mucosal Pathway Gene Expressions in Inflamed and Non-Inflamed Ulcerative Colitis Patients: Potential Relevance to Aetiology and Pathogenesis. Int. J. Mol. Sci. 2022, 23, 6944. https://doi.org/10.3390/ijms23136944
Söderman J, Berglind L, Almer S. Inverse and Concordant Mucosal Pathway Gene Expressions in Inflamed and Non-Inflamed Ulcerative Colitis Patients: Potential Relevance to Aetiology and Pathogenesis. International Journal of Molecular Sciences. 2022; 23(13):6944. https://doi.org/10.3390/ijms23136944
Chicago/Turabian StyleSöderman, Jan, Linda Berglind, and Sven Almer. 2022. "Inverse and Concordant Mucosal Pathway Gene Expressions in Inflamed and Non-Inflamed Ulcerative Colitis Patients: Potential Relevance to Aetiology and Pathogenesis" International Journal of Molecular Sciences 23, no. 13: 6944. https://doi.org/10.3390/ijms23136944