
RNA Cleavage Properties of Nucleobase-Specific RNase MC1 and Cusativin Are Determined by the Dinucleotide-Binding Interactions in the Enzyme-Active Site



Abstract
:
1. Introduction
2. Results
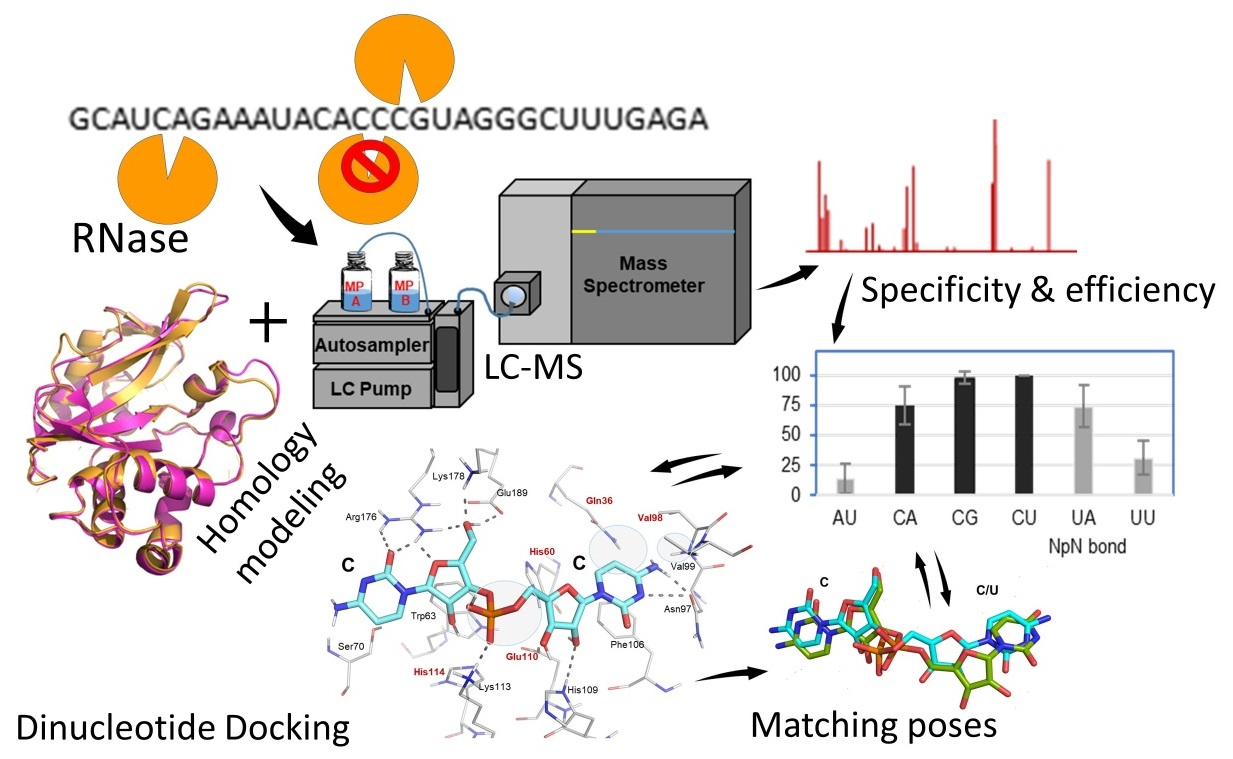
2.1. Dinucleotide-Based Substrate Specificity and Cleavage Efficiency of RNase MC1 and Cusativin
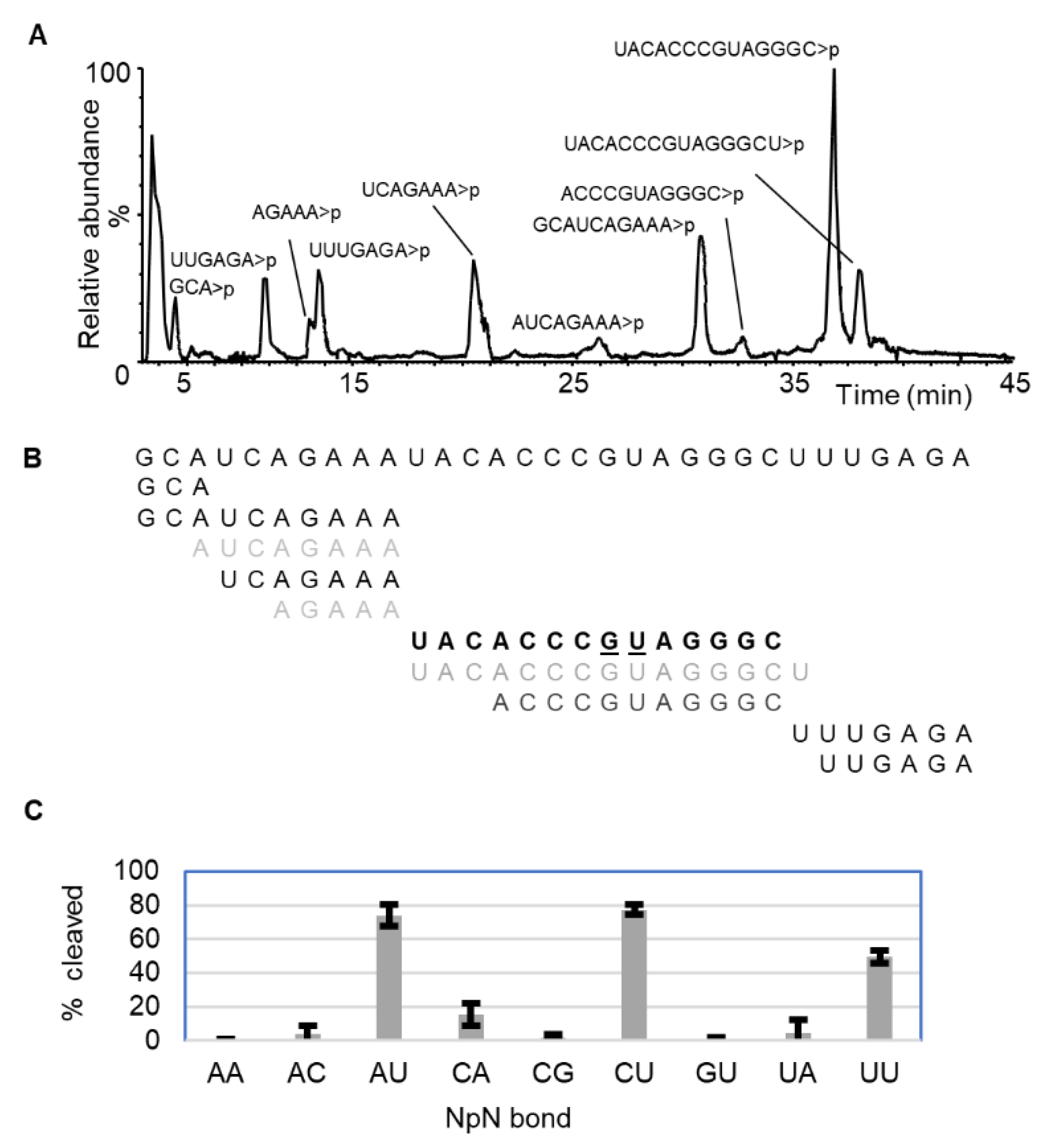
2.1.1. RNase MC1
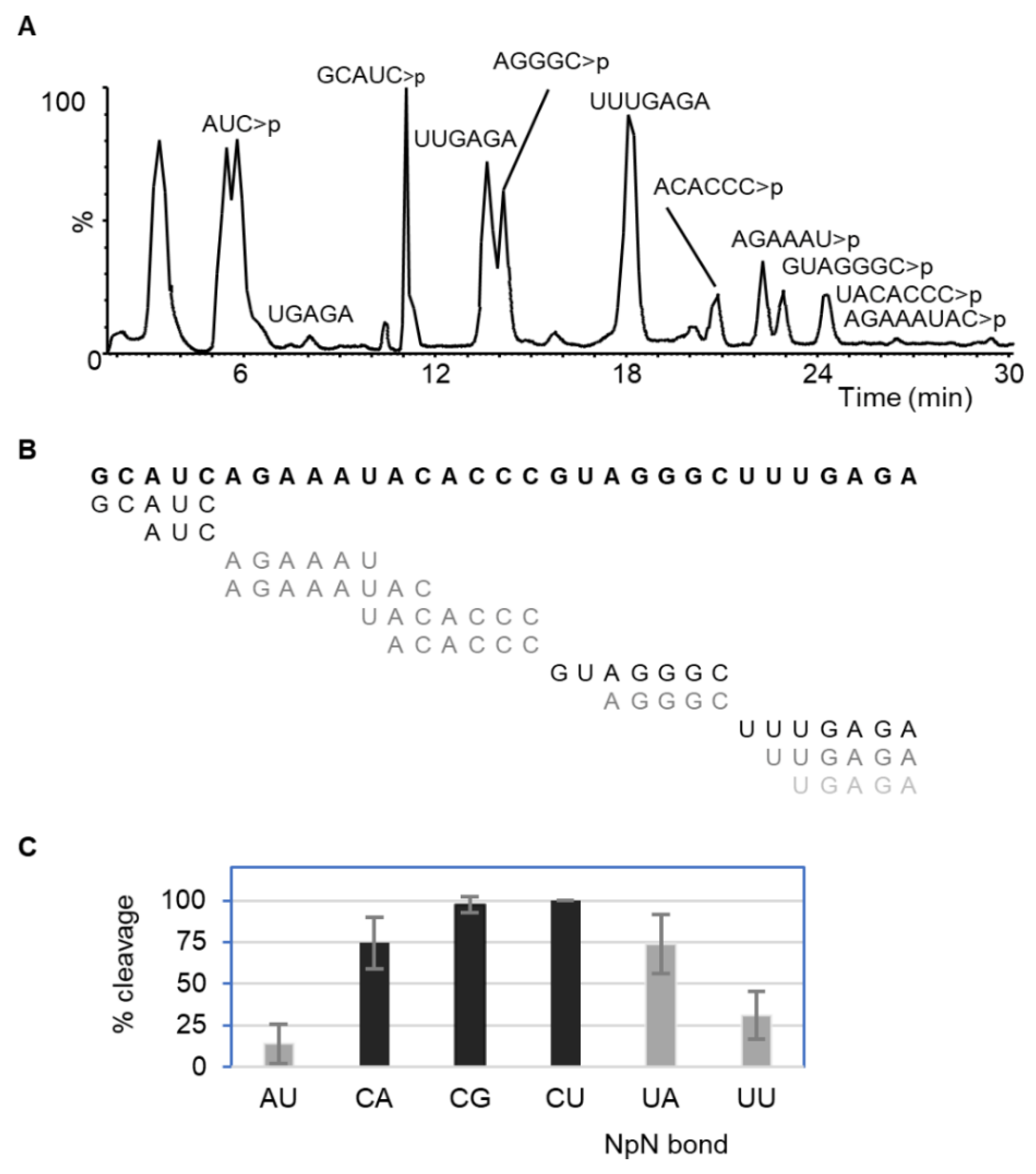
2.1.2. RNase Cusativin
2.2. In Silico Molecular Docking of RNase Active Site with Nucleotide Substrates
2.2.1. Docking RNase T1 with Single Nucleotide Ligands
2.2.2. Docking RNase A with Dinucleotide Ligands
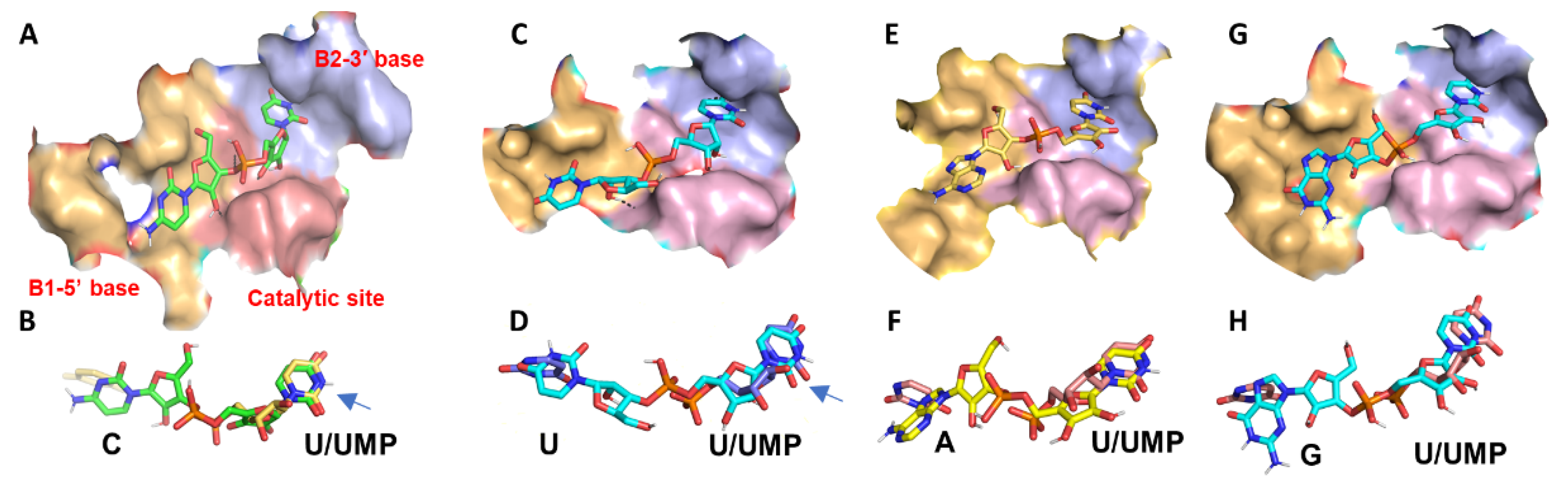
2.2.3. Docking RNase MC1 with Nucleotide Ligands
Docking MC1 with CpU
Docking MC1 with UpU
Docking MC1 with ApU
Docking MC1 with GpU
2.3. Homology Modeling of Cusativin and Docking with Dinucleotide Ligands
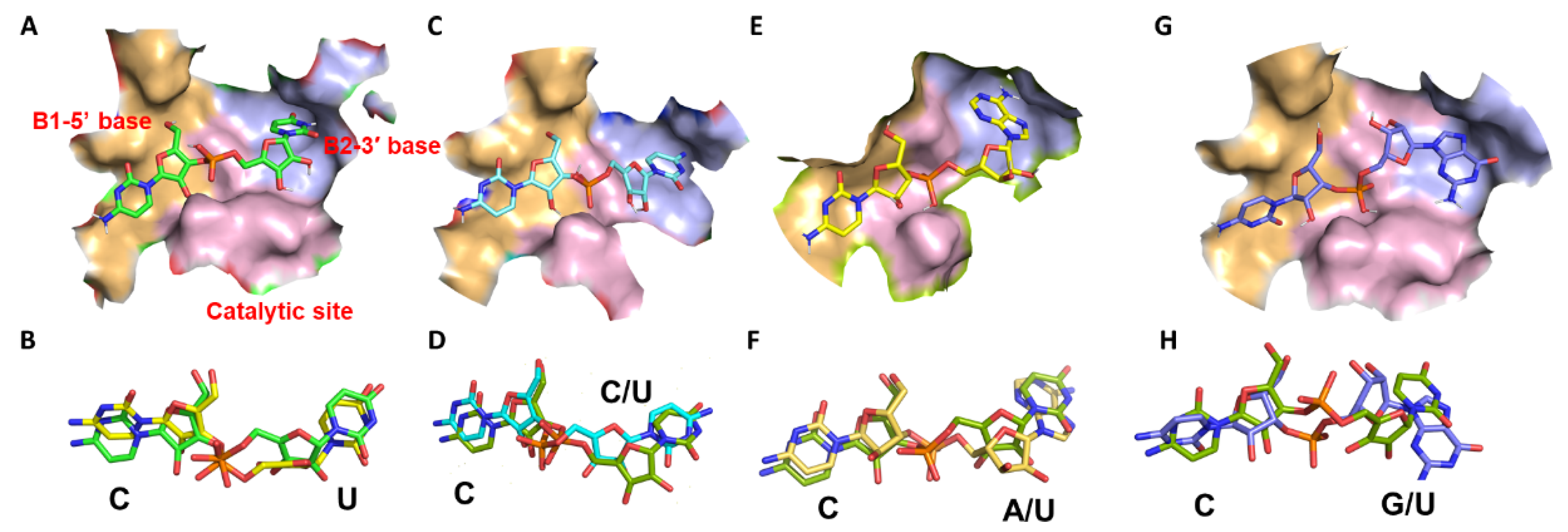
2.3.1. Docking Cusativin with CpU
2.3.2. Docking Cusativin with CpA
2.3.3. Docking Cusativin with CpC
2.3.4. Docking Cusativin with CpG
2.3.5. Docking Cusativin with UpA, ApU, and UpU Ligands
2.4. Effects of the Site-Directed Mutagenesis in the Cusativin-Active Site
2.4.1. Enzyme Kinetic Analysis of Mutants
2.4.2. Docking Interactions of Cusativin Mutants
3. Discussion
3.1. Cleavage Specificity and Efficiency of RNase MC1
3.2. Cleavage Specificity and Efficiency of Cusativin
3.3. Comparison of the Structure and Base Recognition by RNase MC1 and Cusativin
4. Materials and Methods
4.1. RNA
4.2. Cusativin Mutant Protein Preparation
4.3. RNA Digestion by Ribonucleases
4.4. LC-MS Analysis
4.5. Multiple Sequence Alignment
4.6. Protein Modeling and Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Houseley, J.; Tollervey, D. The Many Pathways of RNA Degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, M. Structure-Function Relationships of Acid Ribonucleases: Lysosomal, Vacuolar, and Periplasmic Enzymes. Pharmacol. Ther. 1999, 81, 77–89. [Google Scholar] [CrossRef]
- Luhtala, N.; Parker, R. T2 Family ribonucleases: Ancient enzymes with diverse roles. Trends Biochem. Sci. 2010, 35, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussaoui, M.; Boix, E.; Vilanova, M.; Cuchillo, C.M. The contribution of noncatalytic phosphate-binding subsites to the mechanism of bovine pancreatic ribonuclease A. Cell. Mol. LifeSci. CMLS 1998, 54, 766–774. [Google Scholar] [CrossRef]
- Prats-Ejarque, G.; Lu, L.; Salazar, V.A.; Moussaoui, M.; Boix, E. Evolutionary Trends in RNA Base Selectivity Within the RNase A Superfamily. Front. Pharmacol. 2019, 10, 1170. [Google Scholar] [CrossRef]
- Richards, F.M.; Wyckoff, H.W. 24 Bovine Pancreatic Ribonuclease. In The Enzymes; Boyer, P.D., Ed.; Academic Press: Cambridge, MA, USA, 1971; Volume 4, pp. 647–806. [Google Scholar] [CrossRef]
- Yoshida, H. The Ribonuclease T1 Family. In Methods in Enzymology; Nicholson, A.W., Ed.; Academic Press: Cambridge, MA, USA, 2001; Volume 341, pp. 28–41. [Google Scholar] [CrossRef]
- Raines, R.T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1066. [Google Scholar] [CrossRef]
- Suzukia, A.; Yaoab, M.; Tanakaab, I.; Numatac, T.; Kikukawac, S.; Yamasakic, N.; Kimura, M. Crystal Structures of the Ribonuclease MC1 from Bitter Gourd Seeds, Complexed with 2′-UMP or 3′-UMP, Reveal Structural Basis for Uridine Specificity. Biochem. Biophys. Res. Commun. 2000, 275, 572–576. [Google Scholar] [CrossRef]
- Rojo, M.A.; Arias, F.J.; Iglesias, R.; Ferreras, J.M.; Muñoz, R.; Escarmís, C.; Soriano, F.; López-Fando, J.; Méndez, E.; Girbés, T. Cusativin, a new cytidine-specific ribonuclease accumulated in seeds of Cucumis sativus L. Planta 1994, 194, 328–338. [Google Scholar] [CrossRef]
- Addepalli, B.; Lesner, N.P.; Limbach, P.A. Detection of RNA nucleoside modifications with the uridine-specific ribonuclease MC1 from Momordica charantia. RNA 2015, 21, 1746–1756. [Google Scholar] [CrossRef] [Green Version]
- Addepalli, B.; Venus, S.; Thakur, P.; Limbach, P.A. Novel ribonuclease activity of cusativin from Cucumis sativus for mapping nucleoside modifications in RNA. Anal. Bioanal. Chem. 2017, 409, 5645–5654. [Google Scholar] [CrossRef]
- Addepalli, B.; Limbach, P.A. Pseudouridine in the Anticodon of Escherichia coli tRNATyr(QΨA) Is Catalyzed by the Dual Specificity Enzyme RluF. J. Biol. Chem. 2016, 291, 22327–22337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Limbach, P.; Addepalli, B. Characterization of UVA-Induced Alterations to Transfer RNA Sequences. Biomolecules 2020, 10, 1527. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Estevez, M.; Lobue, P.A.; Limbach, P.A.; Addepalli, B. Improved RNA modification mapping of cellular non-coding RNAs using C- and U-specific RNases. Analyst 2019, 145, 816–827. [Google Scholar] [CrossRef]
- Wong, S.Y.; Javid, B.; Addepalli, B.; Piszczek, G.; Strader, M.B.; Limbach, P.A.; Barry, C.E. Functional Role of Methylation of G518 of the 16S rRNA 530 Loop by GidB in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 6311–6318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünberg, S.; Wolf, E.J.; Jin, J.; Ganatra, M.B.; Becker, K.; Ruse, C.; Taron, C.H.; Corrêa, I.R.; Yigit, E. Enhanced expression and purification of nucleotide-specific ribonucleases MC1 and Cusativin. Protein Expr. Purif. 2021, 190, 105987. [Google Scholar] [CrossRef] [PubMed]
- Boix, E.; Nikolovski, Z.; Moiseyev, G.P.; Rosenberg, H.F.; Cuchillo, C.M.; Nogués, M.V. Kinetic and Product Distribution Analysis of Human Eosinophil Cationic Protein Indicates a Subsite Arrangement That Favors Exonuclease-type Activity. J. Biol. Chem. 1999, 274, 15605–15614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkakoti, N. Enzyme specificity: Base recognition and hydrolysis of RNA by ribonuclease A. FEBS Lett. 1983, 162, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Gohda, K.; Oka, K.I.; Tomita, K.I.; Hakoshima, T. Crystal structure of RNase T1 complexed with the product nucleotide 3’-GMP. Structural evidence for direct interaction of histidine 40 and glutamic acid 58 with the 2’-hydroxyl group of the ribose. J. Biol. Chem. 1994, 269, 17531–17536. [Google Scholar] [CrossRef]
- Pearce, R.; Zhang, Y. Toward the solution of the protein structure prediction problem. J. Biol. Chem. 2021, 297, 100870. [Google Scholar] [CrossRef]
- Zhang, H.; Shen, Y. Template-based prediction of protein structure with deep learning. BMC Genom. 2020, 21, 878. [Google Scholar] [CrossRef]
- Abbasi, K.; Razzaghi, P.; Poso, A.; Ghanbari-Ara, S.; Masoudi-Nejad, A. Deep Learning in Drug Target Interaction Prediction: Current and Future Perspectives. Curr. Med. Chem. 2021, 28, 2100–2113. [Google Scholar] [CrossRef] [PubMed]
- Pakhrin, S.; Shrestha, B.; Adhikari, B.; Kc, D. Deep Learning-Based Advances in Protein Structure Prediction. Int. J. Mol. Sci. 2021, 22, 5553. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, N.; Kim, J.; Murgo, J.-R.; Kissai, M.; Ravichandran, K.R.; Miracco, E.J.; Presnyak, V.; Hua, S. Oligonucleotide Sequence Mapping of Large Therapeutic mRNAs via Parallel Ribonuclease Digestions and LC-MS/MS. Anal. Chem. 2019, 91, 8500–8506. [Google Scholar] [CrossRef] [PubMed]
- Evke, S.; Lin, Q.; Melendez, J.A.; Begley, T.J. Epitranscriptomic Reprogramming Is Required to Prevent Stress and Damage from Acetaminophen. Genes 2022, 13, 421. [Google Scholar] [CrossRef] [PubMed]
- Zegers, I.; Loris, R.; Dehollander, G.; Haikal, A.F.; Poortmans, F.; Steyaert, J.; Wyns, L. Hydrolysis of a slow cyclic thiophosphate substrate of RNase T1 analyzed by time-resolved crystallograph. Nat. Genet. 1998, 5, 280–283. [Google Scholar] [CrossRef]
- Zegers, I.; Haikal, A.; Palmer, R.; Wyns, L. Crystal structure of RNase T1 with 3’-guanylic acid and guanosine. J. Biol. Chem. 1994, 269, 127–133. [Google Scholar] [CrossRef]
- Arni, R.K.; Watanabe, L.; Ward, R.J.; Kreitman, R.J.; Kumar, K.; Walz, F.G. Three-Dimensional Structure of Ribonuclease T1 Complexed with an Isosteric Phosphonate Substrate Analogue of GpU: Alternate Substrate Binding Modes and Catalysis. Biochemistry 1999, 38, 2452–2461. [Google Scholar] [CrossRef]
- Salmas, R.E.; Seeman, P.; Aksoydan, B.; Erol, I.; Kantarcioglu, I.; Stein, M.; Yurtsever, M.; Durdagi, S. Analysis of the Glutamate Agonist LY404,039 Binding to Nonstatic Dopamine Receptor D2 Dimer Structures and Consensus Docking. ACS Chem. Neurosci. 2017, 8, 1404–1415. [Google Scholar] [CrossRef] [Green Version]
- Barai, P.; Raval, N.; Acharya, S.; Borisa, A.; Bhatt, H.; Acharya, N. Neuroprotective effects of bergenin in Alzheimer’s disease: Investigation through molecular docking, in vitro and in vivo studies. Behav. Brain Res. 2018, 356, 18–40. [Google Scholar] [CrossRef]
- Beverly, M.; Hagen, C.; Slack, O. Poly A tail length analysis of in vitro transcribed mRNA by LC-MS. Anal. Bioanal. Chem. 2018, 410, 1667–1677. [Google Scholar] [CrossRef]
- Zegers, I.; Maes, D.; Dao-Thi, M.-H.; Wyns, L.; Poortmans, F.; Palmer, R. The structures of rnase a complexed with 3′-CMP and d(CpA): Active site conformation and conserved water molecules. Protein Sci. 1994, 3, 2322–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, A.; Tanaka, I.; Sakai, R.; Nakashima, T.; Funatsu, G.; Kimura, M. Crystal structure of a ribonuclease from the seeds of bitter gourd (Momordica charantia) at 1.75 Å resolution. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1999, 1433, 253–260. [Google Scholar] [CrossRef]
- Numata, T.; Suzuki, A.; Kakuta, Y.; Kimura, K.; Yao, M.; Tanaka, I.; Yoshida, Y.; Ueda, T.; Kimura, M. Crystal Structures of the Ribonuclease MC1 Mutants N71T and N71S in Complex with 5‘-GMP: Structural Basis for Alterations in Substrate Specificity. Biochemistry 2003, 42, 5270–5278. [Google Scholar] [CrossRef] [PubMed]
- Głów, D.; Nowacka, M.; Skowronek, K.J.; Bujnicki, J.M. Sequence-specific endoribonucleases. Postepy Biochem. 2016, 62, 303–314. [Google Scholar] [CrossRef]
- Masuda, H.; Inouye, M. Toxins of Prokaryotic Toxin-Antitoxin Systems with Sequence-Specific Endoribonuclease Activity. Toxins 2017, 9, 140. [Google Scholar] [CrossRef] [Green Version]
- Numata, T.; Suzuki, A.; Yao, M.; Tanaka, I.; Kimura, M. Amino Acid Residues in Ribonuclease MC1 from Bitter Gourd Seeds Which Are Essential for Uridine Specificity. Biochemistry 2000, 40, 524–530. [Google Scholar] [CrossRef]
- Jisna, V.A.; Jayaraj, P.B. Protein Structure Prediction: Conventional and Deep Learning Perspectives. J. Protein Chem. 2021, 40, 522–544. [Google Scholar] [CrossRef]
- Deshpande, R.; Shankar, V. Ribonucleases from T2 Family. Crit. Rev. Microbiol. 2002, 28, 79–122. [Google Scholar] [CrossRef]
- Thakur, P.; Abernathy, S.; Limbach, P.A.; Addepalli, B. Locating chemical modifications in RNA sequences through ribonucleases and LC-MS based analysis. Methods Enzymol. 2021, 658, 1–24. [Google Scholar] [CrossRef]
- Lobue, P.A.; Yu, N.; Jora, M.; Abernathy, S.; Limbach, P.A. Improved application of RNAModMapper—An RNA modification mapping software tool—For analysis of liquid chromatography tandem mass spectrometry (LC-MS/MS) data. Methods 2018, 156, 128–138. [Google Scholar] [CrossRef]
- Yu, N.; Lobue, P.A.; Cao, X.; Limbach, P.A. RNAModMapper: RNA Modification Mapping Software for Analysis of Liquid Chromatography Tandem Mass Spectrometry Data. Anal. Chem. 2017, 89, 10744–10752. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Sapundzhi, F.; Prodanova, K.; Lazarova, M. Survey of the scoring functions for protein-ligand docking. AIP Conf. Proc. 2019, 2172, 100008. [Google Scholar] [CrossRef]
- Kowalak, J.A.; Pomerantz, S.; Crain, P.F.; McCloskey, J.A. A novel method for the determination of posttranscriptional modification in RNA by mass spectrometry. Nucleic Acids Res. 1993, 21, 4577–4585. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, S.C.; Kowalak, J.A.; McCloskey, J.A. Determination of oligonucleotide composition from mass spectrometrically measured molecular weight. J. Am. Soc. Mass Spectrom. 1993, 4, 204–209. [Google Scholar] [CrossRef] [Green Version]
- Wein, S.; Andrews, B.; Sachsenberg, T.; Santos-Rosa, H.; Kohlbacher, O.; Kouzarides, T.; Garcia, B.A.; Weisser, H. A computational platform for high-throughput analysis of RNA sequences and modifications by mass spectrometry. Nat. Commun. 2020, 11, 926. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NpN | % Cleavage | |
|---|---|---|
| MC1 | Cusativin | |
| ApC | 0–5 | 0 |
| ApU | 70–100 | 14–30 |
| CpA | 10–20 | 70–90 |
| CpG | 0–1 | 80–100 |
| CpU | 80–100 | 80–100 |
| UpA | 0–5 | 70–90 |
| UpU | 45–60 | 14–30 |
| GpU | 1–1.5 | 0 |
| ApA | 0–5 | 0 |
| RNase | Nucleotide Binding Site | |||||
|---|---|---|---|---|---|---|
| B1 Site | B2 Site | |||||
| MC1 | T43 | Q164 | R152 | R181 | L73 | R74 |
| Cusativin | R69 | E189 | K178 | R208 | V99 | T100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thakur, P.; Atway, J.; Limbach, P.A.; Addepalli, B. RNA Cleavage Properties of Nucleobase-Specific RNase MC1 and Cusativin Are Determined by the Dinucleotide-Binding Interactions in the Enzyme-Active Site. Int. J. Mol. Sci. 2022, 23, 7021. https://doi.org/10.3390/ijms23137021
Thakur P, Atway J, Limbach PA, Addepalli B. RNA Cleavage Properties of Nucleobase-Specific RNase MC1 and Cusativin Are Determined by the Dinucleotide-Binding Interactions in the Enzyme-Active Site. International Journal of Molecular Sciences. 2022; 23(13):7021. https://doi.org/10.3390/ijms23137021
Chicago/Turabian StyleThakur, Priti, Jowad Atway, Patrick A. Limbach, and Balasubrahmanyam Addepalli. 2022. "RNA Cleavage Properties of Nucleobase-Specific RNase MC1 and Cusativin Are Determined by the Dinucleotide-Binding Interactions in the Enzyme-Active Site" International Journal of Molecular Sciences 23, no. 13: 7021. https://doi.org/10.3390/ijms23137021