1. Introduction
Osteoarthritis (OA) is a common chronic bone disease characterized by deterioration and inflammation. The primary symptoms of OA include pain and mobility problems. Various variables, including age and trauma, influence the development of OA [
1]. Chondrocytes are the only cells found in the cartilage tissue and play a crucial role in maintaining cartilage stability. Chondrocytes undergo various alterations throughout the onset and progression of OA, including changes in their proliferation, viability, and secretion [
2]. OA is considered a multifaceted illness affecting the entire joint rather than only the cartilage or synovium. This opens new avenues for identifying and developing novel therapies as well as reprofiling candidate medications. Advanced knowledge regarding the pathogenic mechanisms of OA has elucidated the crucial functions of various new pathways that can be targeted. Because OA is a complex disease, a single treatment targeting a specific joint tissue may not be beneficial, and a single multipurpose therapy has yet to be developed [
1,
3].
Detailed knowledge of basic mechanical modifications is required to examine temporal fluctuations in disease progression, such as the transition from high bone resorption in the initial stages to decreased bone resorption in the end stages, and to detect changes in the pain intensity. The selection of suitable medication during specific time points of the illness may assist in the future development of personalized medicine for every patient. In addition, OA may exhibit conflicting endotypes, such as inflammatory pain, which may benefit from the combination of drugs targeting both pain and inflammation [
3,
4]. OA is not a distinct illness with a single pathological mechanism but involves a combination of processes and risk factors that lead to the mechanical problem of the joint. Thus, recognizing initial OA stages can be valuable for the development of efficient personalized therapies. The identification of reliable biomarkers and the development of effective imaging tools and interdisciplinary therapy regimens are critical. Cellular senescence, epigenetic changes, mitochondrial failure, dysregulated nutrition sensing, stem cell depletion, proteostasis loss, and telomere attrition have been proposed as foundational events in aging [
5]. These processes might be interconnected, and some crosstalk might exist among them. Researchers have focused increasing attention on cellular senescence because the elucidation of aging-related processes underlying the development of numerous illnesses can facilitate the development of therapies.
GDF15, a member of the tumor growth factor (TGF)-β superfamily, is involved in various pathological illnesses, including coronary artery disease, diabetes, and several cancer types [
6,
7,
8]. GDF15 was recently described as a potential biomarker of aging [
9]. Furthermore, higher GDF15 levels were observed in older adults. Moreover, individuals with higher GDF15 levels had higher aspartate aminotransferase and total cholesterol (TC)/high-density lipoprotein-cholesterol (HDL-C) levels. These biological features are typically linked with aging-related diseases, such as atherosclerosis [
10]. The aforementioned findings indicate that GDF15 may play a crucial role in driving chondrocyte senescence and the angiogenic microenvironment in OA.
In this study, we combined the bioinformatics approach with clinical, in vitro, and in vivo assays to understand the role of GDF15 in driving senescence in chondrocytes and its effect on angiogenesis. The results of this study revealed that the targeting of GDF15 in OA may constitute a therapeutic strategy.
3. Discussion
Osteoarthritis is a type of arthritis that affects the joints. Significant shifts in the metabolism, functionality, and anatomical structure of varying joints and periarticular tissues clearly define this condition. The damage affects structures including cartilage, meniscus, synovial, and subchondral bone. The most typical symptoms of osteoarthritis are mechanical pain, joint abnormalities and swelling, limitation of range of movement, joint stiffness, and motion cracks. Flaring phases with dominant inflammation, which are commonly characterized by joint swelling, a fast escalating pain, discomfort at rest, and increased stiffness, might impede the natural development of OA [
2,
11,
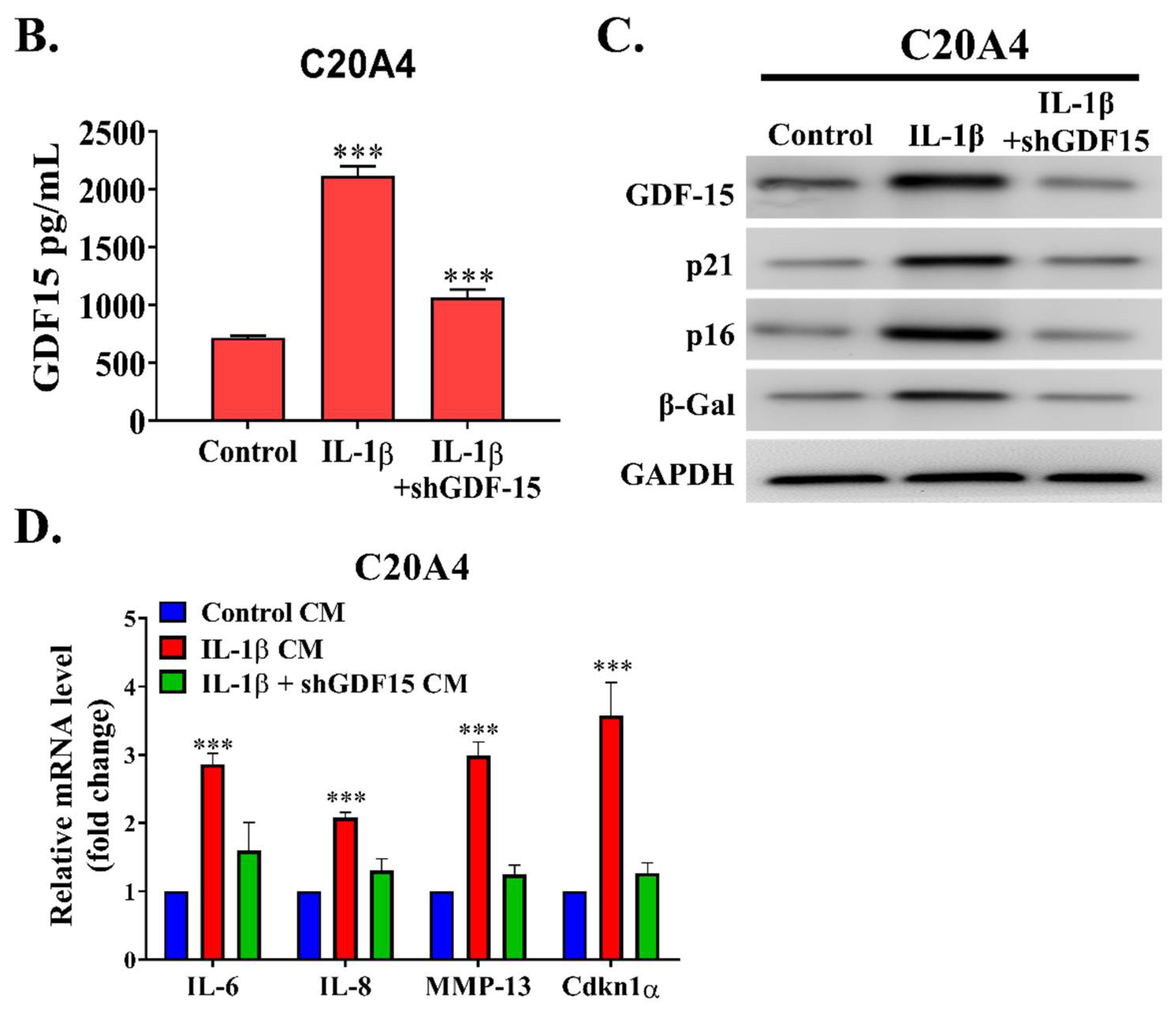
12]. According this study, we demonstrated the role of GDF15 in OA. By employing a bioinformatics approach, we observed that GDF15 is among the most upregulated genes in early OA and that its expression may have induced cellular senescence in chondrocytes affected by OA through MAPK14 activation. We determined that GDF15 is highly secreted in the inflammatory condition and that the utilization of GDF15-rich media induced the expression of SASP in chondrocytes. This secretory phenotype, in turn, resulted in the migration of and tube formation in HUVEC cells, implicating the role of GDF15 in angiogenesis. Because ErbB2 was reported to be expressed in chondrocytes and GDF15 was observed to interact with this receptor in ovarian cancer, we demonstrated that this interaction was conserved in chondrocytes and may have partially explained the mechanism of the GDF15/MAPK14 signaling pathway. Translationally, we noted that GDF15-nAb, a GDF15 neutralizing antibody, effectively hindered the expression of SASP in chondrocytes in vitro and in vivo. From our own perspective, this is a pioneering study highlighting that GDF15 accelerates chondrocyte senescence and might be used as a therapeutic target for OA.
In the early and late phases of OA, inflammation targets SM, causing synovitis. The spread of inflammation in the early stage is restricted to regions near chondropathy sites and is linked with the acceleration of cartilage degeneration (chondrolysis). This finding implies that inflammation is caused by cartilage disintegration. Synovitis occurs throughout the SM in advanced OA, causing fibrosis and villi hypertrophy. Tensile load damages cartilage directly or activates chondrocytes, exacerbating aberrant concentrations of matrix metalloproteinases (MMPs) and causing reactive oxygen species (ROS) to be generated, resulting in cartilage breakdown and the efflux of microcrystals, osteochondral fragments, and extracellular matrix decomposition products in the joint space. Inflamed synovial cells that encompass synoviocytes, macrophages, and lymphocytes secrete cytokines, chemokines, lipid mediators, reactive oxygen species (ROS), and MMPs, which can directly cause deterioration of cartilage matrix components or dysregulate chondrocyte metabolic activity, culminating in a mismatch between cartilage matrix breakdown and production. Cartilaginous degradation products, as well as proinflammatory cytokines produced by chondrocytes and other joint cells, aggravate SM inflammation, creating a continuous devastating loop [
13].
Angiogenesis, or the development of new capillaries from existing blood vessels, has been related to inflammation. The effector cells of the inflammation phase release proangiogenic molecules that facilitate the formation and invasion of new blood vessels, allowing inflammatory cells to invade. Proinflammatory cytokines can directly cause neovascularization or stimulate the synthesis of VEGF. The cytokines, including tumor necrosis factor (TNF-α), IL-1, IL-6, IL-15, IL-17, IL-18, oncostatin M, granulocyte colony-stimulating factor, granulocyte–macrophage colony-stimulating factor, and macrophage migration inhibitory factor play a role in angiogenesis and OA synovitis [
14,
15]. Angiogenesis appears to be a major factor in the persistence of OA because it plays a key role in synovial inflammation and cartilage degradation. Angiogenesis promotes the infiltration of inflammatory cells and the localization of pain receptors. Thus, inhibiting angiogenesis can control inflammation and pain in OA.
Senescence causes metabolic changes in cells, thus promoting the progression of OA over time. Senescent fibroblasts transplanted into mouse knee joints induced cartilage degradation and osteophyte development and reduced mobility, implying that senescent cells modify the synovial milieu and cause OA-like arthropathy [
16]. Senescent joint cells share characteristics such as the erosion of telomeres, the enhanced expressions of p53 and the CDK inhibitors p21 and p16INK4a (p16), the enhanced formation of ROS owing to mitochondrial failure, and the enhanced expression of senescence-associated heterochromatin [
17]. SASP is found in osteocytes, chondrocytes, and synovial fibroblasts. SASP is characterized by the release of proinflammatory cytokines such as IL-1, IL-17, IL-6, TNF-α, and oncostatin M, and various SASP components trigger OA-related alterations such as inflammation, bone development, and extracellular matrix destruction. Thus, discovering the phenotypic ramifications of SASP factors in joint tissues can elucidate the etiology of OA [
18,
19,
20,
21].
GDF15, like other proteins, is regulated at several levels in cells, including transcription, translation, and even translocation. From the cytoplasmic compartment, GDF15 originates as a precursor form as proGDF15 dimer before being processed and secreted as mature form of dimeric GDF15. In addition, the propeptide of GDF15, which is a cleavage product and an unprocessed proGDF15 dimer, can attach to the extracellular matrix and operate as a deposit site. Only mature GDF15 levels in the blood can be easily identified, and they are very low in healthy people; nevertheless, they are significantly raised in cancer, cardiovascular disease, liver and kidney disease, and tissue damage. Additionally, during pregnancy, GDF15 levels in the blood are significantly elevated, while GDF15 levels in the placenta are substantially elevated. Age, smoking, stress, and environmental variables are all risk factors that might elevate GDF15 concentrations. As a result, GDF15 is indicated as a biomarker for a variety of disorders and is thought to be a predictor of all-cause death [
8,
9,
10].
GDF15, together with osteopontin and IL-8, is released by senescent endothelial cells and triggers the ROS accumulation mediated by p16 signaling [
22]. Similar results were reported in senescent adult blood endothelial colony-forming cells that generated more GDF15. This phenomenon shows an advantageous paracrine action on nonsenescent endothelial cells; GDF15 binds to the activin receptor-like kinase 1 receptor and stimulates the Smad1 pathway. This would further trigger senescence across the airway epithelial cells exposed to pollutants, such as cigarette smoke [
23]. The researchers used GDF15 as a mitokine to investigate how it controls age-related inflammatory responses and immunosenescence as elements of an adaptive strategy to ageing. Mitokines are soluble molecules produced under stresses such as mitochondrial stress [
24]. It is known that GDF15 is involved in the inhibition of traditional T-cell stimulation and inflammatory cytokine production during senescence [
25]. Despite increased levels of GDF15 expression in senescent cells, it is unclear whether its paracrine effects affect neighboring cells or whether it affects distant organs by circulating throughout the body.
4. Material & Methods
4.1. Ethical Considerations for Research
The Joint Institutional Review Board of Taipei Medical University accorded the authorization for this research (N202201135). At the time of admission, all patients provided written informed consent before undergoing diagnostic and therapeutic procedures.Throughout this study, the Helsinki Declaration’s principles were implemented. At the timeframes and dosages indicated, cells were exposed to human recombinant GDF15 (PeproTech, Rocky Hill, NJ, USA), MAPK14 inhibitor, or mouse antihuman GDF15 monoclonal antibody (R&D Systems, Minneapolis, MN, USA). Long-course treatment with GDF15 includes centrifuging the cells at 300× g twice weekly and resuspending them in CM containing GDF15.
4.2. Single-Cell RNA Sequencing Dataset Processing
A related single-cell RNA profiling dataset by Nanus et al. that previously observed diversity of cell population, including fibroblast, chondrocyte, endothelial, and other stromal cells that contributed to different stages of osteoarthritis was further analyzed to disclose specific gene expression between those cell clusters [
26]. The dataset from Nanus et al. was archived in the Gene Experiment Omnibus repository with the accession number GSE176308. After downloading individual patient’s file matrix, the Seurat package (version 4.0.6) was enabled in R (version 4.0.1) to construct Seurat objects. Filtering unique characteristics and reducing low-quality mitochondrial genome were used as part of a standard pre-processing procedure. Thereafter, the Seurat object was normalized and scaled, followed by dimensional reduction and cell cluster creation using the t-Distributed Stochastic Neighbor Embedding (tSNE) method. Each cluster’s positive and negative markers were then generated and listed. To depict the amount of expression of interest markers between each cluster, an array of plots consisting of tSNE plots, dot plots, and bar plots were shown.
4.3. Tissue Specimens
The samples were obtained from patients at the institution who had undergone surgery for OA (
n = 12). The patients had a diagnosis of OA on the basis of the guidelines of the Osteoarthritis Research Society International (OARSI) [
27]. The OA grades of the patients were determined using the improved Mankin pathology score [
28]. Normal cartilage specimens were collected as control samples from individuals who had traumatic lower-limb amputation (
n = 6). The clinical characteristics of patients involved in analysis including patients with OA and control (
n total = 18) were summarized in
Table 1. Patients with progressive degeneration, evident osteoporosis, rheumatoid arthritis, and neoplasia lesions were excluded from the control group. The clinical specimens were then analyzed for further tissue staining.
4.4. Specimen Collection
The cartilage’s underlying subchondral bone was retrieved and preserved in formaldehyde. Three days later, the samples were decalcified for 14 days in formic acid solution (30%). The samples were then cut into appropriate sizes, treated with an ethanol gradient series, and placed in paraffin before being serially sectioned (5 μm). Prior to being analyzed, a portion of specimens was air-dried for approximately 6 h in an oven at 60–70 °C. Patients’ blood samples were obtained and combined with a standard anticoagulant. The samples were maintained at room temperature for 2 h, centrifuged (3000 r/min for 10 min), and then placed in a freezing tube for storage (−80 °C).
4.5. Hematoxylin–Eosin Staining
The paraffin-embedded samples were dewaxed with xylene and then re-suspended with graded ethanol solution. The specimens were then cleaned with water. Following coloring with Harris’ hematoxylin and eosin for 5 min, the samples were washed with water for 5 min prior being rinsed with filtered water. The slices were decolored by immersing them in hydrochloric acid and ethanol (0.5 percent) over ten seconds before being rinsed twice with tap water and once with filtered water. After rinsing, Feosin solution was used to stain sections for 40 s, and the slides were mounted using a gradient ethanol series after dehydration.
4.6. Safranin O/Fast Green Staining
Following a dewaxing procedure, the tissue sections were reacted for 1 min with 0.2 percent fast green solution, followed by mixing of 30 s with 1 percent acetic acid solution and 15 min with Safranin O solution. After being rinsed with 95 percent ethanol, the samples were cleaned with xylene and fixed with neutral gum before being dehydrated with a gradient ethanol sequence.
4.7. Immunohistochemistry Staining
The tissue specimens were dewaxed and subsequently blocked for about 30 min with goat serum. Primary antibodies against antirabbit-GDF15 [1:2000], p-MAPK14 [1:1000], CD31 [1:1000], and p16 [1:1000] were bought from Cell Signaling Technology (Beverly, MA, USA) and incubated overnight at 4 °C on tissue specimens. Following that, the specimens were reacted for 20 min at 37 °C using the secondary antibody (goat antirabbit IgG, 1:1000 dilution). Following multiple rinsing with phosphate-buffered solution, the samples were reacted with diaminobenzidine. A yellow–brown color in the nuclei or cytosol portion of cells was regarded as positive expression of GDF15, p-MAPK14, p16, and CD31. The proportion of positively stained cells was calculated as follows: overall number of positive cells/total cell count × 100.
4.8. Enzyme-Linked Immunosorbent Assay
The enzyme-linked immunosorbent assay test (ELISA) was used to detect the level of secreted protein in the preserved serum. The ELISA detection kit for GDF15 was procured from Abcam (cat: ab155432, Boston, MA, USA) and executed pursuant to the manufacturer guidelines. The succeeding day, the serum was retrieved, and the specimens were washed with washing buffer. The specimens were initially incubated for approximately one hour at room temperature in a reaction well before being rinsed. About 0.1 mL of enzyme-labeled antibody was administered to each well and incubated for approximately half to an hour. Each reaction well was filled with tetramethylbenzidine substrate solution, which was then incubated at 37 °C over approximately 30 min. To terminate the reaction, each well received 0.05 mL of 2 M sulfuric acid solution. The light density of each well was then calculated using an ELISA reader under absorbance of 450/630 nm.
4.9. Western Blotting
To isolate the protein content of OA chondrocytes, the sample was collected in an Eppendorf tube and further sonicated three times. In each lane of the gel, we inserted 30 g of the protein isolate and elevated the voltage from 80 through 120 V. The membrane was transferred in 100 V for about 60 min and then blocked for 1 h with 5% skim milk before being incubated overnight at 4 °C with primary antibodies (p16 (1:2000)), MAPK14/pMAPK14 (1:1000), and p21 (1:1000) which was provided by Cell Signaling Technology (Beverly, MA, USA). Other primary antibodies, which including γH2AX [1:5000], ErbB2 [1:1000], and pErbB2 [1:1000], were obtained from Abcam (Boston, MA, USA). Before incubating the membranes with the secondary antibody for an hour, membranes were rinsed three times with PBST detergent for about 5 min. After incubation with secondary antibody, the membranes were washed with PBST and viewed using the chemiluminescence reagent on the Bio-Rad Gel Doc EZ Imager (Bio-rad, Hercules, CA, USA). Image J was employed for subsequent analysis. List of the primary antibody was described in
Supplementary Table S1.
4.10. Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted and kept at −80 °C using the Trizol reagent (Invitrogen, Carlsbad, CA, USA). The absorbance at 260/280 nm and RNA concentration were examined using a UV spectrophotometer. The primers were constructed using VEGF, VEGFR2, VEGFR3, IL-8, IL-6, MMP13, and CDKN1a sequences from Genbank (Invitrogen, Carlsbad, CA, USA). To generate reverse transcription cDNA of the RNAs, a reverse transcription kit was employed (Takara, Tokyo, Japan). The real-time polymerase chain reaction (PCR) system from TransGen Biotech was adopted. For PCR, a 20-μL volume of the master mix was applied. The samples were predenaturated for 10 min before being denatured for 30 s (40 cycles, 95 °C), annealed for 60 s (58 °C), and extended for 30 s (72 °C). The housekeeping control was glyceraldehyde phosphate dehydrogenase (GAPDH). A solubility curve was used to analyze the repeatability of PCR measurement. We calculated the target gene expression level semi-quantitatively by calculating 2−△△Ct from the mean Ct value.
4.11. In Vivo OA Rat Model
Female Wistar rats which were aged 12 weeks old and weighed around 300 g were purchased from BioLASCO Taiwan Co., Ltd. and were pathogen-free. Further investigation was implemented adhering to a protocol accepted by the Taipei Medical University Laboratory Animal Committee (protocol LAC-2020-0146). In our lab, the OA model was established according to the previously evaluated technique. To prepare this model, 200 µL of 4% papain solution and 100 µL of 0.03 mol L-1 L-cysteine solution in distilled water were needed. After mixing, the reaction was incubated for 30 min. The rat’s right knee joint was injected with 25 µL of the mixture. Intra-articular doses were repeated on days 1, 4, and 7 to produce osteoarthritis. Upon successful induction of OA, the rats were randomly separated into two groups of five animals each. The further intervention was randomly assigned to receive either the vehicle (saline) for controls, or neutralizing-antibody therapy (GDF15-nAb) for the treatment group. In this study, GDF15-nAb (10 μg/mL) was injected into the joint through a 50 μL intraarticular injection (or saline alone as a control). Following 8 weeks, all rats were euthanized, and samples of knee tissue from all groups of rats were examined histologically and immunohistochemically for signs of disease.
4.12. Statistics
For each numerical variable, the mean and standard error of the mean were computed. The categorical variables were represented as frequency and percentage. To assess discrepancy across groups, an analysis of variance (ANOVA) was used. Statistical significance was indicated by a p value below 0.05. All tests were conducted in triplicate and analyzed using GraphPad Prism 8.0 (San Diego, CA, USA).



 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}