
Revisiting the Function of p21CDKN1A in DNA Repair: The Influence of Protein Interactions and Stability



Abstract
1. Introduction
1.1. p21CDKN1A, a Multifaceted Protein
1.2. p21 and DNA Repair
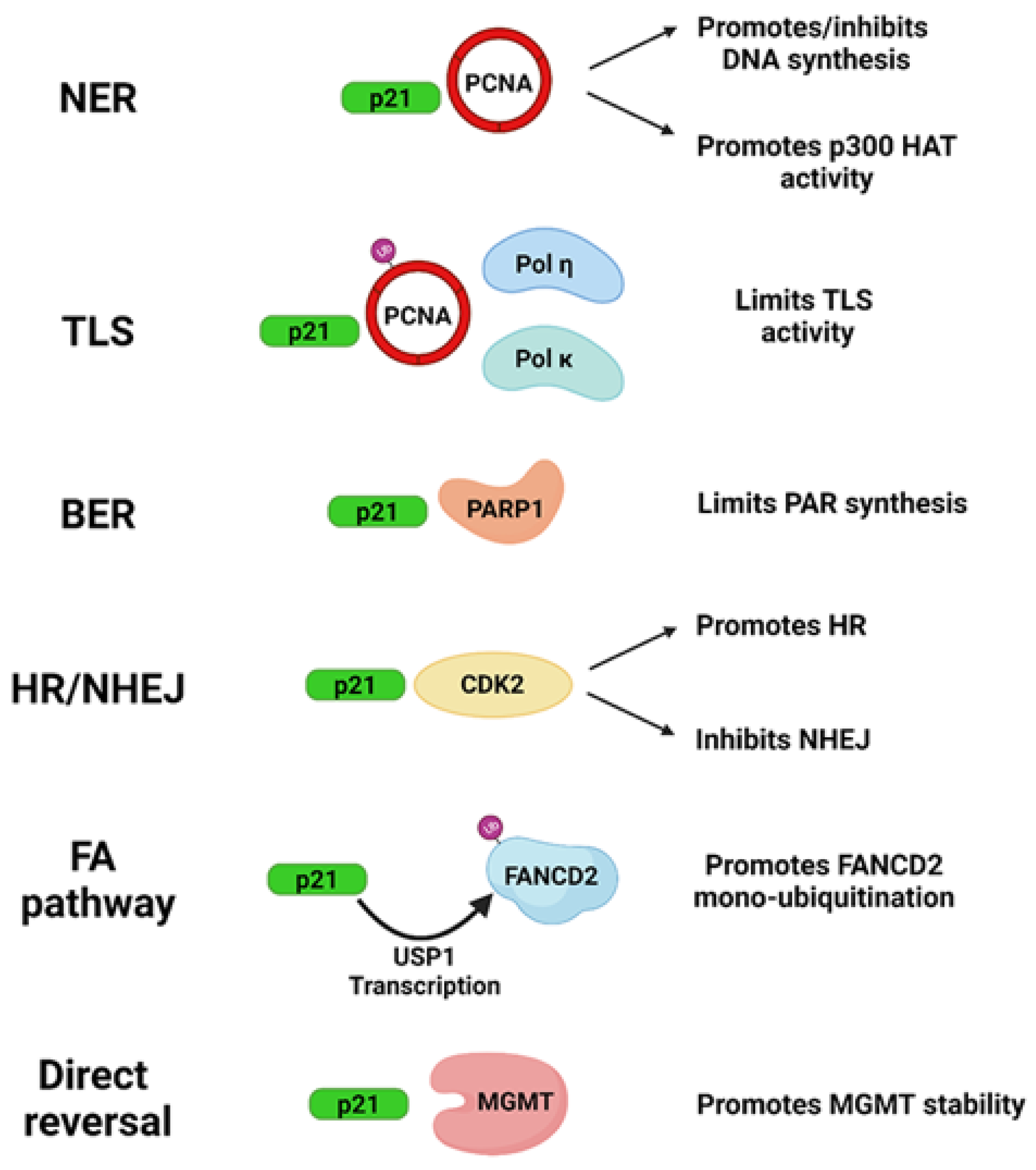
2. Influence of p21 in DNA Repair Systems: Protein Interactions
2.1. Direct Reversal
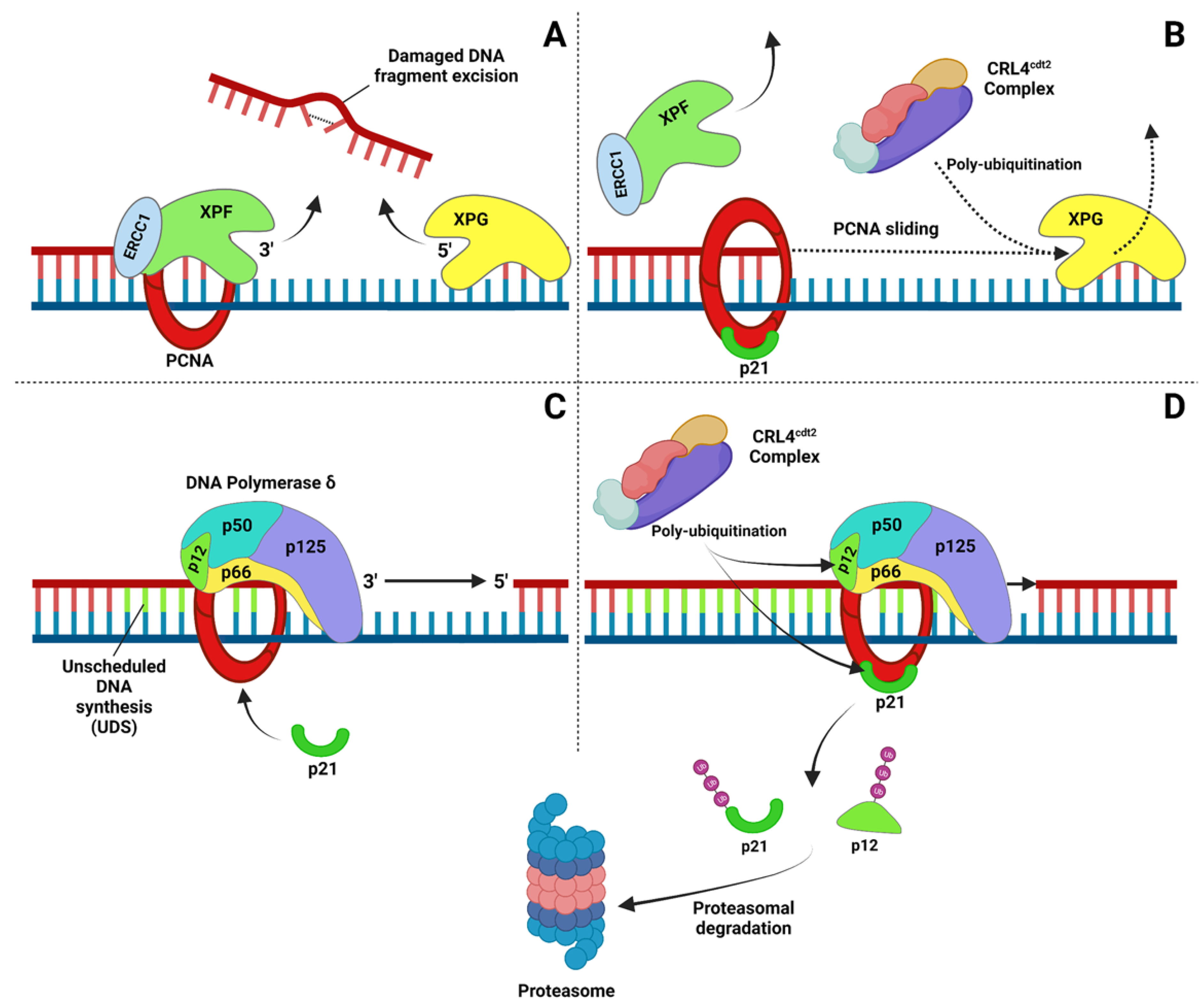
2.2. Nucleotide Excision Repair (NER)
2.3. Base Excision Repair (BER)
2.4. Non-Homologous End Joining (NHEJ)
2.5. Homologous Recombination (HR)

{kind=link}
{kind=link}
{kind=link}
| Year | Protein | Activity | DNA Repair System | Refs. |
|---|---|---|---|---|
| 1994 | PCNA | DNA synthesis | NER | [31,32,33,34,35,36] |
| 1998 | Ku70 | damage recognition | NHEJ | [73] |
| 2001 | p300 | KAT | NER | [56,58,59,60] |
| 2003 | PARP-1 | synthesis of PAR | BER, SSBR | [66,67,68] |
| 2006 | p50 (POLD2) | subunit of DNA pol δ | (not reported) | [84] |
2.6. Mismatch Repair (MMR)
2.7. Translesion DNA Synthesis (TLS)
2.8. Fanconi Anemia (FA) Pathway
3. Influence of p21 Stability in DNA Repair
4. Protein Degradation after DNA Damage: A DNA Repair-Coupled Mechanism
5. p21 Fine Regulation
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21CDKN1A in the DNA damage response. Mutat. Res./Rev. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Dutto, I.; Tillhon, M.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. Biology of the cell cycle inhibitor p21CDKN1A: Molecular mechanisms and relevance in chemical toxicology. Arch. Toxicol. 2015, 89, 155–178. [Google Scholar] [CrossRef]
- Dotto, G.P. p21(WAF1/Cip1): More than a break to the cell cycle? Biochim. Biophys. Acta 2000, 1471, M43–M56. [Google Scholar] [CrossRef]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lee, S.; Zhang, J.; Peters, S.B.; Hannah, J.; Zhang, Y.; Yin, Y.; Koff, A.; Ma, L.; Zhou, P. Cul4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol. Cell 2009, 34, 451–460. [Google Scholar] [CrossRef]
- Perucca, P.; Cazzalini, O.; Madine, M.; Savio, M.; Laskey, R.A.; Vannini, V.; Prosperi, E.; Stivala, L.A. Loss of p21 CDKN1A impairs entry to quiescence and activates a DNA damage response in normal fibroblasts induced to quiescence. Cell Cycle 2009, 8, 105–114. [Google Scholar] [CrossRef]
- Kreis, N.N.; Sanhaji, M.; Rieger, M.A.; Louwen, F.; Yuan, J. p21Waf1/Cip1 deficiency causes multiple mitotic defects in tumor cells. Oncogene 2014, 33, 5716–5728. [Google Scholar] [CrossRef]
- Mansilla, S.F.; Bertolin, A.P.; Bergoglio, V.; Pillaire, M.-J.; González Besteiro, M.A.; Luzzani, C.; Miriuka, S.G.; Cazaux, C.; Hoffmann, J.-S.; Gottifredi, V. Cyclin Kinase-independent role of p21CDKN1A in the promotion of nascent DNA elongation in unstressed cells. elife 2016, 5, e18020. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Science 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Kreis, N.N.; Louwen, F.; Yuan, J. The Multifaceted p21 (Cip1/Waf1/ CDKN1A) in Cell Differentiation, Migration and Cancer Therapy. Cancers 2019, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Kriwacki, R.W.; Hengst, L.; Tennat, L.; Reed, S.I.; Whight, P.E. Structural studies of p21waf1/cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 11504–11509. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; El-Deiry, W.S. p21WAF1 and tumorigenesis: 20 years after. Curr. Opin. Oncol. 2013, 25, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Liao, Y.; Xia, W.; Spohn, B.; Lee, M.H.; Hung, M.C. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 2001, 3, 245–252. [Google Scholar] [CrossRef]
- Cmielová, J.; Rezáčová, M. p21Cip1/Waf1 protein and its function based on a subcellular localization. J. Cell Biochem. 2011, 112, 3502–3506. [Google Scholar] [CrossRef]
- Abukhdeir, A.M.; Park, B.H. p21 and p27, roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med. 2008, 10, e19. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef]
- Mellon, I.; Rajpal, D.K.; Koi, M.; Boland, C.R.; Champe, G.N. Transcription-coupled repair deficiency and mutations in human mismatch repair genes. Science 1996, 272, 557–560. [Google Scholar] [CrossRef]
- Therrien, J.P.; Loignon, M.; Drouin, R.; Drobetsky, E.A. Ablation of p21waf1cip1 expression enhances the capacity of p53-deficient human tumor cells to repair UVB-induced DNA damage. Cancer Res. 2001, 61, 3781–3786. [Google Scholar]
- Tillhon, M.; Cazzalini, O.; Dutto, I.; Stivala, L.A.; Prosperi, E. p21CDKN1A and DNA repair systems: Recent findings and future perspectives. In New Research Directions in DNA Repair; Chen, C., Ed.; InTech: Rijeka, Croatia, 2013; pp. 249–279. [Google Scholar] [CrossRef]
- Flores-Rozas, H.; Kelman, Z.; Dean, F.B.; Pan, Z.Q.; Harper, J.W.; Elledge, S.J.; O’Donnell, M.; Hurwitz, J. Cdk-interacting protein 1 directly binds with proliferating cell nuclear antigen and inhibits DNA replication catalyzed by the DNA polymerase delta holoenzyme. Proc. Natl. Acad. Sci. USA 1994, 91, 8655–8659. [Google Scholar] [CrossRef] [PubMed]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.B.; Shamoo, Y. Structural and thermodynamic analysis of human PCNA with peptides derived from DNA polymerase-delta p66 subunit and flap endonuclease-1. Structure 2004, 12, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef]
- Prosperi, E. The fellowship of the rings: Distinct pools of proliferating cell nuclear antigen (PCNA) trimer at work. FASEB J. 2006, 20, 833–837. [Google Scholar] [CrossRef]
- Choe, K.N.; Moldovan, G.L. Forging Ahead through Darkness: PCNA, Still the Principal Conductor at the Replication Fork. Mol. Cell 2017, 65, 380–392. [Google Scholar] [CrossRef]
- Slade, D. Maneuvers on PCNA Rings during DNA Replication and Repair. Genes 2018, 9, 416. [Google Scholar] [CrossRef]
- Mostofa, A.; Punganuru, S.R.; Rao Madala, H.; Srivenugopal, K.S. S-phase Specific Downregulation of Human O 6-Methylguanine DNA Methyltransferase (MGMT) and its Serendipitous Interactions with PCNA and p21 cip1 Proteins in Glioma Cells. Neoplasia 2018, 20, 305–323. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Li, R.; Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. Differential effects by the p21 CDK inhibitor on PCNA-dependent DNA replication and repair. Nature 1994, 371, 534–537. [Google Scholar] [CrossRef]
- Shivji, M.K.K.; Grey, S.J.; Strausfeld, U.P.; Wood, R.D.; Blow, J.J. Cip1 inhibits DNA replication but not PCNA-dependent nucleotide excision repair. Curr. Biol. 1994, 4, 1062–1068. [Google Scholar] [CrossRef]
- Pan, Z.Q.; Reardon, J.T.; Li, L.; Flores-Rozas, H.; Legerski, R.; Sancar, A.; Hurwitz, J. Inhibition of nucleotide excision repair by cyclin-dependent kinase inhibitor p21. J. Biol. Chem. 1995, 270, 22008–22016. [Google Scholar] [CrossRef] [PubMed]
- Podust, V.N.; Podust, L.; Goubin, F.; Ducommun, B.; Hübscher, H. Mechanism of inhibition of proliferating cell nuclear antigen-dependent DNA synthesis by the cyclin-dependent kinase inhibitor p21. Biochemistry 1995, 34, 8869–8875. [Google Scholar] [CrossRef]
- Shivji, M.K.K.; Ferrari, E.; Ball, K.; Hübscher, U.; Wood, R.D. Resistance of human nucleotide excision repair synthesis in vitro to p21CDKN1. Oncogene 1998, 17, 2827–2838. [Google Scholar] [CrossRef][Green Version]
- Cooper, M.P.; Balajee, A.S.; Bohr, V.A. The C-terminal domain of p21 inhibits nucleotide excision repair in vitro and in vivo. Mol. Cell. Biol. 1999, 10, 2119–2129. [Google Scholar] [CrossRef]
- McDonald, E.R.; Wu, G.S.; Waldman, T.; El-Deiry, W.S. Repair defect of p21waf1/cip1-/- human cancer cells. Cancer Res. 1996, 56, 2250–2255. [Google Scholar] [PubMed]
- Fan, S.; Chang, J.K.; Smith, M.L.; Duba, D.; Fornace, A.J.; O’Connor, P.M. Cells lacking CIP1/WAF1 genes exhibit preferential sensitivity to cisplatin and nitrogen mustard. Oncogene 1997, 14, 2127–2136. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sheikh, M.S.; Chen, Y.Q.; Smith, M.L.; Fornace, A.J. Role of p21waf/cip1/sdi1 in cell death and DNA repair as studied using a tetracycline-inducible system in p53-deficient cells. Oncogene 1997, 14, 1875–1882. [Google Scholar] [CrossRef][Green Version]
- Ruan, S.; Okcu, M.F.; Ren, J.P.; Chiao, P.; Andreeff, M.; Levin, V.; Zhang, W. Overexpressed WAF1/Cip1 renders glioblastoma cells resistant to chemotherapy agents 1,3-bis(2-chloroethyl)-1-nitrosourea and cisplatin. Cancer Res. 1998, 58, 1538–1543. [Google Scholar]
- Adimoolam, S.; Lin, C.X.; Ford, J.M. The p53 regulated Cyclin-dependent kinase inhibitor, p21 (cip1,waf1,sdi1), is not required for global genomic and transcriptional coupled nucleotide excision repair of UV-induced DNA photoproducts. J. Biol. Chem. 2001, 276, 25813–25822. [Google Scholar] [CrossRef]
- Wani, M.A.; Wani, G.; Yao, J.; Zhu, Q.; Wani, A. Human cells deficient in p53 regulated p21waf/cip1 expression exhibit normal nucleotide excision repair of UV-induced DNA damage. Carcinogenesis 2002, 23, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.P.; Wei, W.; Sedivy, J.M. Bypass of senescence after disruption of p21Cip1/Waf1 gene in normal diploid human fibroblasts. Science 1997, 277, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Stivala, L.A.; Riva, F.; Cazzalini, O.; Savio, M.; Prosperi, E. p21waf1/cip1-null human fibroblasts are deficient in nucleotide excision repair downstream the recruitment of PCNA to DNA repair sites. Oncogene 2001, 20, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Smith Ml Chen, I.T.; Zhan, Q.; O’Connor, P.M.; Fornace, A.J., Jr. Involvement of the p53 tumor suppressor in repair of u.v.-type DNA damage. Oncogene 1995, 10, 1053–1059. [Google Scholar]
- Barley, R.D.; Enns, L.; Paterson, M.C.; Mirzayans, R. Aberrant p21WAF1-dependent growth arrest as the possible mechanism of abnormal resistance to ultraviolet light cytotoxicity in Li-Fraumeni syndrome fibroblast strains heterozygous for TP53 mutations. Oncogene 1998, 17, 533–543. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.-L.; Verkaiak, N.S.; Lee, M.Y.; Tam, W.L.; Venkitaraman, A.R.; Lane, D.P.; et al. Rapid recruitment of p53 to DNA damage sites directs DNA repair choice and integrity. Proc. Natl. Acad. Sci. USA 2022, 119, e2113233119. [Google Scholar] [CrossRef]
- Smith, L.; Ford, J.M.; Hollander, M.C.; Bortnick, R.A.; Amounson, S.A.; Seo, Y.R.; Deng, C.; Hanawalt, P.C.; Fornace, A.J. p53-mediated DNA repair responses to UV radiation: Studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol. Cell. Biol. 2000, 20, 3705–3714. [Google Scholar] [CrossRef]
- Maeda, T.; Chong, M.T.; Espino, R.A.; Chua, P.P.; Cao, J.Q.; Chomey, E.G.; Luong, L.; Tron, V.A. Role of p21(Waf-1) in regulating the G1 and G2/M checkpoints in ultraviolet-irradiated keratinocytes. J. Investig. Dermatol. 2002, 119, 513–521. [Google Scholar] [CrossRef]
- Maeda, T.; Espino, R.A.; Chomey, E.G.; Luong, L.; Bano, A.; Meakins, D.; Tron, V.A. Loss of p21WAF1/Cip1 in Gadd45-deficient keratinocytes restores DNA repair capacity. Carcinogenesis 2005, 26, 1804–1810. [Google Scholar] [CrossRef]
- Stoyanova, T.; Yoon, T.; Kopanja, D.; Mokyr, M.B.; Raychaudhuri, P. The xeroderma pigmentosum group E gene product DDB2 activates nucleotide excision repair by regulating the level of p21Waf1/Cip1. Mol. Cell. Biol. 2008, 28, 177–187. [Google Scholar] [CrossRef]
- Stoyanova, T.; Roy, N.; Bhattacharjee, S.; Kopanja, D.; Valli, T.; Bagchi, S.; Raychaudhuri, P. p21 Cooperates with DDB2 Protein in Suppression of Ultraviolet Ray-induced Skin Malignancies. J. Biol. Chem. 2012, 287, 3019–3028. [Google Scholar] [CrossRef] [PubMed]
- Perucca, P.; Cazzalini, O.; Mortusewicz, O.; Necchi, D.; Savio, M.; Nardo, T.; Stivala, L.A.; Leonhardt, H.; Cardoso, M.C.; Prosperi, E. Spatiotemporal dynamics of p21CDKN1A protein recruitment to DNA-damage sites and interaction with proliferating cell nuclear antigen. J. Cell Sci. 2006, 119, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Speroni, J.; Podhajcer, O.L.; Prives, C.; Gottifredi, V. p21 differentially regulates DNA replication and DNA-repair-associated processes after UV irradiation. J. Cell Sci. 2008, 121, 3271–3282. [Google Scholar] [CrossRef] [PubMed]
- Sikder, R.K.; Ellithi, M.; Uzzo, R.N.; Weader, D.J.; Metz, A.L.; Behbahani, A.; McKenzie, E.R.; El-Deiry, W.S.; Abbosh, P.H. Differential Effects of Clinically Relevant N-versus C-Terminal Truncating CDKN1A Mutations on Cisplatin Sensitivity in Bladder Cancer. Mol. Cancer Res. 2021, 19, 403–413. [Google Scholar] [CrossRef]
- Hasan, S.; Hassa, P.O.; Imhof, R.; Hottiger, M.O. Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature 2001, 410, 387–391. [Google Scholar] [CrossRef]
- Gregory, D.J.; Garcia-Wilson, E.; Poole, J.C.; Snowden, A.W.; Roninson, I.B.; Perkins, N.D. Induction of transcription through the CRD1 motif by p21WAF1/CIP1 is core promoter specific and cyclin dependent kinase independent. Cell Cycle 2002, 1, 343–350. [Google Scholar] [CrossRef]
- Cazzalini, O.; Perucca, P.; Savio, M.; Necchi, D.; Bianchi, L.; Stivala, L.A.; Ducommun, B.; Scovassi, A.I.; Prosperi, E. Interaction of p21(CDKN1A) with PCNA regulates the histone acetyltransferase activity of p300 in nucleotide excision repair. Nucleic Acids Res. 2008, 36, 1713–1722. [Google Scholar] [CrossRef]
- Tillhon, M.; Cazzalini, M.; Nardo, T.; Necchi, D.; Sommatis, S.; Stivala, L.A.; Scvassi, A.I.; Prosperi, E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair 2012, 11, 844–852. [Google Scholar] [CrossRef]
- Wang, Q.E.; Han, C.; Zhao, R.; Wani, G.; Zhu, Q.; Gong, L.; Battu, A.; Racoma, I.; Sharma, N.; Wani, A.A. p38 MAPK- and Akt-mediated p300 phosphorylation regulates its degradation to facilitate nucleotide excision repair. Nucleic Acids Res. 2013, 41, 1722–1733. [Google Scholar] [CrossRef]
- Dutto, I.; Scalera, C.; Prosperi, E. CREBBP and p300 lysine acetyl transferases in the DNA damage response. Cell Mol. Life Sci. 2018, 75, 1325–1338. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjooras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Tom, S.; Ranalli, T.A.; Podust, V.N.; Bambara, R.A. Regulatory roles of p21 and apurinic/apyrimidinic endonuclease 1 in base excision repair. J. Biol. Chem. 2001, 276, 48781–48789. [Google Scholar] [CrossRef]
- Dianova, I.I.; Bohr, V.A.; Dianov, G.L. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry 2001, 40, 12639–12644. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.S.; Bloom, L.B.; Narayan, S. Long-patch base excision repair of apurinic/apyrimidinic site DNA is decreased in mouse embryonic fibroblast cell lines treated with plumbagin: Involvement of cyclin-dependent kinase inhibitor p21Waf-1/Cip-1. Oncogene 2002, 21, 5912–5922. [Google Scholar] [CrossRef][Green Version]
- Frouin, I.; Maga, G.; Denegri, M.; Riva, F.; Savio, M.; Spadari, S.; Prosperi, E.; Scovassi, A.I. Human proliferating cell nuclear antigen, Poly(ADP-ribose) polymerase 1, and p21waf1/cip1. A dynamic exchange of partners. J. Biol. Chem. 2003, 278, 39265–39268. [Google Scholar] [CrossRef]
- Cazzalini, O.; Donà, F.; Savio, M.; Tillhon, M.; Maccario, C.; Perucca, P.; Stivala, L.A.; Scovassi, A.I.; Prosperi, E. p21CDKN1A participates in base excision repair by regulating the activity of poly(ADP-ribose) polymerase 1. DNA Repair 2010, 9, 627–635. [Google Scholar] [CrossRef]
- Sukhanova, M.V.; Khodyreva, S.N.; Lebedeva, N.A.; Prasad, R.; Wilson, S.H.; Lavrik, O.I. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase beta and poly(ADPribose) polymerase 1: Interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 2005, 33, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Amé, J.C.; Schreiber, V.; Leonhardt, H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007, 35, 7665–7675. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Sundar, I.K.; Gorbunova, V.; Rahman, I. P21-PARP-1 pathway is involved in cigarette smoke-induced lung DNA damage and cellular senescence. PLoS ONE 2013, 8, e80007. [Google Scholar] [CrossRef]
- Mahopatra, P.; Satapathy, S.R.; Das, D.; Siddarth, S.; Choudhuri, T.; Kundu, C.N. Resveratrol mediated cell death in cigarette smoke transformed breast epithelial cells is through induction of p21Waf1/Cip1 and inhibition of long patch base excision repair pathway. Toxicol. Appl. Pharmacol. 2014, 275, 221–231. [Google Scholar] [CrossRef]
- Dutto, I.; Sukhanova, M.; Tillhon, M.; Cazzalini, O.; Stivala, L.A.; Scovassi, A.I.; Lavrik, O.; Prosperi, E. p21CDKN1A Regulates the Binding of Poly(ADP-Ribose) Polymerase-1 to DNA Repair Intermediates. PLoS ONE 2016, 11, e0146031. [Google Scholar] [CrossRef] [PubMed]
- Kumaravel, T.S.; Bharathy, K.; Kudoh, S.; Tanaka, K.; Kamada, N. Expression, localization and functional interactions of Ku70 subunit of DNA-PK in peripheral lymphocytes and Nalm-19 cells after irradiation. Int. J. Radiat. Biol. 1998, 74, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Scholz, M.; Taucher-Scholz, G. Immediate localized CDKN1A (p21) radiation response after damage produced by heavy-ion tracks. Radiat. Res. 2000, 154, 398–405. [Google Scholar] [CrossRef]
- Jakob, B.; Scholz, M.; Taucher-Scholz, G. Characterization of CDKN1A (p21) binding to sites of heavy-ion-induced damage: Colocalization with proteins involved in DNA repair. Int. J. Radiat. Biol. 2002, 78, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Yutoku, Y.; Koike, A. Accumulation of p21 proteins at DNA damage sites independent of p53 and core NHEJ factors following irradiation. Biochem. Biophys. Res. Commun. 2011, 412, 39–43. [Google Scholar] [CrossRef]
- Stivala, L.A.; Prosperi, E.; Rossi, R.; Bianchi, L. Involvement of proliferating cell nuclear antigen in DNA repair after damage induced by genotoxic agents in human fibroblasts. Carcinogenesis 1993, 14, 2569–2573. [Google Scholar] [CrossRef]
- Balajee, A.S.; Geard, C.R. Chromatin-bound PCNA complex formation triggered by DNA damage occurs independent of the ATM gene product in human cells. Nucleic Acids Res. 2001, 29, 1341–1351. [Google Scholar] [CrossRef][Green Version]
- Shimazaki, N.; Yazaki, T.; Kubota, T.; Sato, A.; Nakamura, A.; Kurei, S.; Toji, S.; Tamai, K.; Koiwai, O. DNA polymerase lambda directly binds to proliferating cell nuclear antigen through its confined C-terminal region. Genes Cells 2005, 10, 705–715. [Google Scholar] [CrossRef]
- Wiese, C.; Rudolph, J.H.; Jakob, B.; Fink, D.; Tobias, F.; Blattner, C.; Taucher-Scholz, G. PCNA-dependent accumulation of CDKN1A into nuclear foci after ionizing irradiation. DNA Repair 2012, 11, 511–521. [Google Scholar] [CrossRef]
- Asaithamby, A.; Hu, B.; Chen, D.J. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8293–8298. [Google Scholar] [CrossRef]
- Scott, M.T.; Morrice, N.; Ball, K.L. Reversible phosphorylation at the C-terminal regulatory domain of p21(Waf1/Cip1) modulates proliferating cell nuclear antigen binding. J. Biol. Chem. 2000, 275, 11529–11537. [Google Scholar] [CrossRef] [PubMed]
- Child, E.S.; Mann, D.J. The intricacies of p21 phosphorylation: Protein/protein interactions, subcellular localization and stability. Cell Cycle 2006, 5, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Mauro, M.; Rego, M.A.; Boisvert, R.A.; Esashi, F.; Cavallo, F.; Jasin, M.; Howlett, N.G. p21 promotes error-free replication-coupled DNA double-strand break repair. Nucleic Acids Res. 2012, 40, 8348–8360. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Stith, C.M.; Burgers, P.; Heyer, W.D. PCNA is required for initiation of recombination-associated DNA synthesis by DNA polymerase. Mol. Cell 2009, 36, 704–713. [Google Scholar] [CrossRef]
- Li, H.; Xie, B.; Rahmeh, A.; Zhou, Y.; Lee, M.Y.W. Direct interaction of p21 with p50, the small subunit of human DNA polymerase delta. Cell Cycle 2006, 5, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Buermeyer, A.B.; Simon, J.A.; Thomas, D.C.; Clark, A.B.; Liskay, R.M.; Kunkel, T.A. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 1996, 87, 65–73. [Google Scholar] [CrossRef]
- Guo, S.; Presnell, S.R.; Yuan, F.; Zhang, Y.; Gu, L.; Li, G.-M. Differential requirement for proliferating cell nuclear antigen in 5′ and 3′ nick-directed excision in human mismatch repair. J. Biol. Chem. 2004, 279, 16912–16917. [Google Scholar] [CrossRef]
- Kleczkowska, H.E.; Marra, G.; Lettieri, T.; Jiricny, J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001, 15, 724–736. [Google Scholar] [CrossRef]
- Jascur, T.; Fotedar, R.; Greene, S.; Hotchkiss, E.; Boland, C.R. N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) triggers MSH2 and Cdt2 protein-dependent degradation of the cell cycle and mismatch repair (MMR) inhibitor protein p21Waf1/Cip1. J. Biol. Chem. 2011, 286, 29531–29539. [Google Scholar] [CrossRef]
- Masih, P.J.; Kunnev, D.; Melendy, T. Mismatch Repair proteins are recruited to replicating DNA through interaction with Proliferating Cell Nuclear Antigen (PCNA). Nucleic Acids Res. 2008, 36, 67–75. [Google Scholar] [CrossRef]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Soria, G.; Podhajcer, O.; Prives, C.; Gottifredi, V. P21Cip1/WAF1 downregulation is required for efficient PCNA ubiquitination after UV irradiation. Oncogene 2006, 25, 2829–2838. [Google Scholar] [CrossRef] [PubMed]
- Avkin, S.; Sevilya, Z.; Toube, L.; Geacintov, N.; Chaney, S.G.; Oren, M.; Livneh, Z. p53 and p21 Regulate Error-Prone DNA Repair to Yield a Lower Mutation Load. Mol. Cell 2006, 22, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, S.F.; Sorian, G.; Vallerga, M.B.; Habif, M.; Martinez-Lopez, W.; Prives, C.; Gottifredi, V. UV-triggered p21 degradation facilitates damaged-DNA replication and preserves genomic stability. Nucleic Acids Res. 2013, 41, 6942–6951. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef]
- Howlett, N.G.; Harney, J.A.; Rego, M.A.; Kolling, F.W.; Glover, T.W. Functional interaction between the Fanconi Anemia D2 protein and proliferating cell nuclear antigen (PCNA) via a conserved putative PCNA interaction motif. J. Biol. Chem. 2009, 284, 28935–28942. [Google Scholar] [CrossRef]
- Rego, M.A.; Harney, J.A.; Mauro, M.; Shen, M.; Howlett, N.G. Regulation of the activation of the Fanconi anemia pathway by the p21 cyclin-dependent kinase inhibitor. Oncogene 2012, 31, 366–375. [Google Scholar] [CrossRef]
- Bornstein, G.; Bloom, J.; Sitry-Shevah, D.; Nakayama, K.; Pagano, M.; Hershko, A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J. Biol. Chem. 2003, 278, 25752–25757. [Google Scholar] [CrossRef]
- Amador, V.; Ge, S.; Santamaría, P.G.; Guardavaccaro, D.; Pagano, M. APC/C(Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Mol. Cell 2007, 27, 462–473. [Google Scholar] [CrossRef]
- Kim, Y.; Starostina, N.G.; Kipreos, E.T. The CRL4Ctdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008, 22, 2507–2519. [Google Scholar] [CrossRef]
- Abbas, T.; Sivaprasad, U.; Terai, K.; Amador, V.; Pagano, M.; Dutta, A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008, 22, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, H.; Shiomi, Y.; Iida, H.; Michishita, M.; Takami, T.; Tsurimoto, T. CDK inhibitor p21 is degraded by a PCNA coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J. Biol. Chem. 2008, 283, 29045–29052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, H.; Li, M.; Agrawal, S.; Chen, X.; Zhang, R. MDM2 is a negative regulator of p21WAF1/CIP1, independent of p53. J. Biol. Chem. 2004, 279, 16000–16006. [Google Scholar] [CrossRef] [PubMed]
- Bendjennat, M.; Boulaire, J.; Jascur, T.; Brickner, H.; Barbier, V.; Sarasin, A.; Fotedar, A.; Fotedar, R. UV irradiation triggers ubiquitin-dependent degradation of p21WAF1 to promote DNA repair. Cell 2003, 114, 599–610. [Google Scholar] [CrossRef]
- Hanawalt, P.C. Controlling the efficiency of excision repair. Mutat. Res. 2001, 485, 3–13. [Google Scholar] [CrossRef]
- Esteve-Puig, R.; Gil, R.; Gonzales-Sanchez, E.; Bech-Serra, J.J.; Grueso, J.; Hernandez-Losa, J.; Moliné, T.; Canals, F.; Ferrer, B.; Cortés, J.; et al. A mouse model uncovers LKB1 as an UVB-induced DNA damage sensor mediating CDKN1A (p21WAF1/CIP1) degradation. PLoS Genet. 2014, 10, e1004721. [Google Scholar] [CrossRef]
- Galanos, P.; Pappas, G.; Polyzos, A.; Kotsinas, A.; Svolaki, I.; Giakoumakis, N.N.; Glytsou, C.; Pateras, I.S.; Swain, U.; Souliotis, V.L.; et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol. 2018, 19, 37. [Google Scholar] [CrossRef]
- Savio, M.; Coppa, T.; Cazzalini, O.; Perucca, P.; Necchi, D.; Nardo, T.; Stivala, L.A.; Prosperi, E. Degradation of p21CDKN1A after DNA damage is independent of type of lesion, and is not required for DNA repair. DNA Repair 2009, 8, 778–785. [Google Scholar] [CrossRef]
- Sheng, C.; Mendler, I.-H.; Rieke, S.; Snyder, P.; Jentsch, M.; Friedrich, D.; Drossel, B.; Loewer, A. PCNA-Mediated Degradation of p21 Coordinates the DNA Damage Response and Cell Cycle Regulation in Individual Cells. Cell Rep. 2019, 27, 48–58. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Patrikidou, A.; Daliani, D.D.; Papandreou, C.N. The ubiquitin-proteasome system in cancer, a major player in DNA repair. Part 1: Post-translational regulation. J. Cell Mol. Med. 2009, 13, 3006–3018. [Google Scholar] [CrossRef]
- Kubota, T.; Nishimura, K.; Kanemaki, M.T.; Donaldson, A.D. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell 2013, 50, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Fu, H.; Aladjem, M.I.; Myung, K. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 2013, 200, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Sommatis, S.; Tillhon, M.; Dutto, I.; Bachi, A.; Rapp, A.; Nardo, T.; Scovassi, A.I.; Necchi, D.; Cardoso, M.C.; et al. CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucleic Acids Res. 2014, 42, 8433–8448. [Google Scholar] [CrossRef] [PubMed]
- Soutoglou, E.; Misteli, T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science 2008, 320, 1507–1510. [Google Scholar] [CrossRef]
- Sabatella, M.; Theil, A.F.; Ribeiro-Silva, C.; Slyskova, J.; Thijssen, K.; Voskamp, C.; Lans, H.; Vermeulen, W. Repair protein persistence at DNA lesions characterizes XPF defect with Cockayne syndrome features. Nucleic Acids Res. 2018, 46, 9563–9577. [Google Scholar] [CrossRef]
- Puumalainen, M.R.; Lessel, D.; Rüthemann, P.; Kaczmarek, N.; Bachmann, K.; Ramadan, K.; Naegeli, H. Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat. Commun. 2014, 5, 3695. [Google Scholar] [CrossRef]
- He, J.; Zhu, Q.; Wani, G.; Sharma, N.; Wani, A.A. Valosin-containing Protein (VCP)/p97 Segregase Mediates Proteolytic Processing of Cockayne Syndrome Group B (CSB) in Damaged Chromatin. J. Biol. Chem. 2016, 291, 7396–7408. [Google Scholar] [CrossRef]
- Havens, C.G.; Walter, J.C. Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol. Cell 2009, 35, 93–104. [Google Scholar] [CrossRef]
- Havens, C.G.; Walter, J.C. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011, 25, 1568–1582. [Google Scholar] [CrossRef]
- Mazian, M.A.; Yamanishi, K.; Rahman, M.Z.A.R.; Ganasen, M.; Nishitani, H. CRL4Cdt2 ubiquitin ligase, a genome caretaker controlled by Cdt2 binding to PCNA and DNA. Genes 2022, 13, 266. [Google Scholar] [CrossRef]
- Li, J.; Wang, Q.-E.; Zhu, Q.; El-Mahdy, M.A.; Wani, G.; Pretorius-Ibba, M.; Wani, A.A. DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. Cancer Res. 2006, 66, 8590–8597. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Wani, G.; Zhao, R.; Qian, J.; Sharma, N.; He, J.; Zhu, Q.; Wang, Q.-E.; Wani, A.A. Cdt2-mediated XPG degradation promotes gap-filling DNA synthesis in nucleotide excision repair. Cell Cycle 2015, 14, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.L. Mechanism and regulation of DNA damage recognition in mammalian nucleotide excision repair. Enzymes 2019, 45, 99–138. [Google Scholar] [PubMed]
- Cazzalini, O.; Perucca, P.; Mocchi, R.; Sommatis, S.; Prosperi, E.; Stivala, L.A. DDB2 association with PCNA is required for its degradation after UV-induced DNA damage. Cell Cycle 2014, 13, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Perucca, P.; Mocchi, R.; Guardamagna, I.; Bassi, E.; Sommatis, S.; Nardo, T.; Prosperi, E.; Stivala, L.A.; Cazzalini, O. A damaged DNA binding protein 2 mutation disrupting interaction with proliferating-cell nuclear antigen affects DNA repair and confers proliferation advantage. Biochem. Biophys. Acta Mol. Cell Res. 2018, 1865, 898–907. [Google Scholar] [CrossRef]
- Lee, M.Y.W.T.; Zhang, S.; Wang, X.; Chao, H.H.; Zhao, H.; Dazynkiewicz, Z.; Zhang, Z.; Lee, E.Y.C. Two forms of human DNA polymerase δ: Who does what and why? DNA Repair 2019, 81, 102656. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, Y.; Trusa, S.; Meng, X.; Lee, E.Y.C.; Lee, M.Y.W.T. A novel DNA damage response: Rapid degradation of the p12 subunit of dna polymerase delta. J. Biol. Chem. 2007, 282, 15330–15340. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, Y.; Sarkenshik, A.; Yates, J.R., 3rd; Thomson, T.M.; Zhang, Z.; Lee, E.Y.C.; Lee, M.Y.W.T. Identification of RNF8 as a ubiquitin ligase involved in targeting the p12 subunit of DNA polymerase δ for degradation in response to DNA damage. J. Biol. Chem. 2013, 288, 2941–2950. [Google Scholar] [CrossRef]
- Terai, K.; Shibata, E.; Abbas, T.; Dutta, A. Degradation of p12 subunit by CRL4Cdt2 E3 ligase inhibits fork progression after DNA damage. J. Biol. Chem. 2013, 288, 30509–30514. [Google Scholar] [CrossRef]
- Bhat, A.; Qin, Z.; Wang, G.; Chen, W.; Xiao, W. Rev7, the regulatory subunit of Polζ, undergoes UV-induced and Cul4-dependent degradation. FEBS J. 2017, 284, 1790–1803. [Google Scholar] [CrossRef]
- Bacquin, A.; Pouvelle, C.; Siaud, N.; Perderiset, M.; Salomé-Desnoulez, S.; Tellier-Lebegue, C.; Lopez, B.; Charbonnier, J.-B.; Kannouche, P.L. The helicase FBH1 is tightly regulated by PCNA via CRL4(Cdt2)-mediated proteolysis in human cells. Nucleic Acids Res. 2013, 41, 6501–6513. [Google Scholar] [CrossRef] [PubMed]
- Matsunuma, R.; Niida, H.; Ohhata, T.; Kitagawa, K.; Sakai, S.; Uchida, C.; Shiotani, B.; Matsumoto, M.; Nakayama, K.I.; Ogura, H.; et al. UV Damage-Induced Phosphorylation of HBO1 Triggers CRL4DDB2-Mediated Degradation To Regulate Cell Proliferation. Mol. Cell Biol. 2015, 36, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Niida, H.; Matsunuma, R.; Horiguchi, C.; Uchida, C.; Nakazawa, Y.; Motegi, A.; Nishimoto, K.; Sakai, S.; Ohhata, T.; Kitagawa, K.; et al. Phosphorylated HBO1 at UV irradiated sites is essential for nucleotide excision repair. Nat. Commun. 2017, 8, 16102. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Liao, Z.; Matsumoto, Y.; Yang, A.; Tomkinson, A.E. Human DNA Ligase I Interacts with and Is Targeted for Degradation by the DCAF7 Specificity Factor of the Cul4-DDB1 Ubiquitin Ligase Complex. J. Biol. Chem. 2016, 291, 21893–21902. [Google Scholar] [CrossRef]
- Slenn, T.J.; Morris, B.; Havens, C.G.; Freeman RMJr Takahashi, T.S.; Walter, J.C. Thymine DNA glycosylase is a CRL4Cdt2 substrate. J. Biol. Chem. 2014, 289, 23043–23055. [Google Scholar] [CrossRef]
- Shibata, E.; Dar, A.; Dutta, A. CRL4Cdt2 E3 ubiquitin ligase and proliferating cell nuclear antigen (PCNA) cooperate to degrade thymine DNA glycosylase in S phase. J. Biol. Chem. 2014, 289, 23056–23064. [Google Scholar] [CrossRef]
- Jung, Y.S.; Liu, G.; Chen, X. Pirh2 E3 ubiquitin ligase targets DNA polymerase eta for 20S proteasomal degradation. Mol. Cell Biol. 2010, 30, 1041–1048. [Google Scholar] [CrossRef]
- Jung, Y.S.; Qian, Y.; Chen, X. DNA polymerase eta is targeted by Mdm2 for polyubiquitination and proteasomal degradation in response to ultraviolet irradiation. DNA Repair 2012, 11, 177–184. [Google Scholar] [CrossRef]
- Tomimatsu, N.; Mukherjee, B.; Harris, J.L.; Boffo, F.L.; Hardebeck, M.C.; Potts, P.R.; Khanna, K.K.; Burma, S. DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair. J. Biol. Chem. 2017, 292, 10779–10790. [Google Scholar] [CrossRef]
- Tsanov, N.; Kermi, C.; Coulombe, P.; Van der Laan, S.; Hodroj, D.; Mairano, D. PIP degron proteins, substrates of CRL4Cdt2, and not PIP boxes, interfere with DNA polymerase η and κ focus formation on UV damage. Nucleic Acids Res. 2014, 42, 3692–3706. [Google Scholar] [CrossRef]
- Mansilla, S.F.; De La Vega, M.B.; Calzetta, N.L.; Siri, S.O.; Gottifredi, V. CDK-independent and PCNA-dependent functions of p21 in DNA replication. Genes 2020, 11, 593. [Google Scholar] [CrossRef] [PubMed]
- Coleman, K.E.; Grant, G.D.; Haggerty, R.A.; Brantley, K.; Shibata, E.; Workman, B.D.; Dutta, A.; Varma, D.; Purvis, J.E.; Cook, J.G. Sequential replication-coupled destruction at G1/S ensures genome stability. Genes Dev. 2015, 29, 1734–1746. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Hayward, R.L.; Meng, L.H.; Takemura, H.; Aune, G.J.; Bonner, W.M.; Aladjem, M.I.; Kohn, K.W.; Pommier, Y. p21CDKN1A allows the repair of replication-mediated DNA double-strand breaks induced by topoisomerase I and is inactivated by the checkpoint kinase inhibitor 7-hydroxystaurosporine. Oncogene 2006, 25, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Hauge, S.; Macurek, L.; Syljuasen, R.G. P21 limits S phase DNA damage caused by the Wee1 inhibitor MK1775. Cell Cycle 2019, 18, 834–847. [Google Scholar] [CrossRef]
- Hutton, R.D.; Craggs, T.D.; White, M.F.; Penedo, J.C. PCNA and XPF cooperate to distort DNA substrates. Nucleic Acids Res. 2010, 38, 1664–1675. [Google Scholar] [CrossRef]
- Staresincic, L.; Fagbemi, A.F.; Enzlin, J.H.; Gourdin, A.M.; Wijgers, N.; Dunand-Sauthier, I.; Giglia-Mari, G.; Clarkson, S.G.; Vermeulen, W.; Schärer, O.D. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009, 28, 1111–1120. [Google Scholar] [CrossRef]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Bagchi, S.; Raychaudhuri, P. DDB2 decides the cell fate following DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 10690–10695. [Google Scholar] [CrossRef]
- Lei, X.; Liu, B.; Han, W.; Ming, M.; He, Y.-Y. UVB-induced p21 degradation promotes apoptosis of human keratinocytes. Photochem. Photobiol. Sci. 2010, 9, 1640–1648. [Google Scholar] [CrossRef]
- Buscemi, G.; Ricci, C.; Zannini, L.; Fontanella, E.; Plevani, P.; Delia, D. Bimodal regulation of p21waf1 protein as function of DNA damage levels. Cell Cycle 2014, 13, 2901–2912. [Google Scholar] [CrossRef][Green Version]
- Origanti, S.; Cai, S.R.; Munir, A.Z.; White, L.S.; Pwinica-Worms, H. Synthetic lethality of Chk1 inhibition combined with p53 and/or p21 loss during a DNA damage response in normal and tumor cells. Oncogene 2013, 32, 577–588. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ticli, G.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. Revisiting the Function of p21CDKN1A in DNA Repair: The Influence of Protein Interactions and Stability. Int. J. Mol. Sci. 2022, 23, 7058. https://doi.org/10.3390/ijms23137058
Ticli G, Cazzalini O, Stivala LA, Prosperi E. Revisiting the Function of p21CDKN1A in DNA Repair: The Influence of Protein Interactions and Stability. International Journal of Molecular Sciences. 2022; 23(13):7058. https://doi.org/10.3390/ijms23137058
Chicago/Turabian StyleTicli, Giulio, Ornella Cazzalini, Lucia A. Stivala, and Ennio Prosperi. 2022. "Revisiting the Function of p21CDKN1A in DNA Repair: The Influence of Protein Interactions and Stability" International Journal of Molecular Sciences 23, no. 13: 7058. https://doi.org/10.3390/ijms23137058
APA StyleTicli, G., Cazzalini, O., Stivala, L. A., & Prosperi, E. (2022). Revisiting the Function of p21CDKN1A in DNA Repair: The Influence of Protein Interactions and Stability. International Journal of Molecular Sciences, 23(13), 7058. https://doi.org/10.3390/ijms23137058