
11β-Hydroxysteroid Dehydrogenase Type 1 within Osteoclasts Mediates the Bone Protective Properties of Therapeutic Corticosteroids in Chronic Inflammation

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Methods
2.1. Human Tibia Bone Biopsies
2.2. Models of Polyarthritis
2.3. Targeted Deletion of 11β-HSD1
2.4. 11β-HSD1 Enzymatic Activity Assay
2.5. Primary Human Osteoblast Culture
2.6. Primary Human Osteoclast Culture
2.7. In Vitro Bone Resorption Assay
2.8. Analysis of P1NP and CTX-1 by ELISA
2.9. MicroCT Morphometry Analysis
2.10. Histological Analysis of Joints and Muscle
2.11. Static Histomorphometry
2.12. Analysis of mRNA Abundance
2.13. Statistical Analysis
3. Results
3.1. Inflammation Upregulates 11β-HSD1 in Bone and in Bone Cells
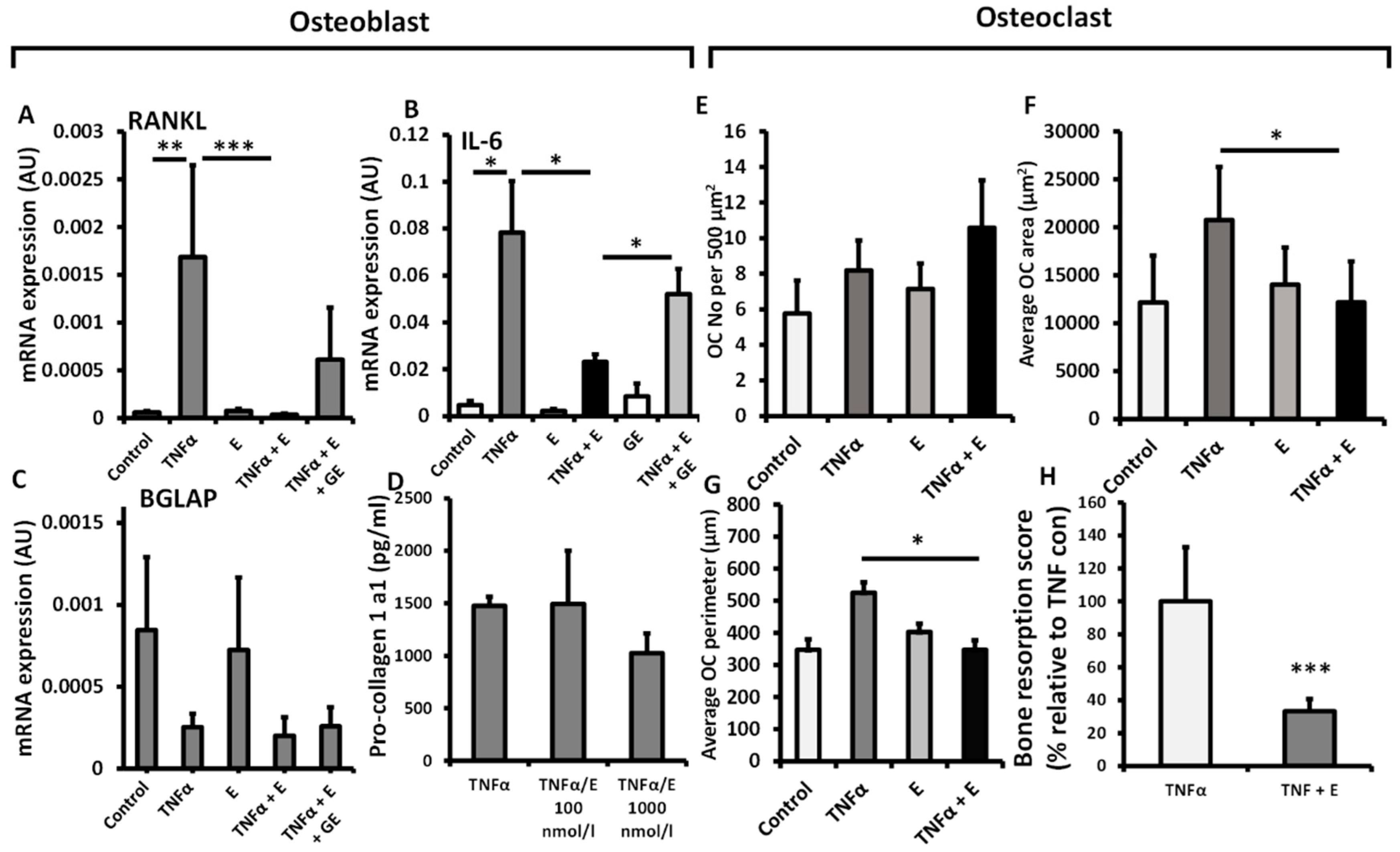
3.2. 11β-HSD1 Influences Bone Metabolism in Osteoclasts but Not Osteoblasts
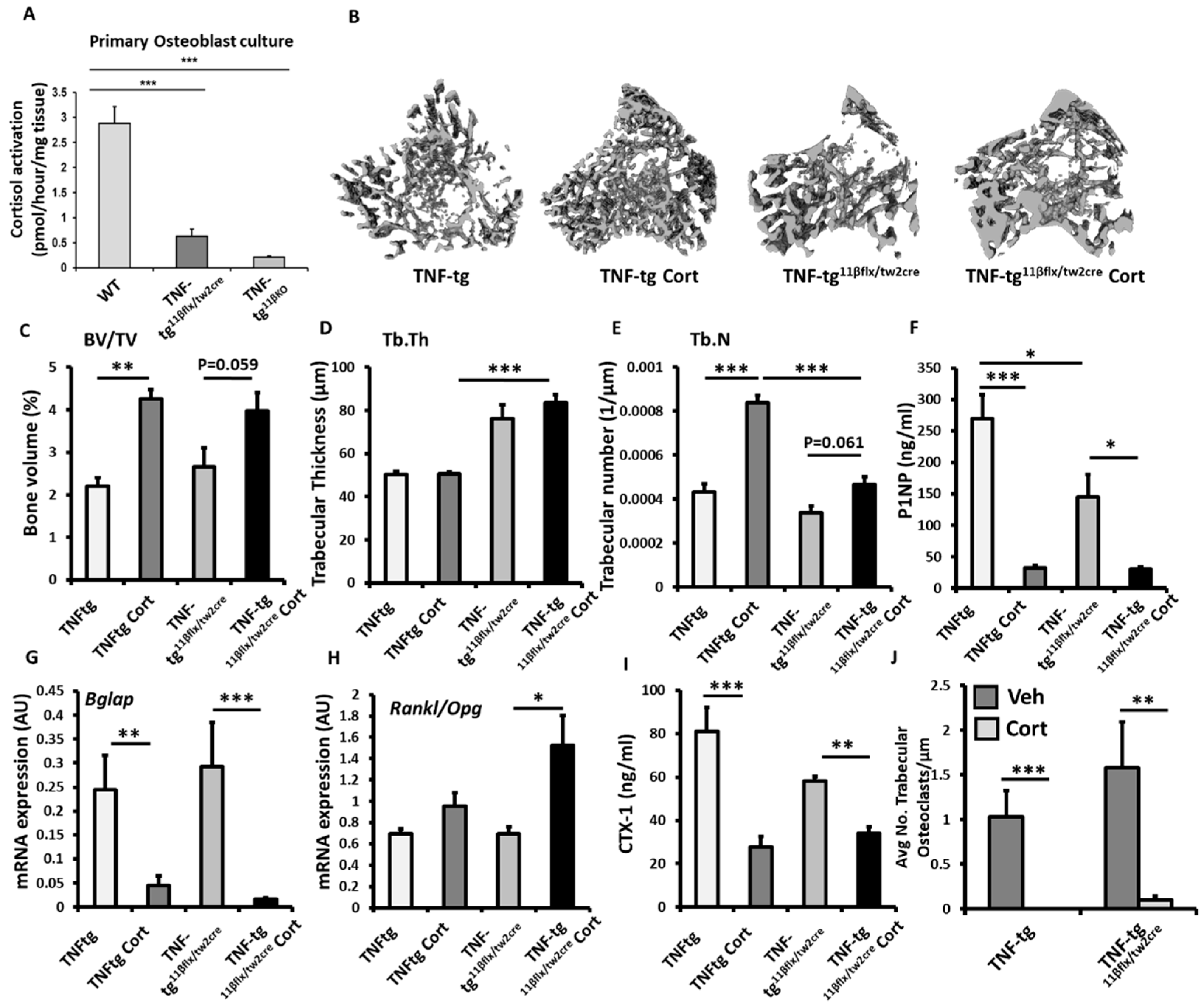
3.3. 11β-HSD1 Mediates Bone Protective Actions of Therapeutic GCs during Inflammation In Vivo
3.4. Animals with Mesenchymal Deletion of 11β-HSD1, Retain Bone Protective Actions of GCs
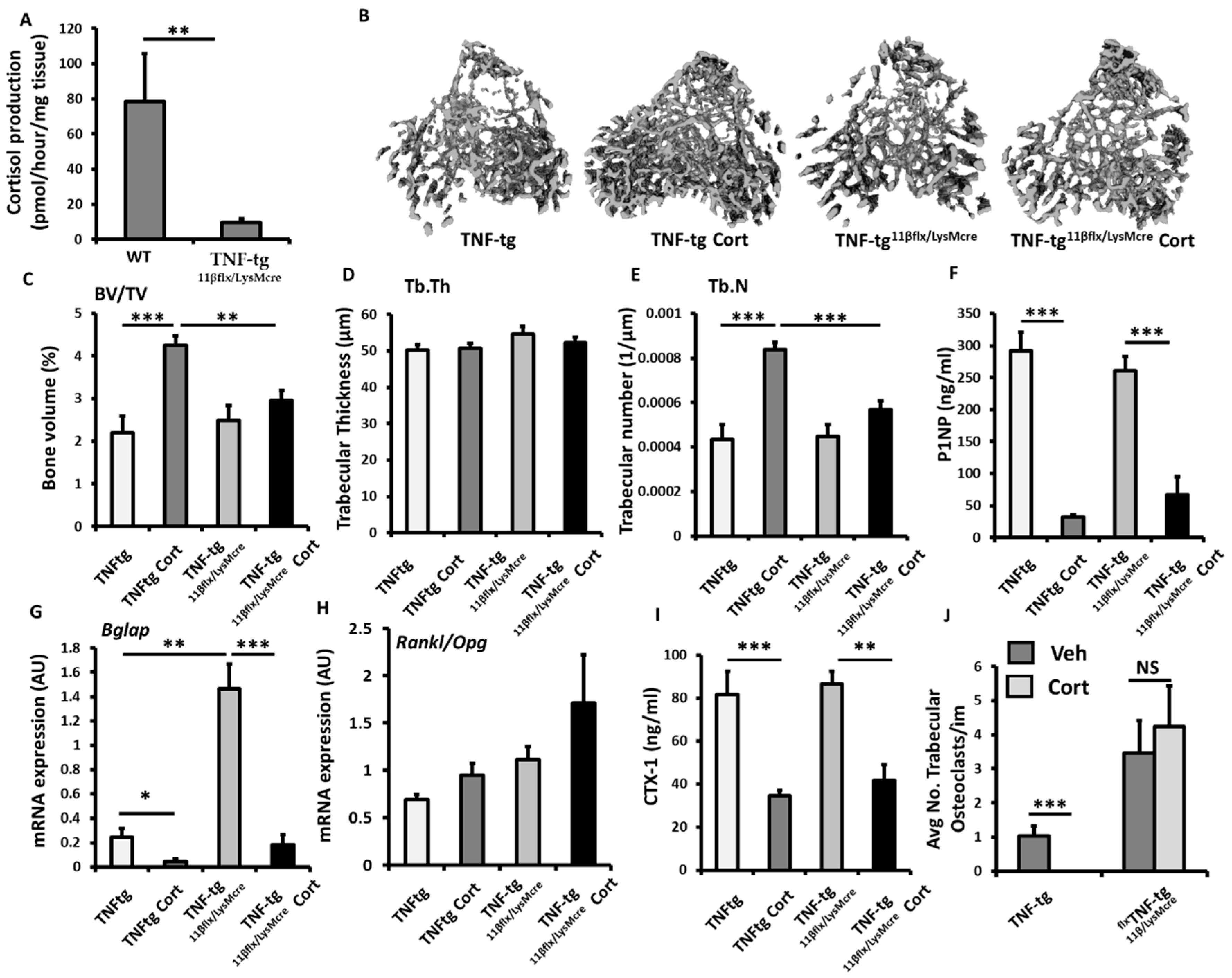
3.5. Myeloid Deletion of 11β-HSD1 Causes a Partial Resistance to the Bone Protective Actions of GCs
4. Discussion
5. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GIOP | Glucocorticoid induced osteoporosis |
| GCs | Glucocorticoids |
| WT | Wild-type |
| 11β-HSD1 | 11β-hydroxysteroid dehydrogenase type 1 |
| BV/TV | Trabecular bone volume to tissue volume |
| Tb.Th | Trabecular thickness |
| Tb.N | Trabecular number |
| 11-DHC | 11-dehydrocorticosterone |
| KO | Knock out |
| Med.A | Endosteal medullary area |
| Per.P | Periosteal perimeter |
| Cort | Corticosterone |
| P1NP | Procollagen type 1 amino-terminal propeptide |
References
- Hardy, R.S.; Raza, K.; Cooper, M.S. Therapeutic glucocorticoids: Mechanisms of actions in rheumatic diseases. Nat. Rev. Rheumatol. 2020, 16, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- RECOVERY Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19—Preliminary Report. N. Engl. J. Med. 2020, 384, 693–704. [Google Scholar] [PubMed]
- Angeli, A.; Guglielmi, G.; Dovio, A.; Capelli, G.; de Feo, D.; Giannini, S.; Giorgino, R.; Moro, L.; Giustina, A. High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: A cross-sectional outpatient study. Bone 2006, 39, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.C.; Elmer, P.J.; Nichols, G.A.; Herson, M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos. Int. 2005, 16, 2168–2174. [Google Scholar] [CrossRef]
- Strehl, C.; Bijlsma, J.W.J.; De Wit, M.; Boers, M.; Caeyers, N.; Cutolo, M.; Dasgupta, B.; Dixon, W.G.; Geenen, R.; Huizinga, T.W.J.; et al. Defining conditions where long-term glucocorticoid treatment has an acceptably low level of harm to facilitate implementation of existing recommendations: Viewpoints from an EULAR task force. Ann. Rheum. Dis. 2016, 75, 952–957. [Google Scholar] [CrossRef] [Green Version]
- Ton, F.N.; Gunawardene, S.C.; Lee, H.; Neer, R.M. Effects of Low-Dose Prednisone on Bone Metabolism. J. Bone Miner. Res. 2005, 20, 464–470. [Google Scholar] [CrossRef]
- Fenton, C.G.; Webster, J.M.; Martin, C.; Fareed, S.; Wehmeyer, C.; Mackie, H.; Jones, R.; Seabright, A.P.; Lewis, J.W.; Lai, Y.-C.; et al. Therapeutic glucocorticoids prevent bone loss but drive muscle wasting when administered in chronic polyarthritis. Arthritis Res. Ther. 2019, 21, 182. [Google Scholar] [CrossRef] [Green Version]
- Fenton, C.G.; Doig, C.L.; Fareed, S.; Naylor, A.; Morrell, A.P.; Addison, O.; Wehmeyer, C.; Buckley, C.D.; Cooper, M.S.; Lavery, G.G.; et al. 11beta-HSD1 plays a critical role in trabecular bone loss associated with systemic glucocorticoid therapy. Arthritis Res. Ther. 2019, 21, 188. [Google Scholar] [CrossRef] [Green Version]
- Zupan, J.; Jeras, M.; Marc, J. Osteoimmunology and the influence of pro-inflammatory cytokines on osteoclasts. Biochem. Med. 2013, 23, 43–63. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Taniguchi, T. Signaling crosstalk between RANKL and interferons in osteoclast differentiation. Arthritis Res. Ther. 2002, 4, S227–S232. [Google Scholar] [CrossRef]
- Kotake, S.; Nanke, Y.; Yago, T.; Kawamoto, M.; Yamanaka, H. Human osteoclastogenic T cells and human osteoclastology. Arthritis Care Res. 2009, 60, 3158–3163. [Google Scholar] [CrossRef]
- Lavery, G.G.; Walker, E.A.; Draper, N.; Jeyasuria, P.; Marcos, J.; Shackleton, C.H.; Parker, K.L.; White, P.C.; Stewart, P.M. Hexose-6-phosphate dehydrogenase knock-out mice lack 11β-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. J. Biol. Chem. 2006, 281, 6546–6551. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.S.; Cooper, M.S.; Hardy, R.S. Endogenous Glucocorticoid Metabolism in Bone: Friend or Foe. Front. Endocrinol. 2021, 12, 733611. [Google Scholar] [CrossRef]
- Fenton, C.; Martin, C.; Jones, R.; Croft, A.; Campos, J.; Naylor, A.J.; Taylor, A.E.; Chimen, M.; Cooper, M.; Lavery, G.G.; et al. Local steroid activation is a critical mediator of the anti-inflammatory actions of therapeutic glucocorticoids. Ann. Rheum. Dis. 2021, 80, 250–260. [Google Scholar] [CrossRef]
- Webster, J.M.; Sagmeister, M.S.; Fenton, C.G.; Seabright, A.P.; Lai, Y.-C.; Jones, S.W.; Filer, A.; Cooper, M.S.; Lavery, G.G.; Raza, K.; et al. Global Deletion of 11β-HSD1 Prevents Muscle Wasting Associated with Glucocorticoid Therapy in Polyarthritis. Int. J. Mol. Sci. 2021, 22, 7828. [Google Scholar] [CrossRef]
- Cooper, M.S.; Walker, E.A.; Bland, R.; Fraser, W.D.; Hewison, M.; Stewart, P.M. Expression and functional consequences of 11β-hydroxysteroid dehydrogenase activity in human bone. Bone 2000, 27, 375–381. [Google Scholar] [CrossRef]
- Eyre, L.J.; Rabbitt, E.H.; Bland, R.; Hughes, S.V.; Cooper, M.S.; Sheppard, M.C.; Stewart, P.M.; Hewison, M. Expression of 11β-hydroxysteroid dehydrogenase in rat osteoblastic cells: Pre-receptor regulation of glucocorticoid responses in bone. J. Cell. Biochem. 2001, 81, 453–462. [Google Scholar] [CrossRef]
- Gottfried-Blackmore, A.; Sierra, A.; McEwen, B.S.; Ge, R.; Bulloch, K. Microglia express functional 11β-hydroxysteroid dehydrogenase type 1. Glia 2010, 58, 1257–1266. [Google Scholar] [CrossRef]
- Kaur, K.; Hardy, R.; Ahasan, M.M.; Eijken, M.; van Leeuwen, J.P.; Filer, A.; Thomas, A.M.; Raza, K.; Buckley, C.D.; Stewart, P.M.; et al. Synergistic induction of local glucocorticoid generation by inflammatory cytokines and glucocorticoids: Implications for inflammation associated bone loss. Ann. Rheum. Dis. 2010, 69, 1185–1190. [Google Scholar] [CrossRef]
- Hardy, R.; Rabbitt, E.H.; Filer, A.; Emery, P.; Hewison, M.; Stewart, P.M.; Gittoes, N.J.; Buckley, C.D.; Raza, K.; Cooper, M.S. Local and systemic glucocorticoid metabolism in inflammatory arthritis. Ann. Rheum. Dis. 2008, 67, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.S.; Filer, A.; Cooper, M.S.; Parsonage, G.; Raza, K.; Hardie, D.L.; Rabbitt, E.H.; Stewart, P.M.; Buckley, C.D.; Hewison, M. Differential expression, function and response to inflammatory stimuli of 11beta-hydroxysteroid dehydrogenase type 1 in human fibroblasts: A mechanism for tissue-specific regulation of inflammation. Arthritis Res. Ther. 2006, 8, R108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [CrossRef] [PubMed]
- Naylor, A.J.; Desanti, G.; Saghir, A.N.; Hardy, R.S. TNFalpha depleting therapy improves fertility and animal welfare in TNFalpha-driven transgenic models of polyarthritis when administered in their routine breeding. Lab. Anim. 2018, 52, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, R.S.; Doig, C.L.; Hussain, Z.; O’Leary, M.; Morgan, S.A.; Pearson, M.J.; Naylor, A.; Jones, S.W.; Filer, A.; Stewart, P.M.; et al. 11β-Hydroxysteroid dehydrogenase type 1 within muscle protects against the adverse effects of local inflammation. J. Pathol. 2016, 240, 472–483. [Google Scholar] [CrossRef]
- Hardy, R.S.; Fenton, C.; Croft, A.P.; Naylor, A.J.; Begum, R.; Desanti, G.; Buckley, C.; Lavery, G.; Cooper, M.; Raza, K. 11 Beta-hydroxysteroid dehydrogenase type 1 regulates synovitis, joint destruction, and systemic bone loss in chronic polyarthritis. J. Autoimmun. 2018, 92, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Xu, J.; Liu, Z.; Sosic, D.; Shao, J.; Olson, E.N.; Towler, D.; Ornitz, D.M. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 2003, 130, 3063–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Hardy, R.; Stoner, S.; Tuckermann, J.; Seibel, M.; Zhou, H. Deletion of Mesenchymal Glucocorticoid Receptor Attenuates Embryonic Lung Development and Abdominal Wall Closure. PLoS ONE 2013, 8, e63578. [Google Scholar] [CrossRef] [Green Version]
- Semjonous, N.M.; Sherlock, M.; Jeyasuria, P.; Parker, K.L.; Walker, E.A.; Stewart, P.M.; Lavery, G.G. Hexose-6-phosphate dehydrogenase contributes to skeletal muscle homeostasis independent of 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 2011, 152, 93–102. [Google Scholar] [CrossRef]
- Abram, C.L.; Roberge, G.L.; Hu, Y.; Lowell, C.A. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 2014, 408, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.W.J.; Burmester, G.R.; Chatzidionysiou, K.; Dougados, M.; Nam, J.L.; Ramiro, S.; Voshaar, M.; Van Vollenhoven, R.F.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef]
- Tang, C.-Y.; Wu, M.; Zhao, D.; Edwards, D.; McVicar, A.; Luo, Y.; Zhu, G.; Wang, Y.; Zhou, H.-D.; Chen, W.; et al. Runx1 is a central regulator of osteogenesis for bone homeostasis by orchestrating BMP and WNT signaling pathways. PLoS Genet. 2021, 17, e1009233. [Google Scholar] [CrossRef]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Förster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef]
- Gilmour, J.S.; Coutinho, A.E.; Cailhier, J.F.; Man, T.Y.; Clay, M.; Thomas, G.; Harris, H.J.; Mullins, J.J.; Seckl, J.R.; Savill, J.S.; et al. Local amplification of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J. Immunol. 2006, 176, 7605–7611. [Google Scholar] [CrossRef] [Green Version]
- Ahasan, M.M.; Hardy, R.; Jones, C.; Kaur, K.; Nanus, D.; Juarez, M.; Morgan, S.A.; Hassan-Smith, Z.; Benezech, C.; Caamano, J.; et al. Inflammatory regulation of glucocorticoid metabolism in mesenchymal stromal cells. Arthritis Care Res. 2012, 64, 2404–2413. [Google Scholar] [CrossRef] [Green Version]
- Døhn, U.M.; Boonen, A.; Hetland, M.L.; Hansen, M.S.; Knudsen, L.S.; Hansen, A.; Madsen, O.R.; Hasselquist, M.; Møller, J.M.; Østergaard, M. Erosive progression is minimal, but erosion healing rare, in patients with rheumatoid arthritis treated with adalimumab. A 1 year investigator-initiated follow-up study using high-resolution computed tomography as the primary outcome measure. Ann. Rheum. Dis. 2009, 68, 1585–1590. [Google Scholar] [CrossRef]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef]
- Arpornmaeklong, P.; Brown, S.E.; Wang, Z.; Krebsbach, P.H. Phenotypic characterization, osteoblastic differentiation, and bone regeneration capacity of human embryonic stem cell-derived mesenchymal stem cells. Stem. Cells Dev. 2009, 18, 955–968. [Google Scholar] [CrossRef]
- Epsley, S.; Tadros, S.; Farid, A.; Kargilis, D.; Mehta, S.; Rajapakse, C.S. The Effect of Inflammation on Bone. Front. Physiol. 2020, 11, 511799. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Jia, D.; Plotkin, L.I.; Bellido, T.; Powers, C.C.; Stewart, S.A.; Manolagas, S.C.; Weinstein, R.S. Glucocorticoids Act Directly on Osteoblasts and Osteocytes to Induce Their Apoptosis and Reduce Bone Formation and Strength. Endocrinology 2004, 145, 1835–1841. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, L.I.; Manolagas, S.C.; Bellido, T. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival: Evidence for inside-out signaling leading to anoikis. J. Biol. Chem. 2007, 282, 24120–24130. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Kar, R.; Gluhak-Heinrich, J.; Yao, W.; Lane, N.E.; Bonewald, L.F.; Biswas, S.K.; Lo, W.-K.; Jiang, J.X. Glucocorticoid-induced autophagy in osteocytes. J. Bone Miner. Res. 2010, 25, 2479–2488. [Google Scholar] [CrossRef]
- Weinstein, R.S.; Hogan, E.A.; Borrelli, M.J.; Liachenko, S.; O’Brien, C.A.; Manolagas, S.C. The Pathophysiological Sequence of Glucocorticoid-Induced Osteonecrosis of the Femoral Head in Male Mice. Endocrinology 2017, 158, 3817–3831. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhou, C.; Gao, M.; Xu, L.; Wen, G.; Zhao, J.; Yan, J. Effect of 11beta-HSD1 inhibitor on bone microstructure and bone density in rats with femoral head necrosis. J. Musculoskelet Neuronal. Interact. 2020, 20, 282–290. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Details | Patients with OA (n = 12) | Patients with RA (n = 10) | Group Comparison (p-Value) |
|---|---|---|---|
| Age (years) | 66.2 + 3.3 | 65.7 + 4.1 | 0.93 |
| CRP (mg/L) | 2.3 + 1.7 | 11.0 + 3.5 | 0.03 |
| ESR (mm/h) | 1.9 + 1.0 | 24.13 + 7.1 | 0.001 |
| Methotrexate (n) | 0 | 5 | na |
| Anti-TNFα therapy (n) | 0 | 2 | na |
| Prednisolone | 0 | 0 | na |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fenton, C.G.; Crastin, A.; Martin, C.S.; Suresh, S.; Montagna, I.; Hussain, B.; Naylor, A.J.; Jones, S.W.; Hansen, M.S.; Gorvin, C.M.; et al. 11β-Hydroxysteroid Dehydrogenase Type 1 within Osteoclasts Mediates the Bone Protective Properties of Therapeutic Corticosteroids in Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 7334. https://doi.org/10.3390/ijms23137334
Fenton CG, Crastin A, Martin CS, Suresh S, Montagna I, Hussain B, Naylor AJ, Jones SW, Hansen MS, Gorvin CM, et al. 11β-Hydroxysteroid Dehydrogenase Type 1 within Osteoclasts Mediates the Bone Protective Properties of Therapeutic Corticosteroids in Chronic Inflammation. International Journal of Molecular Sciences. 2022; 23(13):7334. https://doi.org/10.3390/ijms23137334
Chicago/Turabian StyleFenton, Chloe G, Ana Crastin, Claire S Martin, Saicharan Suresh, Isabella Montagna, Bismah Hussain, Amy J Naylor, Simon W Jones, Morten S Hansen, Caroline M Gorvin, and et al. 2022. "11β-Hydroxysteroid Dehydrogenase Type 1 within Osteoclasts Mediates the Bone Protective Properties of Therapeutic Corticosteroids in Chronic Inflammation" International Journal of Molecular Sciences 23, no. 13: 7334. https://doi.org/10.3390/ijms23137334
APA StyleFenton, C. G., Crastin, A., Martin, C. S., Suresh, S., Montagna, I., Hussain, B., Naylor, A. J., Jones, S. W., Hansen, M. S., Gorvin, C. M., Price, M., Filer, A., Cooper, M. S., Lavery, G. G., Raza, K., & Hardy, R. S. (2022). 11β-Hydroxysteroid Dehydrogenase Type 1 within Osteoclasts Mediates the Bone Protective Properties of Therapeutic Corticosteroids in Chronic Inflammation. International Journal of Molecular Sciences, 23(13), 7334. https://doi.org/10.3390/ijms23137334