
Ca2+ Signalling and Hypoxia/Acidic Tumour Microenvironment Interplay in Tumour Progression



Abstract
1. Introduction
1.1. Cancer Microenvironment: Focus on Tumour Acidic pHe and Hypoxia
1.2. Calcium Signalling
2. Hypoxia and Acidic pHe-Dependent Regulation of Ca2+-Permeable Ion Channels in Normal and Cancer Cells

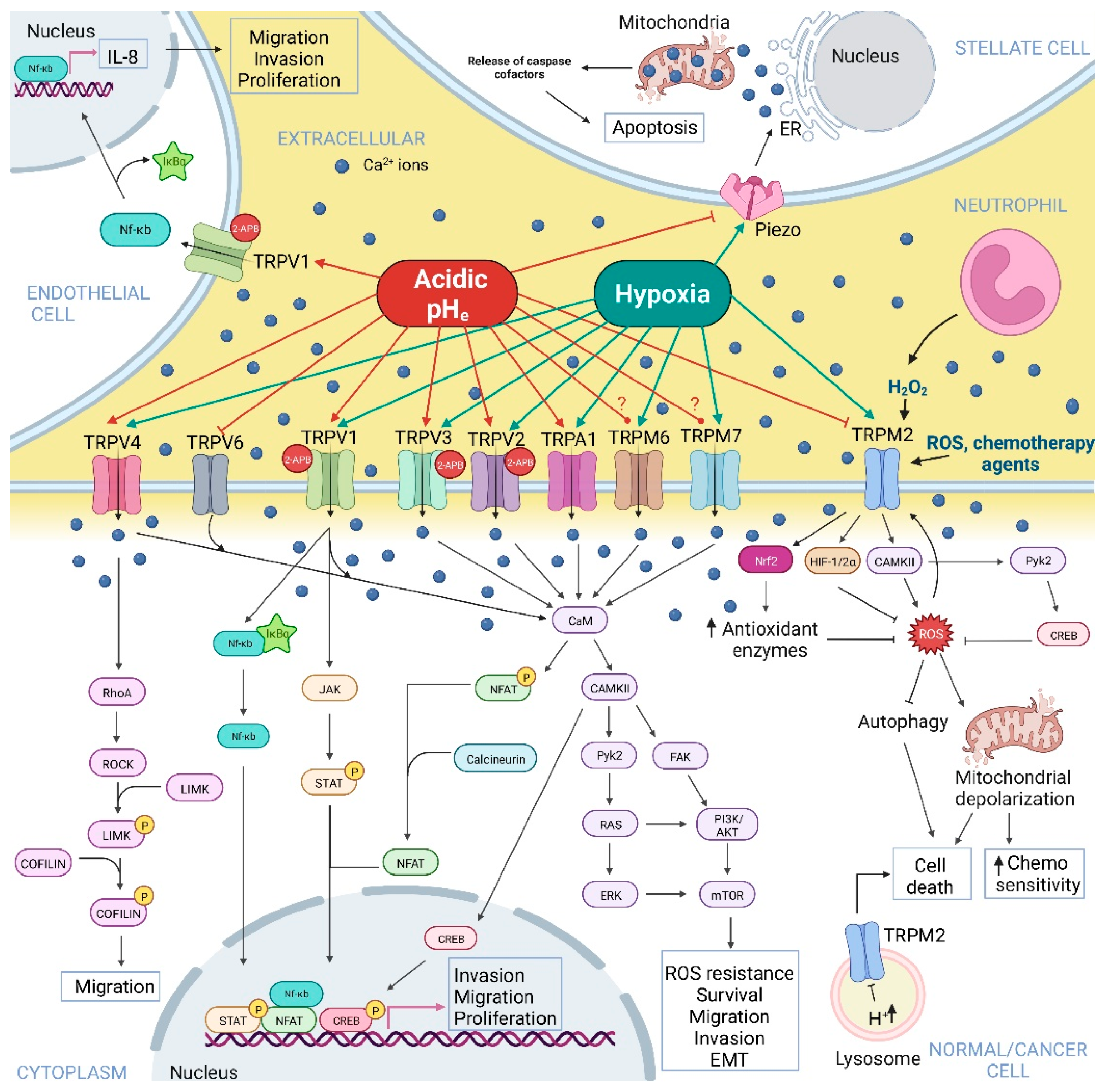{kind=link}
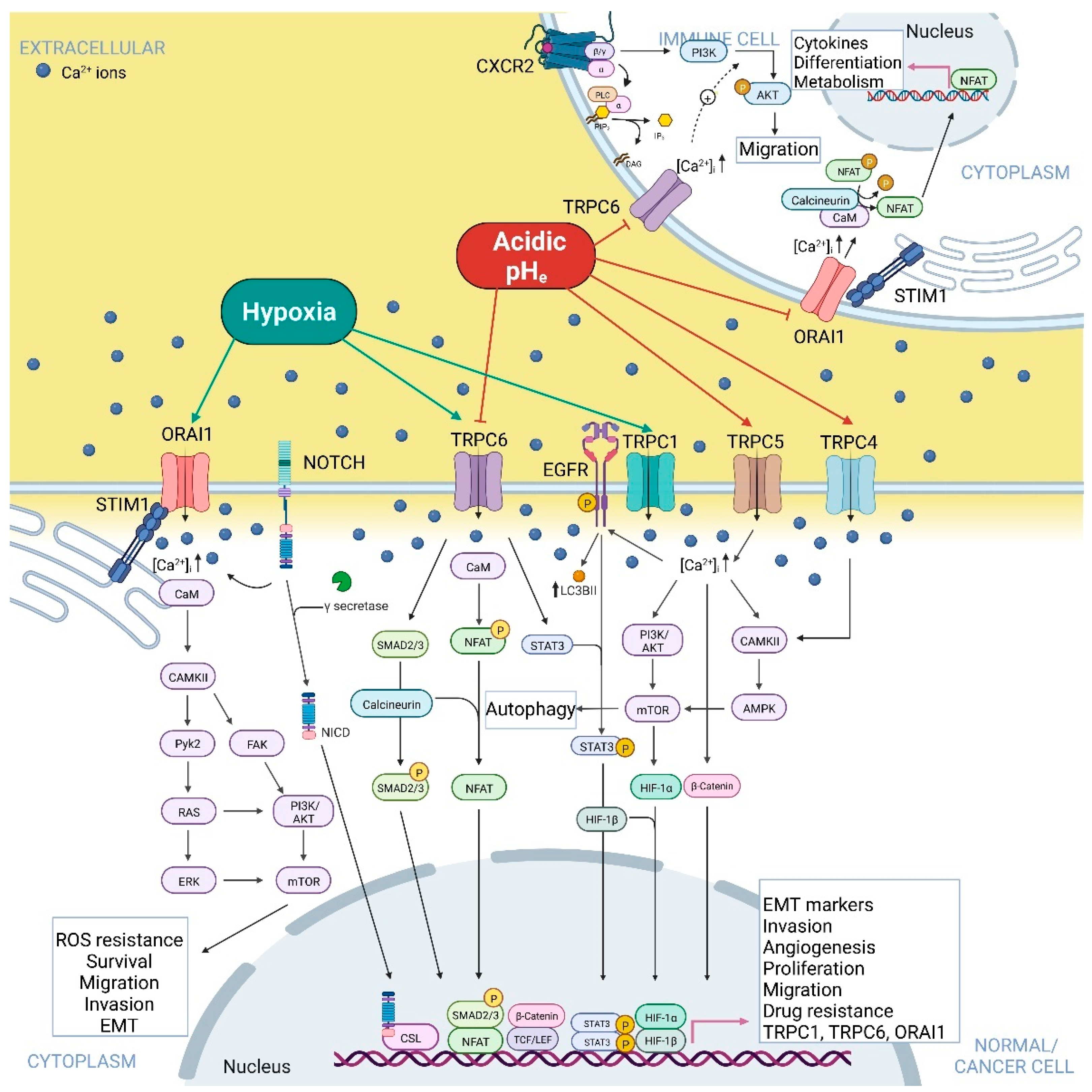{kind=link}
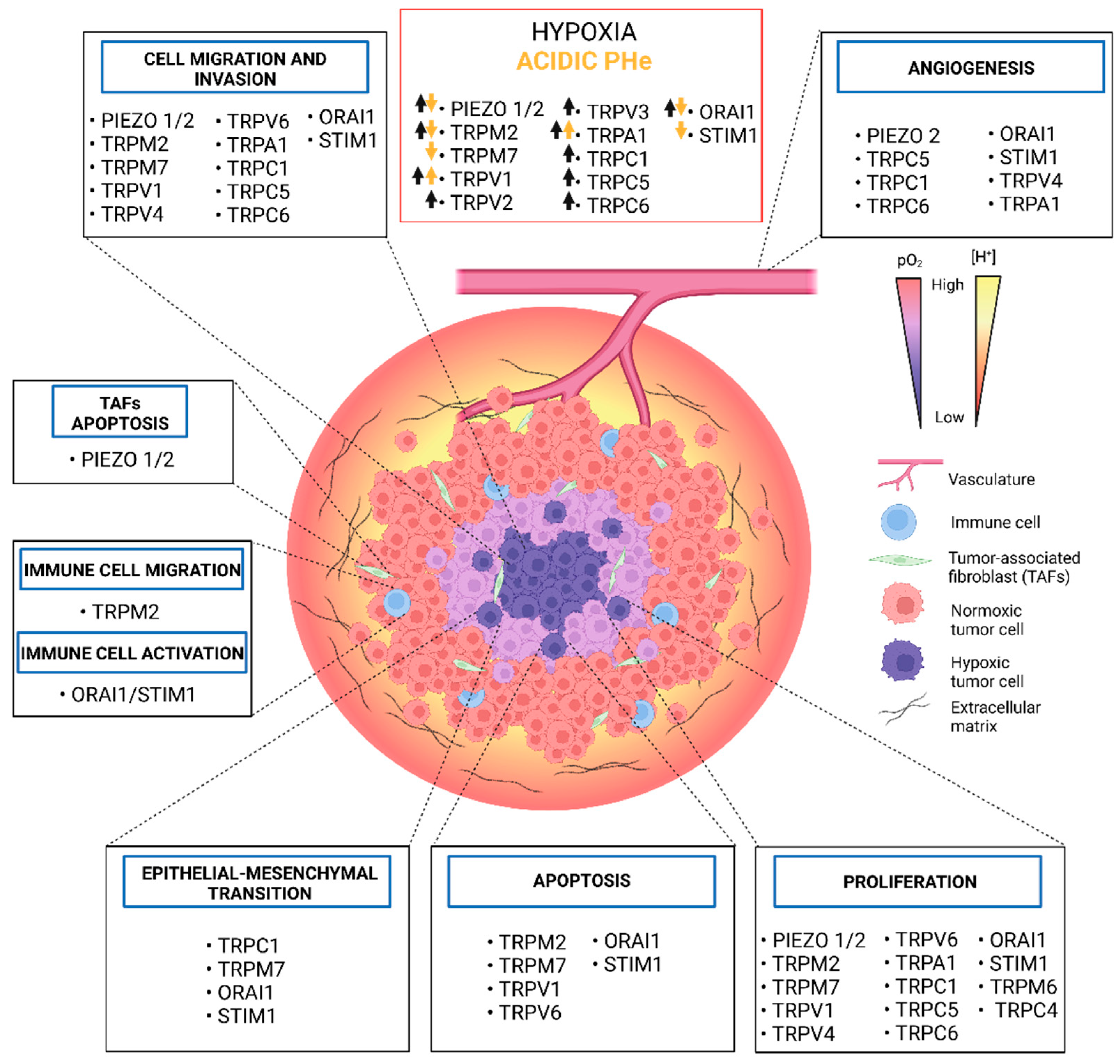{kind=link}
| Ion Channel | Cell Type | Methodology | Acidic pH Value and Treatment Time | Effect of Low pH on Channel’s Activity/Expression | Effect of Low pH on Ca2+ Signals | Cellular Function | Ref. |
|---|---|---|---|---|---|---|---|
| Piezo1 | Piezo1-transiently transfected HEK293 cells | Patch clamp Mn2+ quenching assay | pHe 6.3–6.7, acute treatment | Stabilization of inactivated state, both acidic pHi and pHe inhibit channel’s activity | Decreased Ca2+ influx | Not assessed | [62] |
| Murine pancreatic stellate cells (mPSCs) | Mn2+ quenching assay mPSCs spheroids viability and apoptosis assay | pHe 6.6 and pHi 6.77 (obtained by 30 mM propionate) in acute treatment for Mn2+ quenching assay, while 24 h long treatment for spheroid histology | Acidic pHe do not modify Piezo1 activity, while intracellular acidification inhibits channel’s activity | Acidic pHe do not modify Ca2+ influx, while intracellular acidification decreases Ca2+ influx | Acidic pHe (6.6) impairs PSCs spheroid’s integrity and viability, inducing cell apoptosis | [63] | |
| TRPM2 | Inducible TRPM2-overexpressing HEK293 | Patch clamp | External solution with pH 5–8 superfused for 200 s. Internal solution with pH 6 superfused for 100 s; External solution with pH 3.5–6.5 in acute treatment or more prolonged periods (≥2 min) | Extracellular acidification inactivates the channel in a voltage-dependent manner and [H+]-dependent manner. Intracellular acidification induces channel closure | Not assessed, but recovery from acidic pH-induced inactivation requires external Ca2+ ions | Not assessed | [64] |
| Human neutrophils | Patch clamp | External solution with pH 5 in acute treatment | External acidification negatively affects open probability and single-channel conductance, inducing channel closure | Not assessed | Not assessed | [64] | |
| TRPM2-overexpressing HEK293 | Patch clamp | External solution with pH 3.5–6 in acute treatment | External acidification (up to pH 4.5) reversely decreases mean current amplitude in a [H+]-dependent manner, decreasing single-channel conductance | Not assessed | Not assessed | [65] | |
| TRPM2-overexpressing HEK293 | Patch clamp | External solution with pH 4.0–6.5. Different time exposition based on protocol (from <10 s to ≥2 min) | Acidic pHe inactivates open channels in an irreversible manner. Exposition to pHe 4–5 negatively affects channel activation. | Not assessed | Not assessed | [66] | |
| TRPM2-overexpressing HEK293 | Patch clamp | External solution with pH 5.5, different exposition times (0, 30, 60, 90, and 120 s) | Irreversible inhibition after ≤60 s exposure | Not assessed | Not assessed | [67] | |
| TRPM6 | Pig isolated ventricular myocytes | Patch clamp | External solution with pH 5.5 and pH 6.5, ~5–10 min exposition | External acidification decreases channel’s current amplitude in a pHe-dependent and voltage-independent manner. The inhibitory effect of acidic pHe is prevented by increasing intracellular pH buffering capacity | Not assessed | Not assessed | [68] |
| TRPM6-overexpressing HEK293 cells | Patch clamp | External solution with pH 3–6, ~10 s-long exposition | External acidification increases channel’s current amplitude in a pHe-dependent manner | Not assessed | Not assessed | [69] | |
| TRPM7 | RBL-2H3 cells | Patch clamp | Acidification of intracellular side of membrane with ~200 s long 4–40 mM acetate treatment | Pre-incubation in 40 mM acetate solution inhibits TRPM7 current in a reversible manner | Not assessed | Not assessed | [70] |
| TRPM7-overexpressing Chinese Hamster Ovary (CHO-K1) cells | Patch clamp | Internal and external solution with pH 5.6 and variable exposition (~200–500 s) | Internal and external acidification abolish channels’ current | Not assessed | Not assessed | [70] | |
| TRPM7-overexpressing HEK293 cells | Patch clamp | Internal solution with pH 6.1 and ~10 min exposition | Internal acidification decreases TRPM7 currents’ density | Not assessed | Not assessed | [71] | |
| Mouse hippocampal neurons | Patch clamp | External solution with pH 6.5, 2 min exposition | Extracellular acidification slows down channel’s activation in a voltage-independent way | Not assessed | Not assessed | [72] | |
| TRPM7-overexpressing HEK293T cells | Patch clamp | External solution with pH 4 and pH 6, acute treatment | External acidification increases channel’s current amplitude in a pHe-dependent manner | Not assessed | Not assessed | [69] | |
| TRPM7-overexpressing HEK293T cells | Patch clamp | External solution with pH 3–7, ~50 s-long exposition | External acidification determines a significant increase in TRPM7 inward current in an [H+] in a concentration-dependent manner | Not assessed | Not assessed | [73] | |
| Pig isolated ventricular myocytes | Patch clamp | External solution with pH 5.5 and pH 6.5, ~5–10 min exposition | External acidification decreases channel’s current amplitude in a pHe-dependent and voltage-independent manner. The inhibitory effect of acidic pHe is prevented increasing intracellular pH buffering capacity | Not assessed | Not assessed | [68] | |
| Rat basophilic leukemia cells (RBL) | Patch clamp | External solution with pH 5.5, pH 6 and pH 6.5, ~1-min-long exposition | External acidification decreases channel’s current amplitude in a pHe-dependent manner | Not assessed | Not assessed | [68] | |
| HeLa cells | Patch clamp Cell death assays (fluometric analysis of caspase 3/7 activation, electronic sizing of cell volume, and triple staining with Hoechst/acridine orange and propidium iodide assay. | External solution with pH 4 and pH 6, acute treatment for patch clamp experiments, and 1 h-long treatment with acidic pHe (4 and 6) for cell death assays | External acidification increases channel’s current amplitude in a pHe-dependent manner | Not assessed | Acidosis promotes HeLa necrotic cell death | [74] | |
| Human atrial cardiomyocytes | Patch clamp | External solution with pH 4–6, acute treatment | External acidification increases channel’s current amplitude in presence of divalent cations in the extracellular milieu | Not assessed | Not assessed | [75] | |
| TRPV1 | TRPV1-expressing HEK293 cells | Patch clamp | Acidic solution with pH 5.5 applied intracellularly for ~50 s | Acid treatment does not activate the channel in inside-out patches but potentiates 2-APB-evoked currents from the cytoplasmic side | Not assessed | Not assessed | [76] |
| hTRPV1-transfected HEK293t cells | Calcium imaging | External solution with pH 4.3 and pH 6.1, ~4 min-long exposition | Acidic pHe activates TRPV1 channel | pHe 6.1 determines larger Ca2+ transients with respect to pHe 4.3 in physiological extracellular Ca2+ concentration, while, in presence of low extracellular Ca2+ concentration, cells exposed to pHe 6.1 show reduced Ca2+ entry respect to pHe 4.3 exposition | Not assessed | [77] | |
| Defolliculated Xenopus laevis oocytes, TRPV1-expressing HEK293 cells | Patch clamp | Extracellular solution with pH 6.4, cells pre-treated with acid bath solution for 2 min | Acidic pHe potentiates heat-evoked TRPV1 current in oocytes; potentiation of capsaicin and heat-evoked TRPV1 currents in HEK293 cells | Not assessed | Not assessed | [78] | |
| Primary human adult dermal lymphatic endothelial cell (HDLECs) | Cell viability assay Cell invasion assay in vitro tube formation assay Transwell cell migration assay | 24 h long exposition to pHe 6.4, and 6 h long exposition for in vitro tube formation assay | Acidic pHe activates TRPV1 channel | Not assessed | Acidic pHe affects HDLECs morphology, increasing their migration and invasive abilities, proliferation and promoting lymphangiogenesis via acidosis-induced TRPV1 activation | [79] | |
| TRPV2 | TRPV2-expressing HEK293 cells | Patch clamp | Acute administration of extracellular solution with pHe 5.5 and 6 | Extracellular acidosis potentiates the response of TRPV2 to 2-APB (and analogues) from the cytosolic side, while intracellular acidification and low pHe alone are not able to elicit any detectable current | Not assessed | Not assessed | [80] |
| TRPV3 | TRPV3-expressing HEK293 cells | Patch clamp, calcium imaging | Acute administration of extracellular solution with pHe 5.5 and 6 | Extracellular acidosis potentiates the response of TRPV3 to 2-APB (and analogues) from the cytosolic side. Intracellular acidification activates the channel, eliciting small but detectable currents | Extracellular acidosis increases Ca2+ entry following 2-APB stimulation | Not assessed | [80] |
| TRPV3-expressing HEK293 cells | Patch clamp Cell death assay (PI staining assay) | Intracellular administration of acidic solution with pHe 5.5 and glycolic acid. Extracellular solution with pH 5.5. Intracellular solution with pH 5.5–7. | Glycolic acid-induced intracellular proton release in presence of acidic solution activates the channel in a reversible way. Extracellular acidification does not activate TRPV3, while intracellular acidification alone activates the channel in a pH-dependent manner | Not assessed | Glycolic acid-induced acidification induces cell toxicity and cell death | [81] | |
| Human keratinocytes cells (HaCaT) | Patch clamp, cell death assay (PI staining assay) | Intracellular administration of acidic solution with pHe 5.5 and glycolic acid | Glycolic acid-induced intracellular proton release in presence of acidic solution potentiates the channel’s response to 2-APB in a reversible manner | Not assessed | Glycolic acid-induced acidification induces cell toxicity and cell death | [81] | |
| TRPV4 | Chinese hamster ovary cells | Patch clamp | External solution with pHe 4, 5.5 and 6, acute treatment | Extracellular acidosis activates the channel in a pHe-dependent manner | Not assessed | Not assessed | [82] |
| mTRPV4-overexpressing primary cultured mouse esophageal epithelial cells | Ca2+ imaging | External solution with pHe 5, acute treatment | Not assessed | Extracellular acidic pH decreases Ca2+ entry, lowering cytosolic Ca2+ concentration | Not assessed | [83] | |
| TRPV6 | Jurkat cells | Patch clamp | External solution with pH 6, acute treatment | Extracellular acidosis suppresses TRPV6-mediated currents | Extracellular acidic pH reduces Ca2+ entry, lowering cytosolic Ca2+ concentration | Not assessed | [84] |
| TRPA1 | HEK-293t cells expressing hTRPA1, mTRPA1, or rTRPA1 | Patch clamp Calcium imaging | Acidic solutions with pH 7.0, 6.4, 6.0, and 5.4, 30 s-long treatment in calcium imaging experiments | Extracellular acidosis activates inward currents via hTRPA1 and potentiates acrolein-evoked currents of hTRPA1 in a pHe-dependent and reversible manner, while failing to activate mouse and rodent TRPA1. | Extracellular acidosis increases Ca2+ entry in hTRPA1, no effect on mTRPA1 and rTRPA1. | Not assessed | [85] |
| DRG neurons derived from TRPV1/TRPA1−/− mice and overexpression hTRPA1 | Calcium imaging | Acidic solutions with pH 5, 60 s-long treatment | Not assessed | Acidic pHe induces Ca2+ entry | Not assessed | [85] | |
| Neuroblastoma ND7/23 cells expressing hTRPA1 | Patch clamp | Acidic solution with pH 5, acute treatment | Acidic pHe activates hTRPA1 | Not assessed | Not assessed | [85] | |
| TRPC5 | TRPC5-transiently transfected HEK293 cells | Patch Clamp | External acidic solution with pH 4.2, 5.5, 6.5, 7, ~100 s-long treatment | G protein-activated and spontaneous currents are potentiated by extracellular acidic pH by increasing the channel open probability, with a maximum effect at ~pH 6.5, while more acidic values inhibit the channel | Not assessed | Not assessed | [86] |
| TRPC4 | TRPC4-transiently transfected HEK293 cells | Patch Clamp | External acidic solution with pH 4.2, 5.5, 6.5, 7, ~100 s-long treatment | G protein-activated currents are potentiated by extracellular acidic pH, with a maximum effect at ~pH 6.5 and complete inhibition at pHe 5.5 | Not assessed | Not assessed | [86] |
| mTRPC4-stably transfected HEK293 cells | Patch Clamp | External acidic solution with pH 6.8 | Low pHi (6.75–6.25) accelerates Gi/o-mediated TRPC4 activation, and this requires elevations in intracellular calcium concentration. Intracellular protons inhibit Englerin A-mediated TRPC4 activation | Not assessed | Not assessed | [87] | |
| TRPC6 | TRPC6-transiently transfected HEK293 cells | Patch Clamp | External acidic solution with pH 4.2, 5.5, 6.5, 7, ~100 s-long treatment | Acidic pHe inhibits channel’s inward and outward currents starting from pHe 6.5 and the inhibition is potentiated by more acidic pHe values. | Not assessed | Not assessed | [86] |
| ORAI1/STIM1 | Human macrophages | Patch clamp | External acidic solution with pH 6 and 8, ~200 s-long treatment | Extracellular acidosis inhibits ORAI1 channel in a pHe-dependent and reversible manner | Not assessed | Not assessed | [88] |
| H4IIE rat liver cells overexpressing ORAI1 and STIM1 | Patch clamp | External acidic solutions with pH 5.1 and 5.9 | ORAI1 and STIM1-mediated ICRAC are inhibited by acidic pHe, with maximal effect at pHe 5.5 | Not assessed | Not assessed | [89] | |
| RBL2H3 mast cell line, Jurkat T lymphocytes and heterologous ORAI1-2–3/STIM expressing HEK293 cells | Patch clamp | External and intracellular acidic solutions with pH 6 and 6.6 | External and internal acidification inhibits IP3-induced ICRAC in RBL2H3 mast cell line, Jurkat T lymphocytes, and in heterologous ORAI/STIM-mediated ICRAC in HEK293 cells in a reversible manner | Not assessed | Not assessed | [90] | |
| ORAI1/STIM1-transiently transfected HEK293 cells | Patch Clamp | External acidic solution with pH 5.5 | Acidic pHe inhibits ORAI1-2–3/STIM1 current amplitude in a reversible and pH-dependent manner, with a maximal effect at pHe 4.5 | Not assessed | Not assessed | [91] | |
| ORAI1/STIM1-transiently transfected HEK293 cells | Patch Clamp | Intracellular acidic solution with pH 6.3 | Intracellular acidosis inhibits ORAI1/STIM1 current, regulating the amplitude of the current and the Ca2+-dependent gating of the CRAC channels | Not assessed | Not assessed | [92] | |
| SH-SY5Y human neuroblastoma cells | Ca2+ signals quantification by Mn2+ quench technique | External acidic solution with pH 6.8 and 7 and 7.2. Different treatment time, ranging from ~3–4 min to ~8 min for carbachol-mediated Ca2+ entry and ~7 min for thapsigargin-mediated Ca2+ entry | Not assessed | Tumour acidic pHe inhibits carbachol- and thapsigargin-mediated Ca2+ entry in a reversible manner, while intracellular acidification or alkalinization leads to no effects in carbachol-mediated Ca2+ entry | Not assessed | [93] |
| Ion Channel | Cell Type | Methodology | Hypoxia Technique and Treatment Time | Effect of Hypoxia on Channel’s Activity/Expression | Effect of Hypoxia on Ca2+ Signals | Cellular Function | Ref. |
|---|---|---|---|---|---|---|---|
| Piezo1 | Mouse and human sickle red blood cells (RBCs) | Cell-attached and nystatin-permeabilized patch clamp Calcium imaging | Deoxygenation obtained by exposure with a superfusate gassed 30 min prior to the experiment with 100% N2 | Deoxygenation activates a Ca2+- and cation-permeable conductance in a reversible manner, and this current is sensitive to inhibition by GsMTx-4; 1 mM | Increased Ca2+ influx | Not assessed | [94] |
| Pulmonary arterial endothelial cells (PASMCs) of patients with pulmonary arterial hypertension (PAH) | Calcium imaging EdU and cell counting proliferation assay Western Blot | / | Piezo1 expression and activity are increased in idiopathic pulmonary arterial hypertension and pulmonary arterial smooth muscle cells | Increased Ca2+ influx and increased intracellular Ca2+ release | Increased PAH-PASMCs’ proliferation | [95] | |
| Pulmonary artery smooth muscle cells of mice and rats’ models with experimental chronic hypoxia-induced pulmonary hypertension (PH) Human pulmonary artery endothelial cells (hPAECs) | Western Blot Calcium imaging | Hypoxia induced by incubation in 3% O2 for 4 h–12 h or in 10% O2 for a total of 6 weeks | Piezo1 is significantly upregulated in the lung tissue of PH rats and in chronic hypoxia-induced PH models. Piezo1 protein is transiently upregulated also in hPAECs after 6 h exposition to hypoxic conditions. Hypo-osmotic conditions upregulate Piezo1 protein levels in hPAECs | Hypo-osmotic upregulation of Piezo1 promotes Ca2+ influx, promoting Akt and Erk signalling pathways activation, with downstream upregulation of Notch ligand | GsMTx4-mediated Piezo1 blockade partially reduces the chronic hypoxia-induced PH in mice with chronic hypoxia-induced pulmonary hypertension | [96] | |
| TRPM2 | TRPM2 WT and knockout (KO) neonatal hypoxic-ischemic (HI) brain injury mouse model | Western Blot | Hypoxia damage was induced in ischemic mice models by incubating the pups in a hypoxic chamber for 2 h | TRPM2 is acutely overexpressed 24 h after hypoxia-ischemic injury in brain tissue samples from mouse pups | Not assessed | Brain damage and inflammation are reduced in TRPM2 KO mice 7 days following hypoxic-ischemic brain injury. TRPM2 inhibits cell survival pathways after HI injury | [97] |
| Primary cultures of rat cortical neurons subjected to oxidative stress | Calcium imaging Trypan Blue exclusion assay | Oxidative stress induced by 1 mM or 50 µM H2O2 treatment | Not assessed | H2O2 induces TRPM2-mediated intracellular calcium rise | SiTRPM2 prevents H2O2-mediated neuronal cell death | [98] | |
| TRPM2-overexpressing HEK293 cells | Whole-cell Patch Clamp | Hypoxia induced by cell incubation with gas mixture containing 5% O2 for 30 and 60 min | TRPM2 activation is induced by 30- and 60-min exposure to hypoxic conditions | Not assessed | Hypoxia treatment enhances cell death, probably via TRPM2-mediated Ca2+ influx | [99] | |
| ARPE-19 retinal pigment epithelial cells | Patch Clamp Calcium imaging Propidium iodide cell death assay | Hypoxia induced by CoCl2 (200 μM) for 24 h | Hypoxia induces activation of TRPM2 currents and upregulates TRPM2 protein levels | Hypoxia induces TRPM2-mediated intracellular calcium rise | Hypoxia causes mitochondrial oxidative cell cytotoxicity and cell death via TRPM2-mediated Ca2+ signals | [100] | |
| Primary IGR39 melanoma cells TRPM2-overexpressing HEK293 cells | Patch Clamp Calcium imaging | Treatment with chloramine-T (Chl-T) oxidant agent | Amount of 0.5 mM Chl-T activates TRPM2 in IGR39 and in TRPM2-expressing HEK293 cells | Chl-T treatments induce a significant increase in cytosolic Ca2+ levels | Chl-T-induced TRPM2 activation and increased Ca2+ influx activate BK and KCa3.1 potassium channels | [101] | |
| PC3 prostate cancer cells | Calcium imaging MTT and TUNEL assay | Treatment with 0.5 to 4 mM H2O2 for 6 h | H2O2 induces TRPM2 activation | H2O2 treatment leads to TRPM2-mediated intracellular Ca2+ increase in a concentration-dependent manner | H2O2 induces TRPM2-Ca2+-CaMKII cascade that promotes ROS production, mitochondrial fragmentation, and inhibition of autophagy, inducing cell death | [102] | |
| TRPM2-L and TRPM2-S-expressing SH-SY5Y neuroblastoma cells | Calcium imaging | Treatment with 250 μM H2O2 for 20 min | Not assessed | H2O2 treatment leads to TRPM2-L-mediated intracellular Ca2+ increase and a decrease in TRPM2-S | TRPM2-L-expressing cells show higher HIF-1/2α levels with respect to TRPM2 short isoform and promote tumour growth in vivo | [103] | |
| Human breast cancer cells | Calcium imaging qPCR | Co-culture with neutrophils or H2O2 treatment | Neutrophil-derived H2O2 induces decrease in TRPM2 expression in H2O2-selected tumour cells | Not assessed | TRPM2 activation by neutrophil-derived H2O2 and following Ca2+ entry promotes cancer cells’ death | [104] | |
| TRPM6 | Hepatic ischemia-reperfusion rat model | qPCR | Ischemia was obtained by 60 min clamping the left hepatic artery and the portal vein | TRPM6 expression is increased in liver tissue from ischemia-reperfusion rat model | Not assessed | Not assessed | [105] |
| TRPM7 | TRPM7-overexpressing HEK293T cells Cortical neurons | Ca2+ imaging Patch clamp PI cell death assay | Hypoxia induced by anaerobic chamber containing ˂0.2% O2 atmosphere for 1, 1.5 and 2 h. | Hypoxia induces TRPM7 channel activation | Hypoxia increases Ca2+ entry | Hypoxia-activated TRPM7 mediated-Ca2+ entry determines cell death in cortical neurons | [106] |
| Hepatic ischemia-reperfusion rat model | qPCR | Ischemia was obtained by 60 min clamping the left hepatic artery and the portal vein | TRPM7 expression is increased in liver tissue from ischemia-reperfusion rat model | Not assessed | Not assessed | [105] | |
| TRPV1 | HEK293T cells overexpressing rat TRPV1 | Patch Clamp Calcium imaging | Hypoxic solution obtained by bubbling with 100% N2 gas for at least 20 min before the perfusion (PO2, 3%) | Acute hypoxia weakly increases TRPV1 activity, but negatively affects capsaicin induced TRPV1 currents | Hypoxia leads to a slight increase in cytosolic Ca2+ levels | Not assessed | [107] |
| Rat DRG neurons hTRPV1/rTRPV1-expressing HEK293 cells | Whole-cell patch-clamp | Overnight (18–20 h) exposition to hypoxia (4% O2) | Overnight exposure to hypoxic/high glucose conditions increases TRPV1 mean peak current densities in both cell lines, without affecting its expression | Not assessed | Not assessed | [108] | |
| Rat pulmonary artery smooth muscle cells (PASMCs) | Calcium imaging qPCR Western Blot Wound Healing assay BrdU proliferation assay | 24–48 h long exposition to hypoxia (1% and 10% O2) | Hypoxic conditions do not affect TRPV1 expression, but they increase TRPV1 activity | No assessed | Hypoxia-mediated TRPV1 activation enhances PASMCs migratory abilities and proliferation | [109] | |
| Human pulmonary artery smooth muscle cells (PASMCs) | Calcium imaging qPCR Western Blot Cell count proliferation assay | 72 h long exposition to hypoxia (3% O2) | Chronic hypoxia upregulates both TRPV1 gene and protein levels | Chronic hypoxia increases cytosolic Ca2+ levels | The proliferation of PASMCs is increased under hypoxia | [110] | |
| TRPV2 | HepG2 and Huh-7 human hepatoma cell lines | RT-PCR Western Blot Flow cytometry | 50, 100, 200, and 400 Μm H2O2 treatment for 24 h | H2O2 upregulates the expression of TRPV2 at mRNA and protein levels | Not assessed | Overexpression of TRPV2 promotes H2O2-induced cell death | [111] |
| TRPV3 | Rat myocardial cells | MTT and Edu staining assay Western Blot Caspase-3 and LDH activity assay | 12 h long exposition to hypoxia (1% O2) | TRPV3 is overexpressed in myocardial cells induced by ischemia/hypoxia | Not assessed | TRPV3 silencing protects cardiomyocytes from hypoxia-induced cell death and decreases the secretion of proinflammatory cytokines | [112] |
| Primary rat pulmonary artery smooth muscle cells (PASMCs) | Western Blot Flow cytometry MTT assay | 24 h long exposition to hypoxia (3% O2) | TRPV3 protein expression is enhanced in PASMCs from hypoxic rats | Not assessed | TRPV3 mediates hypoxia-induced PASMCs’ proliferation via PI3K/AKT signalling | [113] | |
| TRPV3-overexpressing HEK293 | Patch Clamp | 12 h long exposition to hypoxia (1% O2) | Pre-incubation in hypoxic conditions potentiates TRPV3 currents in response to 2-APB treatment | Not assessed | Not assessed | [114] | |
| TRPV4 | Rat cardiomyocytes | Western Blot qPCR Calcium imaging | 6 h long exposition to hypoxia (95% N2) in a controlled hypoxic chamber | TRPV4 gene and protein expression levels are increased after 6 h exposure to hypoxia | Hypoxia increases TRPV4-mediated Ca2+ influx responses to 300 nM GSK | Hypoxia-mediated activation of TRPV4 induces cytosolic Ca2+ overload in cardiomyocytes, leading to ROS production and oxidative injury in vitro and in vivo | [115] |
| Adult rat hippocampal astrocytes | Patch Clamp qPCR Western Blot Calcium imaging | Hypoxia/ischemia (H/I) is induced by occlusion of the common carotids in combination with hypoxic conditions (from 1 h up to 7 days, 6% O2) | TRPV4 mRNA and protein expression are significantly increased 1 h after H/I. H/I also activates TRPV4 channel | H/I enhances the response of 4aPDD, inducing TRPV4-mediated Ca2+ oscillations | Not assessed | [116] | |
| TRPA1 | Several breast and lung cancer cell lines | Calcium imaging Cell viability and apoptosis assay via PI and Annexin IV staining | Treatment with 10 µM H2O2 for 15 min for calcium measurements, 1, 20, and 100 µM for 72–96 h-long exposition for cell viability and cell death assays | H2O2 treatment activates TRPA1 channel | H2O2 treatment increases TRPA1-mediated calcium entry | TRPA1-mediated calcium entry promotes cell survival by upregulating anti-apoptotic pathways and promoting oxidative stress resistance | [117] |
| Oligodendrocytes | Calcium imaging | Ischemia inducing solution | Not assessed | Ischemia-induced intracellular acidosis promotes Ca2+ entry via TRPA1 | Ischemia-induced intracellular acidosis and consequent Ca2+ entry via TRPA1 mediate myelin damage | [118] | |
| TRPC1 | U-87 MG glioma cells | qPCR, western blot | Hypoxia induced by exposition to 1% O2 | Not assessed | Not assessed | TRPC1 participates in hypoxia-induced VEGF gene and protein expression | [119] |
| MDA-MB-468 breast cancer cells | qPCR, calcium imaging | Hypoxia induced by exposition to 1% O2 for 24 h | Hypoxia upregulates TRPC1 via HIF1α | siTRPC1 reduces non-stimulated Ca2+ entry and increases Store-Operated Ca2+ entry in hypoxic conditions | TRPC1 overexpression promotes Snail EMT marker upregulation and decrease in claudin-4 epithelial marker in hypoxic conditions. TRPC1 regulates HIF-1α protein levels via Akt-dependent pathway and promotes hypoxia-induced STAT3 and EGFR phosphorylation. TRPC1 also regulates hypoxia-induced LC3BII levels via effects on EGFR. | [120] | |
| TRPC5 | MCF-7/WT and adriamycin-treated (MCF-7/ADM) human breast cancer cells | Western Blot, immunofluorescence, | Not assessed | Not assessed | Not assessed | TRPC5 promotes HIF-1alpha translocation to the nucleus and HIF-1alpha-mediated VEGF expression, boosting tumour angiogenesis | [121] |
| SW620 colon cancer cells | Western blot, transwell invasion, and migration assay, MTT proliferation assay | Not assessed | Not assessed | Not assessed | TRPC5 activates HIF-1alpha-Twist signalling to induce EMT, supporting colon cancer cells’ migration, invasion, and proliferation | [122] | |
| TRPC6 | Murine pancreatic stellate cells (mPSCs) | Time-lapse single-cell random migration assay Bead-based cytokine assay qPCR Western Blot Ca2+ signals quantification by Mn2+ quench technique | 24 h incubation in hypoxic conditions (1% O2, 5%CO2, and 94% N2) or chemically induced hypoxia by pretreatment with 0.5 mmol/l DMOG | Hypoxic conditions enhance TRPC6 expression and activate the channel | Hypoxia stimulates Ca2+ influx mediated by TRPC6 channels | Hypoxia-induced TRPC6 activation enhances mPSCs migration via secretion of pro-migratory factors | [123] |
| lx-2 human hepatic stellate cells (HSCs) | Calcium imaging qPCR Western Blot | Hypoxia induced by 100 μmol/L CoCl2 treatment | Hypoxic conditions enhance TRPC6 expression and activate the channel | Hypoxia stimulates Ca2+ influx mediated by TRPC6 channels | Hypoxia-induced TRPC6 activation and consequent calcium entry promote the synthesis of ECM proteins, which facilitate the fibrotic activation of HSCs | [124] | |
| Huh7 and HepG2 hepatocellular carcinoma cells (HCCs) | Confocal Calcium imaging Western Blot | Hypoxia induced by cell incubation in a low oxygen atmosphere with 1% O2, 5%CO2, and 94% N2 for 6 h | Hypoxic conditions activate the channel | Hypoxia promotes calcium influx | Hypoxia-induced TRPC6-mediated calcium entry promotes HCCs drug resistance via STAT3 pathway | [125] | |
| U373MG and HMEC-1 glioblastoma cell lines | qPCR Western Blot Calcium imaging Proliferation assay Matrigel invasion assay Endothelial cell tube formation assay | Hypoxia induced by 100 μmol/L CoCl2 treatment | Hypoxia enhances TRPC6 expression via Notch pathway | Hypoxia stimulates Ca2+ influx mediated by TRPC6 channels | Hypoxia-induced TRPC6-mediated calcium entry promotes HCCs proliferation, colony formation, and invasion via NFAT pathway | [126] | |
| ORAI1/STIM1 | Primary Aortic Smooth Muscle Cells and HEK293 cells transfected with ORAI1 and STIM1 | Patch Clamp Calcium imaging | Hypoxia was induced with 3 methods: (1) sodium dithionite (Na2S2O4) treatment to 1 mM final concentration, pH adjustment to pH 7.4, and bubbling with 100% N2. (2) cell culture media with 30 min-long bubbling with 100% N2. (3) cell culture media with 30 min-long bubbling with 3% O2 | Intracellular acidification induced by hypoxia in HEK293 cells leads to inhibition of SOCE by disrupting the electrostatic ORAI1/STIM1 binding and closing ORAI1 channel. | Hypoxia-induced intracellular acidification reduces SOCE in Primary Aortic Smooth Muscle Cells and HEK293 cells transfected with ORAI1 and STIM1 | Not assessed | [92] |
| A549 non-small cell lung cancer cells | Western Blot qPCR BrdU cell proliferation assay Calcium imaging Scrape-wound migration assay Matrigel transwell invasion assay | Hypoxia induced by Nicotine treatment for 48 h | Nicotine treatment-induced hypoxia determines ORAI1 overexpression at gene and protein levels | Nicotine treatment-induced hypoxia increases intracellular basal calcium levels and SOCE | Nicotine treatment-induced hypoxia increases A549 cells’ proliferation and migration | [127] | |
| MDA-MB 231 and BT549 breast cancer cell lines and Human Microvascular Endothelial Cell line-1 (HMEC-1) | Western Blot qPCR Calcium imaging Migration assay (Wound healing and transwell migration assay) Matrigel transwell invasion assay Tube formation assay in vitro | Hypoxia induced by cell incubation in low oxygen atmosphere | Hypoxia promotes ORAI1 gene and protein upregulation via activation of Notch1 signalling | Hypoxia increases thapsigargin-induced SOCE, with consequent rise in cytosolic calcium entry | Hypoxia-induced ORAI1 overexpression and consequent increase in SOCE promote NFAT4 activation and enhance neuroblastoma cells’ migration, invasion, and angiogenesis | [128] | |
| HCT-116 and SW480 human colon cancer cells and Human Microvascular Endothelial Cell line-1 (HMEC-1) | Western Blot qPCR Calcium imaging Transwell migration assay Matrigel transwell invasion assay Tube formation assay in vitro Cell attachment and detachment assays | Hypoxia induced by 100 μmol/L CoCl2 treatment | Hypoxia promotes ORAI1 gene and protein upregulation via activation of Notch1 signalling | Hypoxia increases thapsigargin-induced SOCE | Hypoxia-induced ORAI1 overexpression and consequent increase in SOCE promote NFATc3 activation and enhance neuroblastoma cells’ migration, invasion, and angiogenesis | [129] |
2.1. Piezo Channels
2.2. Transient Receptor Potential Channels
2.2.1. TRP Melastatin Subfamily
2.2.2. TRP Vanilloid Subfamily
2.2.3. TRP Ankyrin Subfamily
2.2.4. TRP Canonical Subfamily
2.3. Store-Operated Ca2+ Channels
3. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumour Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. 2020, 2020, 6283796. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef]
- Marchiq, I.; Le Floch, R.; Roux, D.; Simon, M.-P.; Pouyssegur, J. Genetic Disruption of Lactate/H + Symporters (MCTs) and Their Subunit CD147/BASIGIN Sensitizes Glycolytic Tumour Cells to Phenformin. Cancer Res. 2015, 75, 171–180. [Google Scholar] [CrossRef]
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.-M.; et al. Double Genetic Disruption of Lactate Dehydrogenases A and B Is Required to Ablate the “Warburg Effect” Restricting Tumour Growth to Oxidative Metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef]
- Swietach, P.; Hulikova, A.; Vaughan-Jones, R.D.; Harris, A.L. New Insights into the Physiological Role of Carbonic Anhydrase IX in Tumour PH Regulation. Oncogene 2010, 29, 6509–6521. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The Hypoxic Tumour Microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumour Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated PH: A Perfect Storm for Cancer Progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems Analysis of Intracellular PH Vulnerabilities for Cancer Therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef] [PubMed]
- Kazyken, D.; Lentz, S.I.; Fingar, D.C. Alkaline Intracellular PH (PHi) Activates AMPK–MTORC2 Signaling to Promote Cell Survival during Growth Factor Limitation. J. Biol. Chem. 2021, 297, 101100. [Google Scholar] [CrossRef] [PubMed]
- Flinck, M.; Kramer, S.H.; Pedersen, S.F. Roles of PH in Control of Cell Proliferation. Acta Physiol. 2018, 223, e13068. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Tomás, R.; Pérez-Guillén, I. Lactate in the Tumour Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers 2020, 12, 3244. [Google Scholar] [CrossRef] [PubMed]
- Flinck, M.; Kramer, S.H.; Schnipper, J.; Andersen, A.P.; Pedersen, S.F. The Acid-Base Transport Proteins NHE1 and NBCn1 Regulate Cell Cycle Progression in Human Breast Cancer Cells. Cell Cycle 2018, 17, 1056–1067. [Google Scholar] [CrossRef]
- Rengifo Rodriguez, J.E.; Garcia-Perdomo, H.A. Role of Monocarboxylate Transporters in the Diagnosis, Progression, Prognosis, and Treatment of Prostate Cancer. Turk. J. Urol. 2020, 46, 413–418. [Google Scholar] [CrossRef]
- Ward, C.; Meehan, J.; Gray, M.E.; Murray, A.F.; Argyle, D.J.; Kunkler, I.H.; Langdon, S.P. The Impact of Tumour PH on Cancer Progression: Strategies for Clinical Intervention. Explor. Target. Anti-Tumour Ther. 2020, 1, 71–100. [Google Scholar] [CrossRef]
- Mboge, M.; Mahon, B.; McKenna, R.; Frost, S. Carbonic Anhydrases: Role in PH Control and Cancer. Metabolites 2018, 8, 19. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Goncalves, R.L.S.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The Contributions of Respiration and Glycolysis to Extracellular Acid Production. Biochim. Biophys. Acta BBA Bioenerg. 2015, 1847, 171–181. [Google Scholar] [CrossRef]
- Swietach, P. What Is PH Regulation, and Why Do Cancer Cells Need It? Cancer Metastasis Rev. 2019, 38, 5–15. [Google Scholar] [CrossRef]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumour Hypoxia as a Driving Force in Genetic Instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhong, Z.-F.; Wang, S.-P.; Vong, C.-T.; Yu, B.; Wang, Y.-T. HIF-1: Structure, Biology and Natural Modulators. Chin. J. Nat. Med. 2021, 19, 521–527. [Google Scholar] [CrossRef]
- Doe, M.R.; Ascano, J.M.; Kaur, M.; Cole, M.D. Myc Posttranscriptionally Induces HIF1 Protein and Target Gene Expression in Normal and Cancer Cells. Cancer Res. 2012, 72, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Tam, S.Y.; Wu, V.W.C.; Law, H.K.W. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and beyond. Front. Oncol. 2020, 10, 486. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouysségur, J. Hypoxia-Induced Autophagy: Cell Death or Cell Survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Cosse, J.-P.; Michiels, C. Tumour Hypoxia Affects the Responsiveness of Cancer Cells to Chemotherapy and Promotes Cancer Progression. Anti-Cancer Agents Med. Chem. 2008, 8, 790–797. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Luderer, M.; Azab, A.K. Hypoxia Promotes Stem Cell-like Phenotype in Multiple Myeloma Cells. Blood Cancer J. 2014, 4, e262. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, Z.; Zhu, Y.; Chen, J.; Li, W. Role of Hypoxia Inducible Factor-1 in Cancer Stem Cells (Review). Mol. Med. Rep. 2020, 23, 17. [Google Scholar] [CrossRef]
- Lv, Y.; Zhao, S.; Han, J.; Zheng, L.; Yang, Z.; Zhao, L. Hypoxia-Inducible Factor-1α Induces Multidrug Resistance Protein in Colon Cancer. Oncotargets Ther. 2015, 8, 1941–1948. [Google Scholar] [CrossRef]
- Li, D.-W.; Dong, P.; Wang, F.; Chen, X.-W.; Xu, C.-Z.; Zhou, L. Hypoxia Induced Multidrug Resistance of Laryngeal Cancer Cells via Hypoxia-Inducible Factor-1α. Asian Pac. J. Cancer Prev. 2013, 14, 4853–4858. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, X.; Gan, L.; Yang, X.; Miao, Z. Hypoxia Induces Universal but Differential Drug Resistance and Impairs Anticancer Mechanisms of 5-Fluorouracil in Hepatoma Cells. Acta Pharmacol. Sin. 2017, 38, 1642–1654. [Google Scholar] [CrossRef] [PubMed]
- Filatova, A.; Seidel, S.; Böğürcü, N.; Gräf, S.; Garvalov, B.K.; Acker, T. Acidosis Acts through HSP90 in a PHD/VHL-Independent Manner to Promote HIF Function and Stem Cell Maintenance in Glioma. Cancer Res. 2016, 76, 5845–5856. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug Resistance and Cellular Adaptation to Tumour Acidic PH Microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct Role of Macrophages in Different Tumour Microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A.; et al. Neutralization of Tumour Acidity Improves Antitumour Responses to Immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumour Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Gatenbee, C.; Robertson-Tessi, M.; Bravo, R.; Dhillon, J.; Balagurunathan, Y.; Berglund, A.; Vishvakarma, N.; Ibrahim-Hashim, A.; Choi, J.; et al. Acidity Promotes Tumour Progression by Altering Macrophage Phenotype in Prostate Cancer. Br. J. Cancer 2019, 121, 556–566. [Google Scholar] [CrossRef]
- Díaz, F.E.; Dantas, E.; Cabrera, M.; Benítez, C.A.; Delpino, M.V.; Duette, G.; Rubione, J.; Sanjuan, N.; Trevani, A.S.; Geffner, J. Fever-Range Hyperthermia Improves the Anti-Apoptotic Effect Induced by Low PH on Human Neutrophils Promoting a Proangiogenic Profile. Cell Death Dis. 2016, 7, e2437. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Tumour Acidosis: From the Passenger to the Driver’s Seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumour Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Mathiesen, B.; Kindem, K.; Galappathi, K. Acidic Extracellular PH Promotes Experimental Metastasis of Human Melanoma Cells in Athymic Nude Mice. Cancer Res. 2006, 66, 6699–6707. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Novak, I.; Alves, F.; Schwab, A.; Pardo, L.A. Alternating PH Landscapes Shape Epithelial Cancer Initiation and Progression: Focus on Pancreatic Cancer. BioEssays 2017, 39, 1600253. [Google Scholar] [CrossRef] [PubMed]
- Pethő, Z.; Najder, K.; Carvalho, T.; McMorrow, R.; Todesca, L.M.; Rugi, M.; Bulk, E.; Chan, A.; Löwik, C.W.G.M.; Reshkin, S.J.; et al. PH-Channeling in Cancer: How PH-Dependence of Cation Channels Shapes Cancer Pathophysiology. Cancers 2020, 12, 2484. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S. Calcium Signaling. In Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1131, ISBN 978-3-030-12456-4. [Google Scholar]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium Signalling: Dynamics, Homeostasis and Remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The Calcium–Cancer Signalling Nexus. Nat. Rev. Cancer 2017, 17, 373–380. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca 2+ Transport in Cancer: How It Contributes to Cancer Hallmarks? Philos. Trans. R. Soc. B 2014, 369, 20130097. [Google Scholar] [CrossRef]
- Panda, S.; Chatterjee, O.; Roy, L.; Chatterjee, S. Targeting Ca2+ Signaling: A New Arsenal against Cancer. Drug Discov. Today 2022, 27, 923–934. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca2+ Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 2020, 10, 1618. [Google Scholar] [CrossRef]
- Scarpellino, G.; Munaron, L.; Cantelmo, A.R.; Fiorio Pla, A. Calcium-Permeable Channels in Tumour Vascularization: Peculiar Sensors of Microenvironmental Chemical and Physical Cues. In From Malignant Transformation to Metastasis; Stock, C., Pardo, L.A., Eds.; Reviews of Physiology, Biochemistry and Pharmacology; Springer International Publishing: Cham, Switzerland, 2020; Volume 182, pp. 111–137. ISBN 978-3-030-99799-1. [Google Scholar]
- Catacuzzeno, L.; Sforna, L.; Esposito, V.; Limatola, C.; Franciolini, F. Ion Channels in Glioma Malignancy. In Transportome Malfunction in the Cancer Spectrum; Stock, C., Pardo, L.A., Eds.; Reviews of Physiology, Biochemistry and Pharmacology; Springer International Publishing: Cham, Switzerland, 2020; Volume 181, pp. 223–267. ISBN 978-3-030-90919-2. [Google Scholar]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP Ion Channels in Cancer and Tumourigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Seta, K.A.; Yuan, Y.; Spicer, Z.; Lu, G.; Bedard, J.; Ferguson, T.K.; Pathrose, P.; Cole-Strauss, A.; Kaufhold, A.; Millhorn, D.E. The Role of Calcium in Hypoxia-Induced Signal Transduction and Gene Expression. Cell Calcium 2004, 36, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Tajada, S. Calcium Permeable Channels in Cancer Hallmarks. Front. Pharmacol. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Glitsch, M. Protons and Ca2+: Ionic Allies in Tumour Progression? Physiology 2011, 26, 252–265. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef]
- Girault, A.; Ahidouch, A.; Ouadid-Ahidouch, H. Roles for Ca2+ and K+ Channels in Cancer Cells Exposed to the Hypoxic Tumour Microenvironment. Biochim. Biophys. Acta BBA Mol. Cell Res. 2020, 1867, 118644. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Polak, J. Hypoxia. 4. Hypoxia and Ion Channel Function. Am. J. Physiol. Cell Physiol. 2011, 300, C951–C967. [Google Scholar] [CrossRef]
- Glitsch, M. Mechano- and PH-Sensing Convergence on Ca2+-Mobilising Proteins—A Recipe for Cancer? Cell Calcium 2019, 80, 38–45. [Google Scholar] [CrossRef]
- Bae, C.; Sachs, F.; Gottlieb, P.A. Protonation of the Human PIEZO1 Ion Channel Stabilizes Inactivation. J. Biol. Chem. 2015, 290, 5167–5173. [Google Scholar] [CrossRef]
- Kuntze, A.; Goetsch, O.; Fels, B.; Najder, K.; Unger, A.; Wilhelmi, M.; Sargin, S.; Schimmelpfennig, S.; Neumann, I.; Schwab, A.; et al. Protonation of Piezo1 Impairs Cell-Matrix Interactions of Pancreatic Stellate Cells. Front. Physiol. 2020, 11, 89. [Google Scholar] [CrossRef]
- Starkus, J.G.; Fleig, A.; Penner, R. The Calcium-Permeable Non-Selective Cation Channel TRPM2 Is Modulated by Cellular Acidification: Proton-Induced Inhibition of TRPM2. J. Physiol. 2010, 588, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xie, J.; Yue, L. Modulation of TRPM2 by Acidic PH and the Underlying Mechanisms for PH Sensitivity. J. Gen. Physiol. 2009, 134, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zou, J.; Xia, R.; Vaal, M.L.; Seymour, V.A.; Luo, J.; Beech, D.J.; Jiang, L.-H. State-Dependent Inhibition of TRPM2 Channel by Acidic PH. J. Biol. Chem. 2010, 285, 30411–30418. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Yang, W.; Beech, D.J.; Jiang, L.-H. A Residue in the TRPM2 Channel Outer Pore Is Crucial in Determining Species-Dependent Sensitivity to Extracellular Acidic PH. Pflug. Arch. Eur. J. Physiol. 2011, 462, 293–302. [Google Scholar] [CrossRef]
- Gwanyanya, A.; Amuzescu, B.; Zakharov, S.I.; Macianskiene, R.; Sipido, K.R.; Bolotina, V.M.; Vereecke, J.; Mubagwa, K. Magnesium-Inhibited, TRPM6/7-like Channel in Cardiac Myocytes: Permeation of Divalent Cations and PH-Mediated Regulation: Cardiac TRPM6/7-like Channels. J. Physiol. 2004, 559, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, J.; Yue, L. Functional Characterization of Homo- and Heteromeric Channel Kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef]
- Kozak, J.A.; Matsushita, M.; Nairn, A.C.; Cahalan, M.D. Charge Screening by Internal PH and Polyvalent Cations as a Mechanism for Activation, Inhibition, and Rundown of TRPM7/MIC Channels. J. Gen. Physiol. 2005, 126, 499–514. [Google Scholar] [CrossRef]
- Chokshi, R.; Matsushita, M.; Kozak, J.A. Detailed Examination of Mg2+ and PH Sensitivity of Human TRPM7 Channels. Am. J. Physiol. Cell Physiol. 2012, 302, C1004–C1011. [Google Scholar] [CrossRef]
- Chu, X.; Zhu, X.; Wei, W.; Li, G.; Simon, R.P.; MacDonald, J.F.; Xiong, Z. Acidosis Decreases Low Ca2+-induced Neuronal Excitation by Inhibiting the Activity of Calcium-sensing Cation Channels in Cultured Mouse Hippocampal Neurons. J. Physiol. 2003, 550, 385–399. [Google Scholar] [CrossRef]
- Jiang, J.; Li, M.; Yue, L. Potentiation of TRPM7 Inward Currents by Protons. J. Gen. Physiol. 2005, 126, 137–150. [Google Scholar] [CrossRef]
- Numata, T.; Sato-Numata, K.; Okada, Y. TRPM7 Is Involved in Acid-Induced Necrotic Cell Death in a Manner Sensitive to Progesterone in Human Cervical Cancer Cells: Role of TRPM7 in Cervical Cancer Cell Death. Physiol. Rep. 2019, 7, e14157. [Google Scholar] [CrossRef] [PubMed]
- Mačianskienė, R.; Almanaitytė, M.; Jekabsone, A.; Mubagwa, K. Modulation of Human Cardiac TRPM7 Current by Extracellular Acidic PH Depends upon Extracellular Concentrations of Divalent Cations. PLoS ONE 2017, 12, e0170923. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heber, S.; Ciotu, C.I.; Hartner, G.; Gold-Binder, M.; Ninidze, N.; Gleiss, A.; Kress, H.-G.; Fischer, M.J.M. TRPV1 Antagonist BCTC Inhibits PH 6.0-Induced Pain in Human Skin. Pain 2020, 161, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.; Liu, B.; Yao, J.; Fu, Q.; Qin, F. Uncoupling Proton Activation of Vanilloid Receptor TRPV1. J. Neurosci. 2007, 27, 12797–12807. [Google Scholar] [CrossRef]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The Cloned Capsaicin Receptor Integrates Multiple Pain-Producing Stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef]
- Nakanishi, M.; Morita, Y.; Hata, K.; Muragaki, Y. Acidic Microenvironments Induce Lymphangiogenesis and IL-8 Production via TRPV1 Activation in Human Lymphatic Endothelial Cells. Exp. Cell Res. 2016, 345, 180–189. [Google Scholar] [CrossRef]
- Gao, L.; Yang, P.; Qin, P.; Lu, Y.; Li, X.; Tian, Q.; Li, Y.; Xie, C.; Tian, J.; Zhang, C.; et al. Selective Potentiation of 2-APB-Induced Activation of TRPV1–3 Channels by Acid. Sci. Rep. 2016, 6, 20791. [Google Scholar] [CrossRef]
- Cao, X.; Yang, F.; Zheng, J.; Wang, K. Intracellular Proton-Mediated Activation of TRPV3 Channels Accounts for the Exfoliation Effect of α-Hydroxyl Acids on Keratinocytes. J. Biol. Chem. 2012, 287, 25905–25916. [Google Scholar] [CrossRef]
- Suzuki, M.; Mizuno, A.; Kodaira, K.; Imai, M. Impaired Pressure Sensation in Mice Lacking TRPV4. J. Biol. Chem. 2003, 278, 22664–22668. [Google Scholar] [CrossRef] [PubMed]
- Shikano, M.; Ueda, T.; Kamiya, T.; Ishida, Y.; Yamada, T.; Mizushima, T.; Shimura, T.; Mizoshita, T.; Tanida, S.; Kataoka, H.; et al. Acid Inhibits TRPV4-Mediated Ca2+ Influx in Mouse Esophageal Epithelial Cells: Acid-Sensitive TRPV4 in the Esophageal Epithelium. Neurogastroenterol. Motil. 2011, 23, 1020-e497. [Google Scholar] [CrossRef]
- Cherezova, A.L.; Negulyaev, J.A.; Zenin, V.V.; Semenova, S.B. Extracellular PH Regulates the Entry of Calcium into Jurkat T-Cells. Cell Tiss. Biol. 2018, 12, 41–47. [Google Scholar] [CrossRef]
- De la Roche, J.; Eberhardt, M.J.; Klinger, A.B.; Stanslowsky, N.; Wegner, F.; Koppert, W.; Reeh, P.W.; Lampert, A.; Fischer, M.J.M.; Leffler, A. The Molecular Basis for Species-Specific Activation of Human TRPA1 Protein by Protons Involves Poorly Conserved Residues within Transmembrane Domains 5 and 6. J. Biol. Chem. 2013, 288, 20280–20292. [Google Scholar] [CrossRef] [PubMed]
- Semtner, M.; Schaefer, M.; Pinkenburg, O.; Plant, T.D. Potentiation of TRPC5 by Protons. J. Biol. Chem. 2007, 282, 33868–33878. [Google Scholar] [CrossRef] [PubMed]
- Thakur, D.P.; Wang, Q.; Jeon, J.; Tian, J.; Zhu, M.X. Intracellular Acidification Facilitates Receptor-operated TRPC4 Activation through PLCδ1 in a Ca2+-dependent Manner. J. Physiol. 2020, 598, 2651–2667. [Google Scholar] [CrossRef]
- Malayev, A.; Nelson, D.J. Extracellular PH Modulates the Ca2+ Current Activated by Depletion of Intracellular Ca2+ Stores in Human Macrophages. J. Membr. Biol. 1995, 146, 101–111. [Google Scholar] [CrossRef]
- Scrimgeour, N.R.; Wilson, D.P.; Rychkov, G.Y. Glu106 in the Orai1 Pore Contributes to Fast Ca2+-Dependent Inactivation and PH Dependence of Ca2+ Release-Activated Ca2+ (CRAC) Current. Biochem. J. 2012, 441, 743–753. [Google Scholar] [CrossRef]
- Beck, A.; Fleig, A.; Penner, R.; Peinelt, C. Regulation of Endogenous and Heterologous Ca2+ Release-Activated Ca2+ Currents by PH. Cell Calcium 2014, 56, 235–243. [Google Scholar] [CrossRef]
- Tsujikawa, H.; Yu, A.S.; Xie, J.; Yue, Z.; Yang, W.; He, Y.; Yue, L. Identification of Key Amino Acid Residues Responsible for Internal and External PH Sensitivity of Orai1/STIM1 Channels. Sci. Rep. 2015, 5, 16747. [Google Scholar] [CrossRef]
- Mancarella, S.; Wang, Y.; Deng, X.; Landesberg, G.; Scalia, R.; Panettieri, R.A.; Mallilankaraman, K.; Tang, X.D.; Madesh, M.; Gill, D.L. Hypoxia-Induced Acidosis Uncouples the STIM-Orai Calcium Signaling Complex. J. Biol. Chem. 2011, 286, 44788–44798. [Google Scholar] [CrossRef]
- Laskay, G.; Kálmán, K.; Van Kerkhove, E.; Steels, P.; Ameloot, M. Store-Operated Ca2+-Channels Are Sensitive to Changes in Extracellular PH. Biochem. Biophys. Res. Commun. 2005, 337, 571–579. [Google Scholar] [CrossRef]
- Vandorpe, D.H.; Xu, C.; Shmukler, B.E.; Otterbein, L.E.; Trudel, M.; Sachs, F.; Gottlieb, P.A.; Brugnara, C.; Alper, S.L. Hypoxia Activates a Ca2+-Permeable Cation Conductance Sensitive to Carbon Monoxide and to GsMTx-4 in Human and Mouse Sickle Erythrocytes. PLoS ONE 2010, 5, e8732. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Lu, W.; Chen, Y.; Duan, X.; Zhang, C.; Luo, X.; Lin, Z.; Chen, J.; Liu, S.; Yan, H.; et al. Upregulation of Piezo1 (Piezo Type Mechanosensitive Ion Channel Component 1) Enhances the Intracellular Free Calcium in Pulmonary Arterial Smooth Muscle Cells from Idiopathic Pulmonary Arterial Hypertension Patients. Hypertension 2021, 77, 1974–1989. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Babicheva, A.; Jain, P.P.; Rodriguez, M.; Ayon, R.J.; Ravellette, K.S.; Wu, L.; Balistrieri, F.; Tang, H.; et al. Endothelial Upregulation of Mechanosensitive Channel Piezo1 in Pulmonary Hypertension. Am. J. Physiol. Cell Physiol. 2021, 321, C1010–C1027. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Turlova, E.; Li, F.; Bao, M.; Szeto, V.; Wong, R.; Abussaud, A.; Wang, H.; Zhu, S.; Gao, X.; et al. Transient Receptor Potential Melastatin 2 Channels (TRPM2) Mediate Neonatal Hypoxic-Ischemic Brain Injury in Mice. Exp. Neurol. 2017, 296, 32–40. [Google Scholar] [CrossRef]
- Kaneko, S.; Kawakami, S.; Hara, Y.; Wakamori, M.; Itoh, E.; Minami, T.; Takada, Y.; Kume, T.; Katsuki, H.; Mori, Y.; et al. A Critical Role of TRPM2 in Neuronal Cell Death by Hydrogen Peroxide. J. Pharmacol. Sci. 2006, 101, 66–76. [Google Scholar] [CrossRef]
- Duzgun Ergun, D.; Dursun, S.; Pastaci Ozsobaci, N.; Hatırnaz Ng, O.; Naziroglu, M.; Ozcelik, D. The Potential Protective Roles of Zinc, Selenium and Glutathione on Hypoxia-Induced TRPM2 Channel Activation in Transfected HEK293 Cells. J. Recept. Signal Transduct. 2020, 40, 521–530. [Google Scholar] [CrossRef]
- Özkaya, D.; Nazıroğlu, M.; Vanyorek, L.; Muhamad, S. Involvement of TRPM2 Channel on Hypoxia-Induced Oxidative Injury, Inflammation, and Cell Death in Retinal Pigment Epithelial Cells: Modulator Action of Selenium Nanoparticles. Biol. Trace Elem. Res. 2021, 199, 1356–1369. [Google Scholar] [CrossRef]
- Ferrera, L.; Barbieri, R.; Picco, C.; Zuccolini, P.; Remigante, A.; Bertelli, S.; Fumagalli, M.R.; Zifarelli, G.; La Porta, C.A.M.; Gavazzo, P.; et al. TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase. Int. J. Mol. Sci. 2021, 22, 8359. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, L.; Yue, J. Oxidative Stress Activates the TRPM2-Ca2+-CaMKII-ROS Signaling Loop to Induce Cell Death in Cancer Cells. Biochim. Biophys. Acta BBA Mol. Cell Res. 2017, 1864, 957–967. [Google Scholar] [CrossRef]
- Chen, S.; Hoffman, N.E.; Shanmughapriya, S.; Bao, L.; Keefer, K.; Conrad, K.; Merali, S.; Takahashi, Y.; Abraham, T.; Hirschler-Laszkiewicz, I.; et al. A Splice Variant of the Human Ion Channel TRPM2 Modulates Neuroblastoma Tumour Growth through Hypoxia-Inducible Factor (HIF)-1/2α. J. Biol. Chem. 2014, 289, 36284–36302. [Google Scholar] [CrossRef]
- Gershkovitz, M.; Caspi, Y.; Fainsod-Levi, T.; Katz, B.; Michaeli, J.; Khawaled, S.; Lev, S.; Polyansky, L.; Shaul, M.E.; Sionov, R.V.; et al. TRPM2 Mediates Neutrophil Killing of Disseminated Tumour Cells. Cancer Res. 2018, 78, 2680–2690. [Google Scholar] [CrossRef] [PubMed]
- Bilecik, T.; Karateke, F.; Elkan, H.; Gokce, H. The Effects of TRPM2, TRPM6, TRPM7 and TRPM8 Gene Expression in Hepatic Ischemia Reperfusion Injury. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3088–3095. [Google Scholar] [PubMed]
- Aarts, M.; Iihara, K.; Wei, W.-L.; Xiong, Z.-G.; Arundine, M.; Cerwinski, W.; MacDonald, J.F.; Tymianski, M. A Key Role for TRPM7 Channels in Anoxic Neuronal Death. Cell 2003, 115, 863–877. [Google Scholar] [CrossRef]
- Kim, K.S.; Yoo, H.Y.; Park, K.S.; Kim, J.K.; Zhang, Y.-H.; Kim, S.J. Differential Effects of Acute Hypoxia on the Activation of TRPV1 by Capsaicin and Acidic PH. J. Physiol. Sci. 2012, 62, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Ristoiu, V.; Shibasaki, K.; Uchida, K.; Zhou, Y.; Ton, B.-H.T.; Flonta, M.-L.; Tominaga, M. Hypoxia-Induced Sensitization of Transient Receptor Potential Vanilloid 1 Involves Activation of Hypoxia-Inducible Factor-1 Alpha and PKC. Pain 2011, 152, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Parpaite, T.; Cardouat, G.; Mauroux, M.; Gillibert-Duplantier, J.; Robillard, P.; Quignard, J.-F.; Marthan, R.; Savineau, J.-P.; Ducret, T. Effect of Hypoxia on TRPV1 and TRPV4 Channels in Rat Pulmonary Arterial Smooth Muscle Cells. Pflug. Arch. Eur. J. Physiol. 2016, 468, 111–130. [Google Scholar] [CrossRef]
- Wang, Y.X.; Wang, J.; Wang, C.; Liu, J.; Shi, L.P.; Xu, M.; Wang, C. Functional Expression of Transient Receptor Potential Vanilloid-Related Channels in Chronically Hypoxic Human Pulmonary Arterial Smooth Muscle Cells. J. Membr. Biol. 2008, 223, 151–159. [Google Scholar] [CrossRef]
- Ma, W.; Li, C.; Yin, S.; Liu, J.; Gao, C.; Lin, Z.; Huang, R.; Huang, J.; Li, Z. Novel Role of TRPV2 in Promoting the Cytotoxicity of H2O2-Mediated Oxidative Stress in Human Hepatoma Cells. Free. Radic. Biol. Med. 2015, 89, 1003–1013. [Google Scholar] [CrossRef]
- Wang, J.; Chen, X.; Huang, W. MicroRNA-369 Attenuates Hypoxia-Induced Cardiomyocyte Apoptosis and Inflammation via Targeting TRPV3. Braz. J. Med. Biol. Res. 2021, 54, e10550. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, Y.; Luo, Q.; Wang, P.; Shi, P.; Song, C.; Mingyao, E.; Ren, J.; Fu, B.; Sun, H. The Transient Receptor Potential Vanilloid-3 Regulates Hypoxia-Mediated Pulmonary Artery Smooth Muscle Cells Proliferation via PI3K/AKT Signaling Pathway. Cell Prolif. 2018, 51, e12436. [Google Scholar] [CrossRef]
- Karttunen, S.; Duffield, M.; Scrimgeour, N.R.; Squires, L.; Lim, W.L.; Dallas, M.L.; Scragg, J.L.; Chicher, J.; Dave, K.A.; Whitelaw, M.L.; et al. Oxygen-Dependent Hydroxylation by Factor Inhibiting HIF (FIH) Regulates the TRPV3 Ion Channel. J. Cell Sci. 2015, 128, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.-F.; Qian, C.; Zhao, N.; Dong, Q.; Li, J.; Wang, B.-B.; Chen, L.; Yu, L.; Han, B.; Du, Y.-M.; et al. Activation of Transient Receptor Potential Vanilloid 4 Involves in Hypoxia/Reoxygenation Injury in Cardiomyocytes. Cell Death Dis. 2017, 8, e2828. [Google Scholar] [CrossRef] [PubMed]
- Butenko, O.; Dzamba, D.; Benesova, J.; Honsa, P.; Benfenati, V.; Rusnakova, V.; Ferroni, S.; Anderova, M. The Increased Activity of TRPV4 Channel in the Astrocytes of the Adult Rat Hippocampus after Cerebral Hypoxia/Ischemia. PLoS ONE 2012, 7, e39959. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.-Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer Cells Co-Opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018, 33, 985–1003.e7. [Google Scholar] [CrossRef]
- Hamilton, N.B.; Kolodziejczyk, K.; Kougioumtzidou, E.; Attwell, D. Proton-Gated Ca2+-Permeable TRP Channels Damage Myelin in Conditions Mimicking Ischaemia. Nature 2016, 529, 523–527. [Google Scholar] [CrossRef]
- Wang, B.; Li, W.; Meng, X.; Zou, F. Hypoxia Up-Regulates Vascular Endothelial Growth Factor in U-87 MG Cells: Involvement of TRPC1. Neurosci. Lett. 2009, 459, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Milevskiy, M.J.G.; Kaemmerer, E.; Turner, D.; Yapa, K.T.D.S.; Brown, M.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. TRPC1 Is a Differential Regulator of Hypoxia-Mediated Events and Akt Signalling in PTEN-Deficient Breast Cancer Cells. J. Cell Sci. 2017, 130, 2292–2305. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pan, Q.; Meng, H.; Jiang, Y.; Mao, A.; Wang, T.; Hua, D.; Yao, X.; Jin, J.; Ma, X. Enhancement of Vascular Endothelial Growth Factor Release in Long-Term Drug-Treated Breast Cancer via Transient Receptor Potential Channel 5-Ca2+-Hypoxia-Inducible Factor 1α Pathway. Pharmacol. Res. 2015, 93, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhu, Y.; Dong, Y.; Zhang, P.; Han, X.; Jin, J.; Ma, X. Overexpression of TrpC5 Promotes Tumour Metastasis via the HIF-1α–Twist Signaling Pathway in Colon Cancer. Clin. Sci. 2017, 131, 2439–2450. [Google Scholar] [CrossRef]
- Nielsen, N.; Kondratska, K.; Ruck, T.; Hild, B.; Kovalenko, I.; Schimmelpfennig, S.; Welzig, J.; Sargin, S.; Lindemann, O.; Christian, S.; et al. TRPC6 Channels Modulate the Response of Pancreatic Stellate Cells to Hypoxia. Pflug. Arch. Eur. J. Physiol. 2017, 469, 1567–1577. [Google Scholar] [CrossRef]
- Iyer, S.C.; Kannan, A.; Gopal, A.; Devaraj, N.; Halagowder, D. Receptor Channel TRPC6 Orchestrate the Activation of Human Hepatic Stellate Cell under Hypoxia Condition. Exp. Cell Res. 2015, 336, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Liang, C.; Chen, E.; Chen, W.; Liang, F.; Zhi, X.; Wei, T.; Xue, F.; Li, G.; Yang, Q.; et al. Regulation of Multi-Drug Resistance in Hepatocellular Carcinoma Cells Is TRPC6/Calcium Dependent. Sci. Rep. 2016, 6, 23269. [Google Scholar] [CrossRef] [PubMed]
- Chigurupati, S.; Venkataraman, R.; Barrera, D.; Naganathan, A.; Madan, M.; Paul, L.; Pattisapu, J.V.; Kyriazis, G.A.; Sugaya, K.; Bushnev, S.; et al. Receptor Channel TRPC6 Is a Key Mediator of Notch-Driven Glioblastoma Growth and Invasiveness. Cancer Res. 2010, 70, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, J.; Jiang, H.; Zhang, Q.; Yang, H.; Xu, X.; Zhang, C.; Xu, C.; Wang, J.; Lu, W. Nicotine Enhances Store-operated Calcium Entry by Upregulating HIF-1α and SOCC Components in Non-small Cell Lung Cancer Cells. Oncol. Rep. 2018. [Google Scholar] [CrossRef]
- Liu, X.; Wang, T.; Wang, Y.; Chen, Z.; Hua, D.; Yao, X.; Ma, X.; Zhang, P. Orai1 Is Critical for Notch-Driven Aggressiveness under Hypoxic Conditions in Triple-Negative Breast Cancers. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 975–986. [Google Scholar] [CrossRef]
- Liu, X.; Wan, X.; Kan, H.; Wang, Y.; Yu, F.; Feng, L.; Jin, J.; Zhang, P.; Ma, X. Hypoxia-Induced Upregulation of Orai1 Drives Colon Cancer Invasiveness and Angiogenesis. Eur. J. Pharmacol. 2018, 832, 1–10. [Google Scholar] [CrossRef]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef]
- Parpaite, T.; Coste, B. Piezo Channels. Curr. Biol. 2017, 27, R250–R252. [Google Scholar] [CrossRef]
- De Felice, D.; Alaimo, A. Mechanosensitive Piezo Channels in Cancer: Focus on Altered Calcium Signaling in Cancer Cells and in Tumour Progression. Cancers 2020, 12, 1780. [Google Scholar] [CrossRef]
- Pardo-Pastor, C.; Rubio-Moscardo, F.; Vogel-González, M.; Serra, S.A.; Afthinos, A.; Mrkonjic, S.; Destaing, O.; Abenza, J.F.; Fernández-Fernández, J.M.; Trepat, X.; et al. Piezo2 Channel Regulates RhoA and Actin Cytoskeleton to Promote Cell Mechanobiological Responses. Proc. Natl. Acad. Sci. USA 2018, 115, 1925–1930. [Google Scholar] [CrossRef]
- Chen, X.; Wanggou, S.; Bodalia, A.; Zhu, M.; Dong, W.; Fan, J.J.; Yin, W.C.; Min, H.-K.; Hu, M.; Draghici, D.; et al. A Feedforward Mechanism Mediated by Mechanosensitive Ion Channel PIEZO1 and Tissue Mechanics Promotes Glioma Aggression. Neuron 2018, 100, 799–815.e7. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, C.; Zhou, R.-M.; Yao, J.; Li, X.-M.; Shen, Y.; Cheng, H.; Yuan, J.; Yan, B.; Jiang, Q. Piezo2 Protein: A Novel Regulator of Tumour Angiogenesis and Hyperpermeability. Oncotarget 2016, 7, 44630–44643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, Y.; Huang, T.; Wu, F.; Liu, L.; Kwan, J.S.H.; Cheng, A.S.L.; Yu, J.; To, K.F.; Kang, W. PIEZO1 Functions as a Potential Oncogene by Promoting Cell Proliferation and Migration in Gastric Carcinogenesis. Mol. Carcinog. 2018, 57, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Dombroski, J.A.; Hope, J.M.; Sarna, N.S.; King, M.R. Channeling the Force: Piezo1 Mechanotransduction in Cancer Metastasis. Cells 2021, 10, 2815. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Pedersen, S.F.; Schwab, A.; Stock, C. Intracellular PH Gradients in Migrating Cells. Am. J. Physiol. Cell Physiol. 2011, 300, C490–C495. [Google Scholar] [CrossRef]
- Hertz, L.; Huisjes, R.; Llaudet-Planas, E.; Petkova-Kirova, P.; Makhro, A.; Danielczok, J.G.; Egee, S.; del Mar Mañú-Pereira, M.; van Wijk, R.; Vives Corrons, J.-L.; et al. Is Increased Intracellular Calcium in Red Blood Cells a Common Component in the Molecular Mechanism Causing Anemia? Front. Physiol. 2017, 8, 673. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A. Therapeutic Potential of KCa3.1 Blockers: Recent Advances and Promising Trends. Expert Rev. Clin. Pharmacol. 2010, 3, 385–396. [Google Scholar] [CrossRef]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-Ribose and Hydrogen Peroxide Synergize with ADP-Ribose in the Activation of TRPM2 Channels. Mol. Cell 2005, 18, 61–69. [Google Scholar] [CrossRef]
- McHugh, D.; Flemming, R.; Xu, S.-Z.; Perraud, A.-L.; Beech, D.J. Critical Intracellular Ca2+ Dependence of Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel Activation. J. Biol. Chem. 2003, 278, 11002–11006. [Google Scholar] [CrossRef]
- Perraud, A.-L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of Free ADP-Ribose from Mitochondria Mediates Oxidative Stress-Induced Gating of TRPM2 Cation Channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [PubMed]
- Turlova, E.; Feng, Z.; Sun, H. The Role of TRPM2 Channels in Neurons, Glial Cells and the Blood-Brain Barrier in Cerebral Ischemia and Hypoxia. Acta Pharmacol. Sin. 2018, 39, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.; Mankoo, H.; Wei, L.; An, X.; Li, C.; Li, D.; Jiang, L. TRPM2 Channel: A Novel Target for Alleviating Ischaemia-reperfusion, Chronic Cerebral Hypo-perfusion and Neonatal Hypoxic-ischaemic Brain Damage. J. Cell. Mol. Med. 2020, 24, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef]
- Zhao, L.-Y.; Xu, W.-L.; Xu, Z.-Q.; Qi, C.; Li, Y.; Cheng, J.; Liu, L.-K.; Wu, Y.-N.; Gao, J.; Ye, J.-H. The Overexpressed Functional Transient Receptor Potential Channel TRPM2 in Oral Squamous Cell Carcinoma. Sci. Rep. 2016, 6, 38471. [Google Scholar] [CrossRef]
- Zeng, X.; Sikka, S.C.; Huang, L.; Sun, C.; Xu, C.; Jia, D.; Abdel-Mageed, A.B.; Pottle, J.E.; Taylor, J.T.; Li, M. Novel Role for the Transient Receptor Potential Channel TRPM2 in Prostate Cancer Cell Proliferation. Prostate Cancer Prostatic Dis. 2010, 13, 195–201. [Google Scholar] [CrossRef]
- Hopkins, M.M.; Feng, X.; Liu, M.; Parker, L.P.; Koh, D.W. Inhibition of the Transient Receptor Potential Melastatin-2 Channel Causes Increased DNA Damage and Decreased Proliferation in Breast Adenocarcinoma Cells. Int. J. Oncol. 2015, 46, 2267–2276. [Google Scholar] [CrossRef]
- Almasi, S.; Sterea, A.M.; Fernando, W.; Clements, D.R.; Marcato, P.; Hoskin, D.W.; Gujar, S.; El Hiani, Y. TRPM2 Ion Channel Promotes Gastric Cancer Migration, Invasion and Tumour Growth through the AKT Signaling Pathway. Sci. Rep. 2019, 9, 4182. [Google Scholar] [CrossRef]
- Almasi, S.; Kennedy, B.E.; El-Aghil, M.; Sterea, A.M.; Gujar, S.; Partida-Sánchez, S.; El Hiani, Y. TRPM2 Channel–Mediated Regulation of Autophagy Maintains Mitochondrial Function and Promotes Gastric Cancer Cell Survival via the JNK-Signaling Pathway. J. Biol. Chem. 2018, 293, 3637–3650. [Google Scholar] [CrossRef]
- Guler, Y.; Ovey, I.S. Synergic and Comparative Effect of 5-Fluorouracil and Leucoverin on Breast and Colon Cancer Cells through TRPM2 Channels. Bratisl. Lek. Listy 2018, 119, 692–700. [Google Scholar] [CrossRef]
- Lange, I.; Yamamoto, S.; Partida-Sanchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 Functions as a Lysosomal Ca2+-Release Channel in Cells. Sci. Signal. 2009, 2, ra23. [Google Scholar] [CrossRef] [PubMed]
- Hantute-Ghesquier, A.; Haustrate, A.; Prevarskaya, N.; Lehen’kyi, V. TRPM Family Channels in Cancer. Pharmaceuticals 2018, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, F.; Zhao, Y.; Wang, J.; Pei, L.; Sun, N.; Shi, J. Hypoxia Induces an Increase in Intracellular Magnesium via Transient Receptor Potential Melastatin 7 (TRPM7) Channels in Rat Hippocampal Neurons In Vitro. J. Biol. Chem. 2011, 286, 20194–20207. [Google Scholar] [CrossRef]
- Mori, Y.; Takahashi, N.; Polat, O.K.; Kurokawa, T.; Takeda, N.; Inoue, M. Redox-Sensitive Transient Receptor Potential Channels in Oxygen Sensing and Adaptation. Pflug. Arch. Eur. J. Physiol. 2016, 468, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired Nociception and Pain Sensation in Mice Lacking the Capsaicin Receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Reeh, P.W.; Fischer, M.J.M. Nobel Somatosensations and Pain. Pflug. Arch. Eur. J. Physiol. 2022, 474, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Storozhuk, M.V.; Moroz, O.F.; Zholos, A.V. Multifunctional TRPV1 Ion Channels in Physiology and Pathology with Focus on the Brain, Vasculature, and Some Visceral Systems. BioMed Res. Int. 2019, 2019, 5806321. [Google Scholar] [CrossRef]
- Hellwig, N.; Plant, T.D.; Janson, W.; Schäfer, M.; Schultz, G.; Schaefer, M. TRPV1 Acts as Proton Channel to Induce Acidification in Nociceptive Neurons. J. Biol. Chem. 2004, 279, 34553–34561. [Google Scholar] [CrossRef]
- Li, L.; Chen, C.; Chiang, C.; Xiao, T.; Chen, Y.; Zhao, Y.; Zheng, D. The Impact of TRPV1 on Cancer Pathogenesis and Therapy: A Systematic Review. Int. J. Biol. Sci. 2021, 17, 2034–2049. [Google Scholar] [CrossRef]
- Siveen, K.S.; Nizamuddin, P.B.; Uddin, S.; Al-Thani, M.; Frenneaux, M.P.; Janahi, I.A.; Steinhoff, M.; Azizi, F. TRPV2: A Cancer Biomarker and Potential Therapeutic Target. Dis. Markers 2020, 2020, 8892312. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Q.; Fan, K.; Li, B.; Li, H.; Qi, H.; Guo, J.; Cao, Y.; Sun, H. Overexpression of TRPV3 Correlates with Tumour Progression in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2016, 17, 437. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, P.; Lu, Y.; Wang, J.; Jeon, J.; Wang, Q.; Tian, J.-B.; Zang, B.; Yu, Y.; Zhu, M.X. Mechanisms of Proton Inhibition and Sensitization of the Cation Channel TRPV3. J. Gen. Physiol. 2021, 153, e202012663. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Choong, L.; Jin, T.; Mon, N.; Chong, S.; Liew, C.; Putti, T.; Lu, S.; Harteneck, C.; Lim, Y. TRPV4 Plays a Role in Breast Cancer Cell Migration via Ca2+-Dependent Activation of AKT and Downregulation of E-Cadherin Cell Cortex Protein. Oncogenesis 2017, 6, e338. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Choong, L.Y.; Mon, N.N.; Lu, S.; Lin, Q.; Pang, B.; Yan, B.; Krishna, V.S.R.; Singh, H.; Tan, T.Z.; et al. TRPV4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Sci. Rep. 2016, 6, 27903. [Google Scholar] [CrossRef]
- Fiorio Pla, A.; Ong, H.L.; Cheng, K.T.; Brossa, A.; Bussolati, B.; Lockwich, T.; Paria, B.; Munaron, L.; Ambudkar, I.S. TRPV4 Mediates Tumour-Derived Endothelial Cell Migration via Arachidonic Acid-Activated Actin Remodeling. Oncogene 2012, 31, 200–212. [Google Scholar] [CrossRef]
- Vellino, S.; Oddou, C.; Rivier, P.; Boyault, C.; Hiriart-Bryant, E.; Kraut, A.; Martin, R.; Coute, Y.; Knölker, H.-J.; Valverde, M.A.; et al. Crosstalk between the Calcium Channel TRPV4 and Reactive Oxygen Species Interlocks Adhesive and Degradative Functions of Invadosomes. J. Cell Biol. 2021, 220, e201910079. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, B.; Wang, X.; Mao, J.; Li, W.; Sun, Y.; Yuan, Y.; Ben, Q.; Hua, L.; Qian, A. TRPV4 Overexpression Promotes Metastasis through Epithelial–Mesenchymal Transition in Gastric Cancer and Correlates with Poor Prognosis. Oncotargets Ther. 2020, 13, 8383–8394. [Google Scholar] [CrossRef]
- Li, X.; Cheng, Y.; Wang, Z.; Zhou, J.; Jia, Y.; He, X.; Zhao, L.; Dong, Y.; Fan, Y.; Yang, X.; et al. Calcium and TRPV4 Promote Metastasis by Regulating Cytoskeleton through the RhoA/ROCK1 Pathway in Endometrial Cancer. Cell Death Dis. 2020, 11, 1009. [Google Scholar] [CrossRef]
- Thoppil, R.J.; Cappelli, H.C.; Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Thodeti, C.K. TRPV4 Channels Regulate Tumour Angiogenesis via Modulation of Rho/Rho Kinase Pathway. Oncotarget 2016, 7, 25849–25861. [Google Scholar] [CrossRef]
- Azimi, I.; Robitaille, M.; Armitage, K.; So, C.L.; Milevskiy, M.J.G.; Northwood, K.; Lim, H.F.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Activation of the Ion Channel TRPV4 Induces Epithelial to Mesenchymal Transition in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9417. [Google Scholar] [CrossRef]
- Peters, A.A.; Jamaludin, S.Y.N.; Yapa, K.T.D.S.; Chalmers, S.; Wiegmans, A.P.; Lim, H.F.; Milevskiy, M.J.G.; Azimi, I.; Davis, F.M.; Northwood, K.S.; et al. Oncosis and Apoptosis Induction by Activation of an Overexpressed Ion Channel in Breast Cancer Cells. Oncogene 2017, 36, 6490–6500. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Guo, M.; Lv, X.; Wang, Z.; Yang, J.; Li, Y.; Yu, F.; Wen, X.; Feng, L.; Zhou, T. Role of Transient Receptor Potential Vanilloid 4 in Vascular Function. Front. Mol. Biosci. 2021, 8, 677661. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.M. TRPV6 as A Target for Cancer Therapy. J. Cancer 2020, 11, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Lehen’kyi, V.; Flourakis, M.; Skryma, R.; Prevarskaya, N. TRPV6 Channel Controls Prostate Cancer Cell Proliferation via Ca2+/NFAT-Dependent Pathways. Oncogene 2007, 26, 7380–7385. [Google Scholar] [CrossRef] [PubMed]
- Raphaël, M.; Lehen’kyi, V.; Vandenberghe, M.; Beck, B.; Khalimonchyk, S.; Vanden Abeele, F.; Farsetti, L.; Germain, E.; Bokhobza, A.; Mihalache, A.; et al. TRPV6 Calcium Channel Translocates to the Plasma Membrane via Orai1-Mediated Mechanism and Controls Cancer Cell Survival. Proc. Natl. Acad. Sci. USA 2014, 111, E3870–E3879. [Google Scholar] [CrossRef] [PubMed]
- Kärki, T.; Rajakylä, E.K.; Acheva, A.; Tojkander, S. TRPV6 Calcium Channel Directs Homeostasis of the Mammary Epithelial Sheets and Controls Epithelial Mesenchymal Transition. Sci. Rep. 2020, 10, 14683. [Google Scholar] [CrossRef] [PubMed]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High Expression of Transient Receptor Potential Channels in Human Breast Cancer Epithelial Cells and Tissues: Correlation with Pathological Parameters. Cell. Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [CrossRef]
- Song, H.; Dong, M.; Zhou, J.; Sheng, W.; Li, X.; Gao, W. Expression and Prognostic Significance of TRPV6 in the Development and Progression of Pancreatic Cancer. Oncol. Rep. 2018, 39, 1432–1440. [Google Scholar] [CrossRef]
- Xue, H.; Wang, Y.; MacCormack, T.J.; Lutes, T.; Rice, C.; Davey, M.; Dugourd, D.; Ilenchuk, T.T.; Stewart, J.M. Inhibition of Transient Receptor Potential Vanilloid 6 Channel, Elevated in Human Ovarian Cancers, Reduces Tumour Growth in a Xenograft Model. J. Cancer 2018, 9, 3196–3207. [Google Scholar] [CrossRef]
- Sutoo, S.; Maeda, T.; Suzuki, A.; Kato, Y. Adaptation to Chronic Acidic Extracellular PH Elicits a Sustained Increase in Lung Cancer Cell Invasion and Metastasis. Clin. Exp. Metastasis 2020, 37, 133–144. [Google Scholar] [CrossRef]
- Peleg, S.; Sellin, J.H.; Wang, Y.; Freeman, M.R.; Umar, S. Suppression of Aberrant Transient Receptor Potential Cation Channel, Subfamily V, Member 6 Expression in Hyperproliferative Colonic Crypts by Dietary Calcium. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G593–G601. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; An, B.-S.; Choi, K.-C.; Jeung, E.-B. Change of Genes in Calcium Transport Channels Caused by Hypoxic Stress in the Placenta, Duodenum, and Kidney of Pregnant Rats1. Biol. Reprod. 2013, 88, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.R.; Chun, J.N.; So, I.; Kim, H.J.; Baek, S.; Jeon, J.-H.; Shin, S.-Y. Data-Driven Analysis of TRP Channels in Cancer: Linking Variation in Gene Expression to Clinical Significance. Cancer Genom. Proteom. 2016, 13, 83–90. [Google Scholar]
- Takahashi, N.; Kuwaki, T.; Kiyonaka, S.; Numata, T.; Kozai, D.; Mizuno, Y.; Yamamoto, S.; Naito, S.; Knevels, E.; Carmeliet, P.; et al. TRPA1 Underlies a Sensing Mechanism for O2. Nat. Chem. Biol. 2011, 7, 701–711. [Google Scholar] [CrossRef]
- Berrout, J.; Kyriakopoulou, E.; Moparthi, L.; Hogea, A.S.; Berrout, L.; Ivan, C.; Lorger, M.; Boyle, J.; Peers, C.; Muench, S.; et al. TRPA1–FGFR2 Binding Event Is a Regulatory Oncogenic Driver Modulated by MiRNA-142-3p. Nat. Commun. 2017, 8, 947. [Google Scholar] [CrossRef]
- Derouiche, S.; Mariot, P.; Warnier, M.; Vancauwenberghe, E.; Bidaux, G.; Gosset, P.; Mauroy, B.; Bonnal, J.-L.; Slomianny, C.; Delcourt, P.; et al. Activation of TRPA1 Channel by Antibacterial Agent Triclosan Induces VEGF Secretion in Human Prostate Cancer Stromal Cells. Cancer Prev. Res. 2017, 10, 177–187. [Google Scholar] [CrossRef]
- Roberts, E.; Cossigny, D.A.F.; Quan, G.M.Y. The Role of Vascular Endothelial Growth Factor in Metastatic Prostate Cancer to the Skeleton. Prostate Cancer 2013, 2013, 418340. [Google Scholar] [CrossRef]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient Receptor Potential Channel Expression Signatures in Tumour-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers 2019, 11, 956. [Google Scholar] [CrossRef]
- Samanta, A.; Hughes, T.E.T.; Moiseenkova-Bell, V.Y. Transient Receptor Potential (TRP) Channels. In Membrane Protein Complexes: Structure and Function; Harris, J.R., Boekema, E.J., Eds.; Springer: Singapore, 2018; Volume 87, pp. 141–165. ISBN 978-981-10-7756-2. [Google Scholar]
- Dong, H.; Shim, K.-N.; Li, J.M.J.; Estrema, C.; Ornelas, T.A.; Nguyen, F.; Liu, S.; Ramamoorthy, S.L.; Ho, S.; Carethers, J.M.; et al. Molecular Mechanisms Underlying Ca2+-Mediated Motility of Human Pancreatic Duct Cells. Cell Physiol. 2010, 299, 11. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Geerts, D.; Ay, A.-S.; Potier-Cartereau, M.; Ahidouch, A.; Ouadid-Ahidouch, H. Functional Cooperation between KCa3.1 and TRPC1 Channels in Human Breast Cancer: Role in Cell Proliferation and Patient Prognosis. Oncotarget 2016, 7, 36419–36435. [Google Scholar] [CrossRef]
- El Hiani, Y.; Ahidouch, A.; Lehen’kyi, V.; Hague, F.; Gouilleux, F.; Mentaverri, R.; Kamel, S.; Lassoued, K.; Brûlé, G.; Ouadid-Ahidouch, H. Extracellular Signal-Regulated Kinases 1 and 2 and TRPC1 Channels Are Required for Calcium-Sensing Receptor-Stimulated MCF-7 Breast Cancer Cell Proliferation. Cell. Physiol. Biochem. 2009, 23, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Bomben, V.C.; Sontheimer, H. Disruption of Transient Receptor Potential Canonical Channel 1 Causes Incomplete Cytokinesis and Slows the Growth of Human Malignant Gliomas: TRPC1 Channels Are Essential for Glioma Proliferation. Glia 2010, 58, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-N.; Zeng, B.; Zhang, Y.; Daskoulidou, N.; Fan, H.; Qu, J.-M.; Xu, S.-Z. Involvement of TRPC Channels in Lung Cancer Cell Differentiation and the Correlation Analysis in Human Non-Small Cell Lung Cancer. PLoS ONE 2013, 8, e67637. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.A.; Azimi, I.; Marcial, D.; Peters, A.A.; Chalmers, S.B.; Yapa, K.T.D.S.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Differential Engagement of ORAI1 and TRPC1 in the Induction of Vimentin Expression by Different Stimuli. Lab. Investig. 2020, 100, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a Key Mediator of Angiogenesis in Cancer. Oncology 2005, 69, 4–10. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, X.; Li, H.; Chen, Z.; Yao, X.; Jin, J.; Ma, X. TRPC5-Induced Autophagy Promotes Drug Resistance in Breast Carcinoma via CaMKKβ/AMPKα/MTOR Pathway. Sci. Rep. 2017, 7, 3158. [Google Scholar] [CrossRef]
- Ma, X.; Cai, Y.; He, D.; Zou, C.; Zhang, P.; Lo, C.Y.; Xu, Z.; Chan, F.L.; Yu, S.; Chen, Y.; et al. Transient Receptor Potential Channel TRPC5 Is Essential for P-Glycoprotein Induction in Drug-Resistant Cancer Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16282–16287. [Google Scholar] [CrossRef]
- Wang, T.; Chen, Z.; Zhu, Y.; Pan, Q.; Liu, Y.; Qi, X.; Jin, L.; Jin, J.; Ma, X.; Hua, D. Inhibition of Transient Receptor Potential Channel 5 Reverses 5-Fluorouracil Resistance in Human Colorectal Cancer Cells. J. Biol. Chem. 2015, 290, 448–456. [Google Scholar] [CrossRef]
- Wang, T.; Ning, K.; Sun, X.; Zhang, C.; Jin, L.; Hua, D. Glycolysis Is Essential for Chemoresistance Induced by Transient Receptor Potential Channel C5 in Colorectal Cancer. BMC Cancer 2018, 18, 207. [Google Scholar] [CrossRef]
- Plant, T.D.; Schaefer, M. TRPC4 and TRPC5: Receptor-Operated Ca2+-Permeable Nonselective Cation Channels. Cell Calcium 2003, 33, 441–450. [Google Scholar] [CrossRef]
- Veliceasa, D.; Ivanovic, M.; Hoepfner, F.T.-S.; Thumbikat, P.; Volpert, O.V.; Smith, N.D. Transient Potential Receptor Channel 4 Controls Thrombospondin-1 Secretion and Angiogenesis in Renal Cell Carcinoma: Thrombospondin-1 Loss in Renal Cancer. FEBS J. 2007, 274, 6365–6377. [Google Scholar] [CrossRef] [PubMed]
- Song, H.B.; Jun, H.-O.; Kim, J.H.; Fruttiger, M.; Kim, J.H. Suppression of Transient Receptor Potential Canonical Channel 4 Inhibits Vascular Endothelial Growth Factor-Induced Retinal Neovascularization. Cell Calcium 2015, 57, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Yuan, C.; Yang, X.; Atkin, S.L.; Xu, S.-Z. TRPC Channels and Their Splice Variants Are Essential for Promoting Human Ovarian Cancer Cell Proliferation and Tumourigenesis. Curr. Cancer Drug Targets 2013, 13, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.; Raman, P.; Tullai, J.; Xu, L.; Henault, M.; Thomas, E.; Yeola, S.; Lao, J.; McPate, M.; Verkuyl, J.M.; et al. Englerin A Agonizes the TRPC4/C5 Cation Channels to Inhibit Tumour Cell Line Proliferation. PLoS ONE 2015, 10, e0127498. [Google Scholar] [CrossRef]
- Lindemann, O.; Umlauf, D.; Frank, S.; Schimmelpfennig, S.; Bertrand, J.; Pap, T.; Hanley, P.J.; Fabian, A.; Dietrich, A.; Schwab, A. TRPC6 Regulates CXCR2-Mediated Chemotaxis of Murine Neutrophils. J. Immunol. 2013, 190, 5496–5505. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumour-Associated Neutrophils in Cancer: Going Pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef]
- Prakriya, M. Store-Operated Orai Channels. In Current Topics in Membranes; Elsevier: Amsterdam, The Netherlands, 2013; Volume 71, pp. 1–32. ISBN 978-0-12-407870-3. [Google Scholar]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 Mediate SOCE and Contribute to Apoptotic Resistance of Pancreatic Adenocarcinoma. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 2263–2269. [Google Scholar] [CrossRef]
- Didiasova, M.; Zakrzewicz, D.; Magdolen, V.; Nagaraj, C.; Bálint, Z.; Rohde, M.; Preissner, K.T.; Wygrecka, M. STIM1/ORAI1-Mediated Ca2+ Influx Regulates Enolase-1 Exteriorization. J. Biol. Chem. 2015, 290, 11983–11999. [Google Scholar] [CrossRef]
- Huang, H.-K.; Lin, Y.-H.; Chang, H.-A.; Lai, Y.-S.; Chen, Y.-C.; Huang, S.-C.; Chou, C.-Y.; Chiu, W.-T. Chemoresistant Ovarian Cancer Enhances Its Migration Abilities by Increasing Store-Operated Ca2+ Entry-Mediated Turnover of Focal Adhesions. J Biomed. Sci. 2020, 27, 36. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai Mediate Constitutive Ca2+ Entry and Control Endoplasmic Reticulum Ca2+ Refilling in Primary Cultures of Colorectal Carcinoma Cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef]
- Singh, A.K. Orai-1 and Orai-2 Regulate Oral Cancer Cell Migration and Colonisation by Suppressing Akt/MTOR/NF-κB Signalling. Life Sci. 2020, 261, 118372. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.-C.; Kim, M.; et al. STIM1- and Orai1-Mediated Ca2+ Oscillation Orchestrates Invadopodium Formation and Melanoma Invasion. J. Cell Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, S.; Niu, H.; Ye, Y.; Hu, F.; Chen, S.; Li, X.; Luo, X.; Jiang, S.; Liu, Y.; et al. STIM1 Accelerates Cell Senescence in a Remodeled Microenvironment but Enhances the Epithelial-to-Mesenchymal Transition in Prostate Cancer. Sci. Rep. 2015, 5, 11754. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Elevated Orai1 and STIM1 Expressions Upregulate MACC1 Expression to Promote Tumour Cell Proliferation, Metabolism, Migration, and Invasion in Human Gastric Cancer. Cancer Lett. 2016, 381, 31–40. [Google Scholar] [CrossRef]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 Calcium Channel and Resistance to Chemotherapy in Breast Cancer Cells: The P53 Connection. Cell Death Differ. 2018, 25, 693–707. [Google Scholar] [CrossRef]
- Tang, B.-D.; Xia, X.; Lv, X.-F.; Yu, B.-X.; Yuan, J.-N.; Mai, X.-Y.; Shang, J.-Y.; Zhou, J.-G.; Liang, S.-J.; Pang, R.-P. Inhibition of Orai1-Mediated Ca2+ Entry Enhances Chemosensitivity of HepG2 Hepatocarcinoma Cells to 5-Fluorouracil. J. Cell. Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef]
- Taylor, J.; Azimi, I.; Monteith, G.; Bebawy, M. Ca2+ Mediates Extracellular Vesicle Biogenesis through Alternate Pathways in Malignancy. J. Extracell. Vesicles 2020, 9, 1734326. [Google Scholar] [CrossRef]
- Gavriliouk, D.; Scrimgeour, N.R.; Grigoryev, S.; Ma, L.; Zhou, F.H.; Barritt, G.J.; Rychkov, G.Y. Regulation of Orai1/STIM1 Mediated ICRAC by Intracellular PH. Sci. Rep. 2017, 7, 9829. [Google Scholar] [CrossRef]
- Li, S.; Hao, B.; Lu, Y.; Yu, P.; Lee, H.-C.; Yue, J. Intracellular Alkalinization Induces Cytosolic Ca2+ Increases by Inhibiting Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA). PLoS ONE 2012, 7, e31905. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels and the Hallmarks of Cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumour Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Bogeski, I.; Kummerow, C.; Al-Ansary, D.; Schwarz, E.C.; Koehler, R.; Kozai, D.; Takahashi, N.; Peinelt, C.; Griesemer, D.; Bozem, M.; et al. Differential Redox Regulation of ORAI Ion Channels: A Mechanism to Tune Cellular Calcium Signaling. Sci. Signal. 2010, 3, ra24. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of Channel-Forming ORAI Proteins Determines an Oncogenic Switch in Prostate Cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Pan, H.; Yao, J.; Zhou, Y.; Han, W. SOCE and Cancer: Recent Progress and New Perspectives. Int. J. Cancer 2016, 138, 2067–2077. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Audero, M.M.; Prevarskaya, N.; Fiorio Pla, A. Ca2+ Signalling and Hypoxia/Acidic Tumour Microenvironment Interplay in Tumour Progression. Int. J. Mol. Sci. 2022, 23, 7377. https://doi.org/10.3390/ijms23137377
Audero MM, Prevarskaya N, Fiorio Pla A. Ca2+ Signalling and Hypoxia/Acidic Tumour Microenvironment Interplay in Tumour Progression. International Journal of Molecular Sciences. 2022; 23(13):7377. https://doi.org/10.3390/ijms23137377
Chicago/Turabian StyleAudero, Madelaine Magalì, Natalia Prevarskaya, and Alessandra Fiorio Pla. 2022. "Ca2+ Signalling and Hypoxia/Acidic Tumour Microenvironment Interplay in Tumour Progression" International Journal of Molecular Sciences 23, no. 13: 7377. https://doi.org/10.3390/ijms23137377
APA StyleAudero, M. M., Prevarskaya, N., & Fiorio Pla, A. (2022). Ca2+ Signalling and Hypoxia/Acidic Tumour Microenvironment Interplay in Tumour Progression. International Journal of Molecular Sciences, 23(13), 7377. https://doi.org/10.3390/ijms23137377