
The Possible Role of Neural Cell Apoptosis in Multiple Sclerosis


{kind=link}
{kind=link}
Abstract
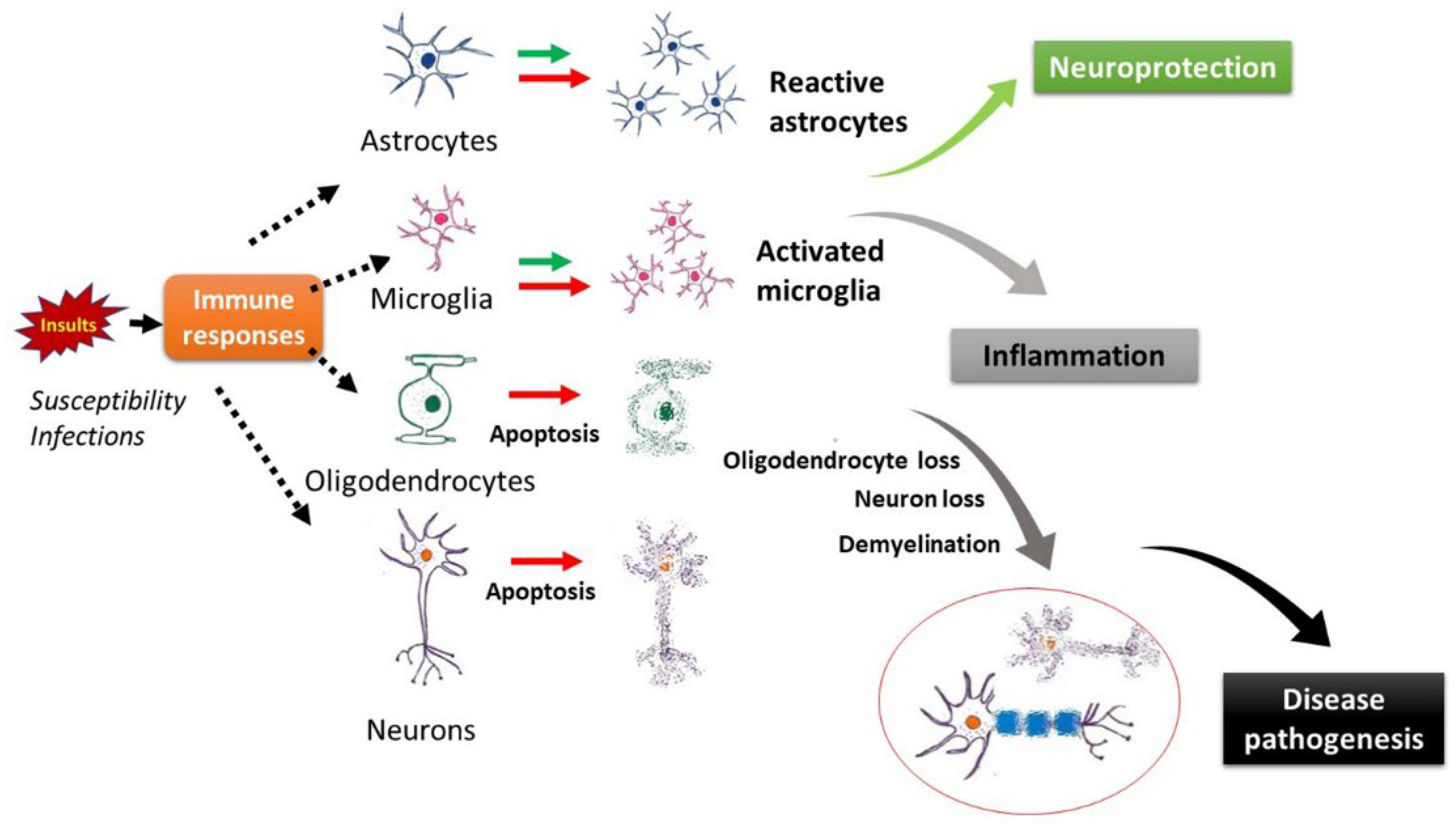
:1. Introduction
2. In MS, Demyelination and Neuron Loss May Possibly Occur Simultaneously
3. Apoptosis/Programmed Cell Death
4. Oligodendrocyte Apoptosis and Demyelination
4.1. Myelin-Directed Autoimmunity Triggers Oligodendrocyte Apoptosis in Animal Models of MS
4.2. Theiler’s Murine Encephalomyelitis Virus-Induced Demyelinating Disease and Other Virus-Induced Animal Models
4.3. Oligodendrocyte Apoptosis in Patients with MS
5. Apoptosis in Neurons in Demyelinating Diseases
5.1. In MS, Neuronal Death Is Prominent and Correlates with Disease Progression
5.2. Neuron Apoptosis in MS
5.3. Other Clinical Evidence for the Possible Role of Neuronal Damage in MS
6. Neuronal Apoptosis and Microglia
7. Implications of Apoptosis in Demyelination and MS Disease Pathogenesis
7.1. Possible Roles of Apoptosis in Demyelination and Disease Activities
7.2. Failure of Remyelination
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patsopoulos, N.; Barcellos, L.F.; Hintzen, R.Q.; Schaefer, C.; Van Duijn, C.M.; Noble, J.A.; Raj, T.; Gourraud, P.-A.; Stranger, B.; Oksenberg, J.; et al. Fine-Mapping the Genetic Association of the Major Histocompatibility Complex in Multiple Sclerosis: HLA and Non-HLA Effects. PLoS Genet. 2013, 9, e1003926. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.-S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.-S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis: Where do we stand? Continuum 2013, 19, 901–921. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal Transection in the Lesions of Multiple Sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, L.; Velmeshev, D.; Holmqvist, S.; Kaufmann, M.; Werneburg, S.; Jung, D.; Vistnes, S.; Stockley, J.H.; Young, A.; Steindel, M.; et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019, 573, 75–82. [Google Scholar] [CrossRef]
- Ferguson, B.; Matyszak, M.K.; Esiri, M.M.; Perry, V.H. Axonal damage in acute multiple sclerosis lesions. Brain 1997, 120, 393–399. [Google Scholar] [CrossRef] [PubMed]
- San Francisco MS-EPIC Team of University of California; Cree, B.A.; Hollenbach, J.A.; Bove, R.; Kirkish, G.; Sacco, S.; Caverzasi, E.; Bischof, A.; Gundel, T.; Zhu, A.H.; et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann. Neurol. 2019, 85, 653–666. [Google Scholar] [PubMed]
- Stassart, R.M.; Möbius, W.; Nave, K.-A.; Edgar, J.M. The Axon-Myelin Unit in Development and Degenerative Disease. Front. Neurosci. 2018, 12, 467. [Google Scholar] [CrossRef] [Green Version]
- Trapp, B.D.; Vignos, M.; Dudman, J.; Chang, A.; Fisher, E.; Staugaitis, S.M.; Battapady, H.; Mork, S.; Ontaneda, D.; E Jones, S.; et al. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: A retrospective study. Lancet Neurol. 2018, 17, 870–884. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, M.; Atzori, M.; Bernardi, V.; Morra, A.; Romualdi, C.; Rinaldi, L.; McAuliffe, M.J.M.; Barachino, L.; Perini, P.; Fischl, B.; et al. Cortical atrophy is relevant in multiple sclerosis at clinical onset. J. Neurol. 2007, 254, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Bozzali, M.; Rovaris, M.; Gonen, O.; Kesavadas, C.; Ghezzi, A.; Martinelli, V.; Grossman, R.I.; Scotti, G.; Comi, G.; et al. Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis. Brain 2003, 126, 433–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapeller, P.; McLean, M.A.; Griffin, C.M.; Chard, D.; Parker, G.J.; Barker, G.J.; Thompson, A.J.; Miller, D.H. Preliminary evidence for neuronal damage in cortical grey matter and normal appearing white matter in short duration relapsing-remitting multiple sclerosis: A quantitative MR spectroscopic imaging study. J. Neurol. 2001, 248, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Kurnellas, M.; Donahue, K.; Elkabes, S. Mechanisms of neuronal damage in multiple sclerosis and its animal models: Role of calcium pumps and exchangers. Biochem. Soc. Trans. 2007, 35, 923–926. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Kennedy, P.G. Viruses, apoptosis, and neuroinflammation—A double-edged sword. J. Neurovirol. 2015, 21, 1–7. [Google Scholar] [CrossRef]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Wyllie, A.H. Apoptosis: An overview. Br. Med Bull. 1997, 53, 451–465. [Google Scholar] [CrossRef]
- A Thornberry, N. The caspase family of cysteine proteases. Br. Med Bull. 1997, 53, 478–490. [Google Scholar] [CrossRef]
- Crowley, L.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of apoptosis and necrosis by annexin V binding, propidium iodide uptake, and flow cytometry. Cold Spring Harbor Protocols. 2016, 2016. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubert, M.; Pomeranz, L.E.; Blaho, J.A. Herpes simplex virus blocks apoptosis by precluding mitochondrial cytochrome c release independent of caspase activation in infected human epithelial cells. Apoptosis 2006, 12, 19–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.-M.; MacFarlane, M.; Zhuang, J.; Wolf, B.B.; Green, D.R.; Cohen, G.M. Distinct Caspase Cascades Are Initiated in Receptor-mediated and Chemical-induced Apoptosis. J. Biol. Chem. 1999, 274, 5053–5060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akassoglou, K.; Bauer, J.; Kassiotis, G.; Pasparakis, M.; Lassmann, H.; Kollias, G.; Probert, L. Oligodendrocyte Apoptosis and Primary Demyelination Induced by Local TNF/p55TNF Receptor Signaling in the Central Nervous System of Transgenic Mice: Models for Multiple Sclerosis with Primary Oligodendrogliopathy. Am. J. Pathol. 1998, 153, 801–813. [Google Scholar] [CrossRef]
- Guire, C.M.; Beyaert, R.; van Loo, G. Death receptor signalling in central nervous system inflammation and demyelination. Trends Neurosci. 2011, 34, 619–628. [Google Scholar] [CrossRef]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of Necroptosis in Multiple Sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef] [Green Version]
- Hisahara, S.; Yuan, J.; Momoi, T.; Okano, H.; Miura, M. Caspase-11 Mediates Oligodendrocyte Cell Death and Pathogenesis of Autoimmune-Mediated Demyelination. J. Exp. Med. 2001, 193, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Hövelmeyer, N.; Hao, Z.; Kranidioti, K.; Kassiotis, G.; Buch, T.; Frommer, F.; Von Hoch, L.; Kramer, D.; Minichiello, L.; Kollias, G.; et al. Apoptosis of Oligodendrocytes via Fas and TNF-R1 Is a Key Event in the Induction of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2005, 175, 5875–5884. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Tao, T.; Yan, W.; Chen, C.S.; Qin, X. Curcumin Inhibits Mitochondrial Injury and Apoptosis from the Early Stage in EAE Mice. Oxidative Med. Cell. Longev. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Caprariello, A.V.; Mangla, S.; Miller, R.H.; Selkirk, S.M. Apoptosis of oligodendrocytes in the central nervous system results in rapid focal demyelination. Ann. Neurol. 2012, 72, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Caprariello, A.V.; Batt, C.E.; Zippe, I.; Romito-DiGiacomo, R.R.; Karl, M.; Miller, R.H. Apoptosis of Oligodendrocytes during Early Development Delays Myelination and Impairs Subsequent Responses to Demyelination. J. Neurosci. 2015, 35, 14031–14041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajoohesh-Ganji, A.; Miller, R.H. Targeted Oligodendrocyte Apoptosis in Optic Nerve Leads to Persistent Demyelination. Neurochem. Res. 2019, 45, 580–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, J.P.; Yauch, R.L.; Lang, S.; Kim, B.S. Potential role of CD4+ T cell-mediated apoptosis of activated astrocytes in Theiler’s virus-induced demyelination. J. Immunol. 1999, 162, 6543–6551. [Google Scholar] [PubMed]
- Weissenböck, H.; Bakonyi, T.; Chvala, S.; Nowotny, N. Experimental Usutu virus infection of suckling mice causes neuronal and glial cell apoptosis and demyelination. Acta Neuropathol. 2004, 108, 453–460. [Google Scholar] [CrossRef]
- Barnett, M.H.; Prineas, J.W. Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesion. Ann. Neurol. 2004, 55, 458–468. [Google Scholar] [CrossRef]
- Henderson, A.P.D.; Barnett, M.H.; Parratt, J.D.E.; Prineas, J.W. Multiple sclerosis: Distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 2009, 66, 739–753. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Hagman, S.; Raunio, M.; Rossi, M.; Dastidar, P.; Elovaara, I. Disease-associated inflammatory biomarker profiles in blood in different subtypes of multiple sclerosis: Prospective clinical and MRI follow-up study. J. Neuroimmunol. 2011, 234, 141–147. [Google Scholar] [CrossRef]
- Rinta, S.; Kuusisto, H.; Raunio, M.; Paalavuo, R.; Levula, M.; Lehtimäki, T.; Elovaara, I. Apoptosis-related molecules in blood in multiple sclerosis. J. Neuroimmunol. 2008, 205, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Reuss, R.; Mistarz, M.; Mirau, A.; Kraus, J.; Bödeker, R.-H.; Oschmann, P. FADD Is Upregulated in Relapsing Remitting Multiple Sclerosis. Neuroimmunomodulation 2014, 21, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Hagman, S.; Kolasa, M.; Basnyat, P.; Helminen, M.; Kähönen, M.; Dastidar, P.; Lehtimäki, T.; Elovaara, I. Analysis of apoptosis-related genes in patients with clinically isolated syndrome and their association with conversion to multiple sclerosis. J. Neuroimmunol. 2015, 280, 43–48. [Google Scholar] [CrossRef]
- Artemiadis, A.K.; Anagnostouli, M.C. Apoptosis of Oligodendrocytes and Post-Translational Modifications of Myelin Basic Protein in Multiple Sclerosis: Possible Role for the Early Stages of Multiple Sclerosis. Eur. Neurol. 2010, 63, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J.; Singhrao, S.K.; Neal, J.W.; Lassmann, H.; Morgan, B.P. The Membrane Attack Complex of Complement Causes Severe Demyelination Associated with Acute Axonal Injury. J. Immunol. 2002, 168, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Damjanovic, D.; Valsasina, P.; Rocca, M.; Stromillo, M.; Gallo, A.; Enzinger, C.; Hulst, H.; Rovira, A.; Muhlert, N.; De Stefano, N.; et al. Hippocampal and Deep Gray Matter Nuclei Atrophy Is Relevant for Explaining Cognitive Impairment in MS: A Multicenter Study. Am. J. Neuroradiol. 2016, 38, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Eshaghi, A.; Prados, F.; Brownlee, W.J.; Altmann, D.R.; Tur, C.; Cardoso, M.J.; De Angelis, F.; van de Pavert, S.H.; Cawley, N.; De Stefano, N.; et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann. Neurol. 2018, 83, 210–222. [Google Scholar] [CrossRef] [Green Version]
- Scalfari, A.; Romualdi, C.; Nicholas, R.S.; Mattoscio, M.; Magliozzi, R.; Morra, A.; Monaco, S.; Muraro, P.A.; Calabrese, M. The cortical damage, early relapses, and onset of the progressive phase in multiple sclerosis. Neurology 2018, 90, e2107–e2118. [Google Scholar] [CrossRef] [Green Version]
- Andica, C.; Hagiwara, A.; Kamagata, K.; Yokoyama, K.; Shimoji, K.; Saito, A.; Takenaka, Y.; Nakazawa, M.; Hori, M.; Cohen-Adad, J.; et al. Gray Matter Alterations in Early and Late Relapsing-Remitting Multiple Sclerosis Evaluated with Synthetic Quantitative Magnetic Resonance Imaging. Sci. Rep. 2019, 9, 8147. [Google Scholar] [CrossRef] [Green Version]
- Picon, C.; Jayaraman, A.; James, R.; Beck, C.; Gallego, P.; Witte, M.E.; van Horssen, J.; Mazarakis, N.D.; Reynolds, R. Neuron-specific activation of necroptosis signaling in multiple sclerosis cortical grey matter. Acta Neuropathol. 2021, 141, 585–604. [Google Scholar] [CrossRef]
- Okouchi, M.; Ekshyyan, O.; Maracine, M.; Aw, T.Y. Neuronal Apoptosis in Neurodegeneration. Antioxid. Redox Signal. 2007, 9, 1059–1096. [Google Scholar] [CrossRef] [PubMed]
- Psenicka, M.W.; Smith, B.C.; Tinkey, R.A.; Williams, J.L. Connecting Neuroinflammation and Neurodegeneration in Multiple Sclerosis: Are Oligodendrocyte Precursor Cells a Nexus of Disease? Front. Cell. Neurosci. 2021, 15, 654284. [Google Scholar] [CrossRef] [PubMed]
- Alcázar, A.; Regidor, I.; Masjuan, J.; Salinas, M.; Álvarez-Cermeño, J. Axonal damage induced by cerebrospinal fluid from patients with relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2000, 104, 58–67. [Google Scholar] [CrossRef]
- Meyer, R.; Weissert, R.; Diem, R.; Storch, M.K.; De Graaf, K.L.; Kramer, B.; Bähr, M. Acute Neuronal Apoptosis in a Rat Model of Multiple Sclerosis. J. Neurosci. 2001, 21, 6214–6220. [Google Scholar] [CrossRef] [PubMed]
- Cid, C.; Álvarez-Cermeño, J.; Regidor, I.; Plaza, J.; Salinas, M.; Alcázar, A. Caspase inhibitors protect against neuronal apoptosis induced by cerebrospinal fluid from multiple sclerosis patients. J. Neuroimmunol. 2003, 136, 119–124. [Google Scholar] [CrossRef]
- Lisak, R.P.; Nedelkoska, L.; Benjamins, J.A.; Schalk, D.; Bealmear, B.; Touil, H.; Li, R.; Muirhead, G.; Bar-Or, A. B cells from patients with multiple sclerosis induce cell death via apoptosis in neurons in vitro. J. Neuroimmunol. 2017, 309, 88–99. [Google Scholar] [CrossRef]
- Green, A.J.; McQuaid, S.; Hauser, S.L.; Allen, I.V.; Lyness, R. Ocular pathology in multiple sclerosis: Retinal atrophy and inflammation irrespective of disease duration. Brain 2010, 133, 1591–1601. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski-Treska, J.; Ulrich, G.; Chasserot-Golaz, S.; Zwiller, J.; Revel, M.-O.; Aunis, D.; Bader, M.-F. Mechanisms Underlying Neuronal Death Induced by Chromogranin A-activated Microglia. J. Biol. Chem. 2001, 276, 13113–13120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magliozzi, R.; Pezzini, F.; Pucci, M.; Rossi, S.; Facchiano, F.; Marastoni, D.; Montagnana, M.; Lippi, G.; Reynolds, R.; Calabrese, M. Changes in Cerebrospinal Fluid Balance of TNF and TNF Receptors in Naïve Multiple Sclerosis Patients: Early Involvement in Compartmentalised Intrathecal Inflammation. Cells 2021, 10, 1712. [Google Scholar] [CrossRef]
- Kraft, A.D.; McPherson, C.A.; Harry, G.J. Heterogeneity of microglia and TNF signaling as determinants for neuronal death or survival. NeuroToxicology 2009, 30, 785–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guadagno, J.; Xu, X.; Karajgikar, M.; Brown, A.; Cregan, S. Microglia-derived TNFalpha induces apoptosis in neural precursor cells via transcriptional activation of the Bcl-2 family member Puma. Cell Death Dis. 2013, 4, e538. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2014, 77, 75–99. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Dénes, Á. Shaping Neuronal Fate: Functional Heterogeneity of Direct Microglia-Neuron Interactions. Neuron 2020, 109, 222–240. [Google Scholar] [CrossRef]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Anderson, S.R.; Roberts, J.M.; Ghena, N.; A Irvin, E.; Schwakopf, J.; Cooperstein, I.B.; Bosco, A.; Vetter, M.L. Author response: Neuronal apoptosis drives remodeling states of microglia and shifts in survival pathway dependence. Elife 2022. [Google Scholar] [CrossRef]
- Gilden, D.H. Infectious causes of multiple sclerosis. Lancet Neurol. 2005, 4, 195–202. [Google Scholar] [CrossRef]
- Wang, Z.; Kennedy, P.G.; Dupree, C.; Wang, M.; Lee, C.; Pointon, T.; Langford, T.D.; Graner, M.W.; Yu, X. Antibodies from Multiple Sclerosis Brain Identified Epstein-Barr Virus Nuclear Antigen 1 & 2 Epitopes which Are Recognized by Oligoclonal Bands. J. Neuroimmune Pharmacol. 2020, 16, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L. The enigmatic links between Epstein-Barr virus infection and multiple sclerosis. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Fujinami, R.S. Inside-Out versus Outside-In models for virus induced demyelination: Axonal damage triggering demyelination. Springer Semin. Immunopathol. 2002, 24, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M.; Bratton, D.L. Antiinflammatory effects of apoptotic cells. J. Clin. Investig. 2013, 123, 2773–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassmann, H.; Brück, W.; Lucchinetti, C.; Rodriguez, M. Remyelination in multiple sclerosis. Mult. Scler. 1997, 3, 133–136. [Google Scholar] [CrossRef]
- Franklin, R.J. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 2002, 3, 705–714. [Google Scholar] [CrossRef]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Brück, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006, 129 Pt 12, 3165–3172. [Google Scholar] [CrossRef] [Green Version]
- Pluchino, S.; Muzio, L.; Imitola, J.; Deleidi, M.; Alfaro-Cervello, C.; Salani, G.; Porcheri, C.; Brambilla, E.; Cavasinni, F.; Bergamaschi, A.; et al. Persistent inflammation alters the function of the endogenous brain stem cell compartment. Brain 2008, 131, 2564–2578. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, P.G.E.; George, W.; Yu, X. The Possible Role of Neural Cell Apoptosis in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 7584. https://doi.org/10.3390/ijms23147584
Kennedy PGE, George W, Yu X. The Possible Role of Neural Cell Apoptosis in Multiple Sclerosis. International Journal of Molecular Sciences. 2022; 23(14):7584. https://doi.org/10.3390/ijms23147584
Chicago/Turabian StyleKennedy, Peter G. E., Woro George, and Xiaoli Yu. 2022. "The Possible Role of Neural Cell Apoptosis in Multiple Sclerosis" International Journal of Molecular Sciences 23, no. 14: 7584. https://doi.org/10.3390/ijms23147584
APA StyleKennedy, P. G. E., George, W., & Yu, X. (2022). The Possible Role of Neural Cell Apoptosis in Multiple Sclerosis. International Journal of Molecular Sciences, 23(14), 7584. https://doi.org/10.3390/ijms23147584