
Integrated mRNA and Small RNA Sequencing Reveals a microRNA Regulatory Network Associated with Starch Biosynthesis in Lotus (Nelumbo nucifera Gaertn.) Rhizomes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Starch Content and Starch Granule Morphology of Lotus Rhizome
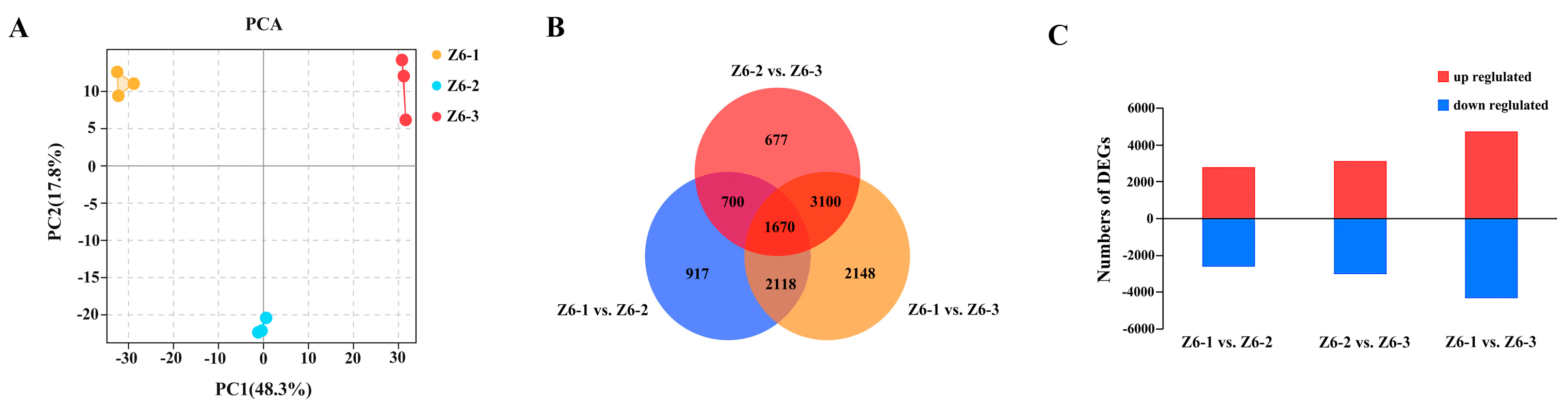
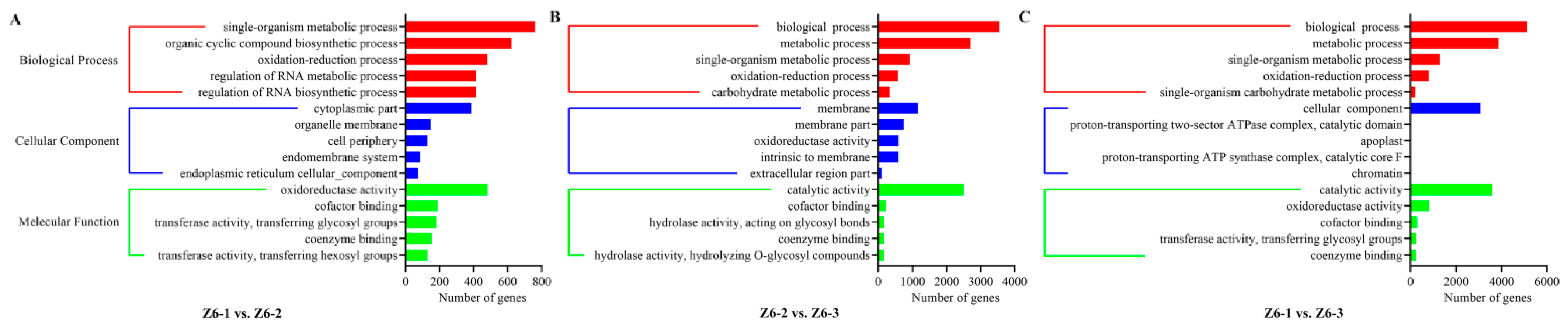
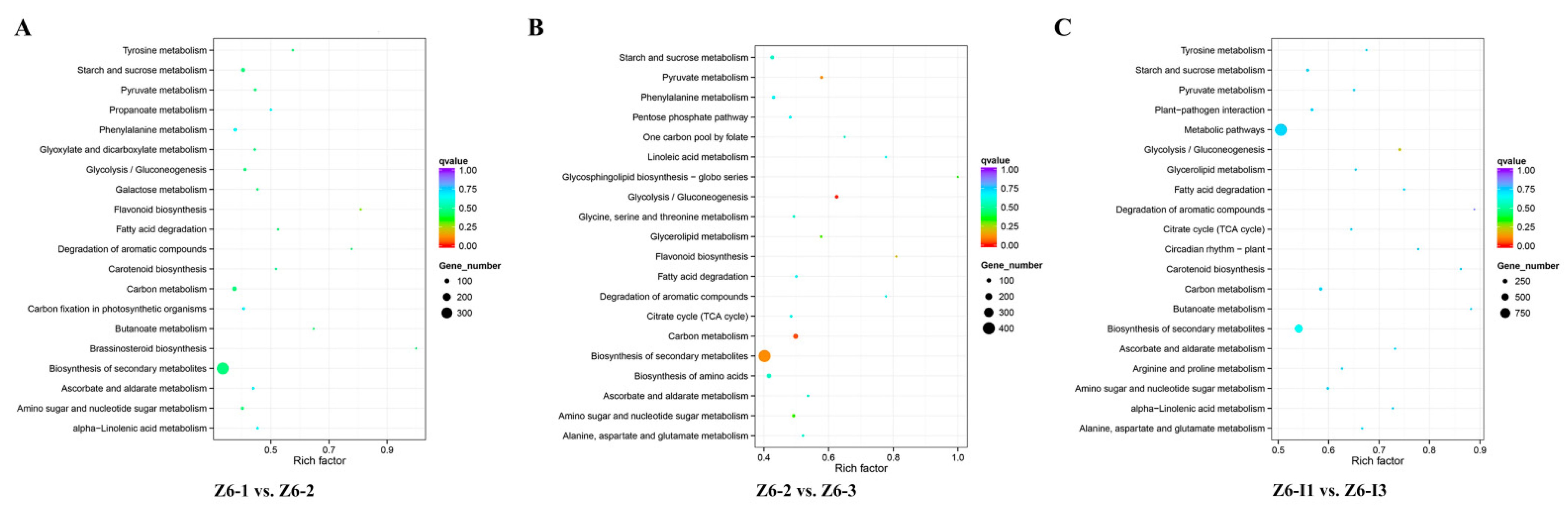
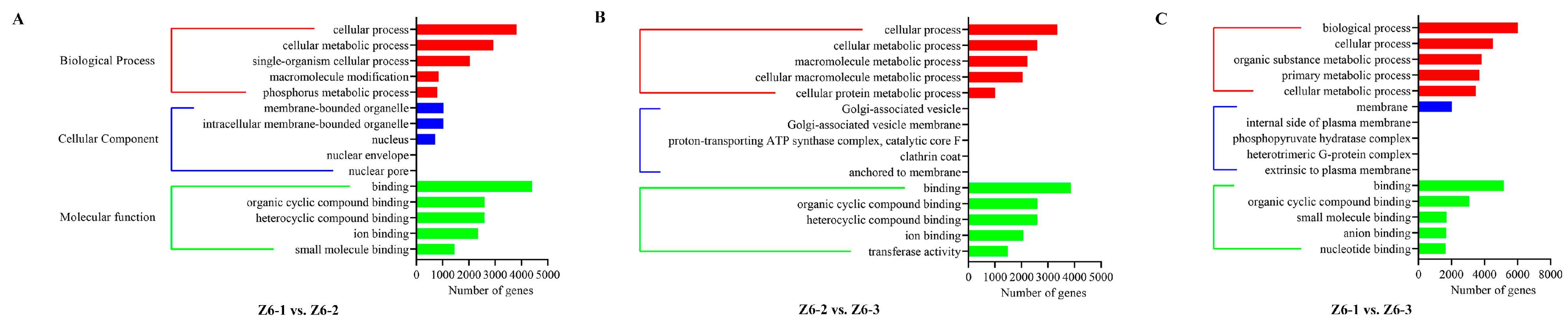
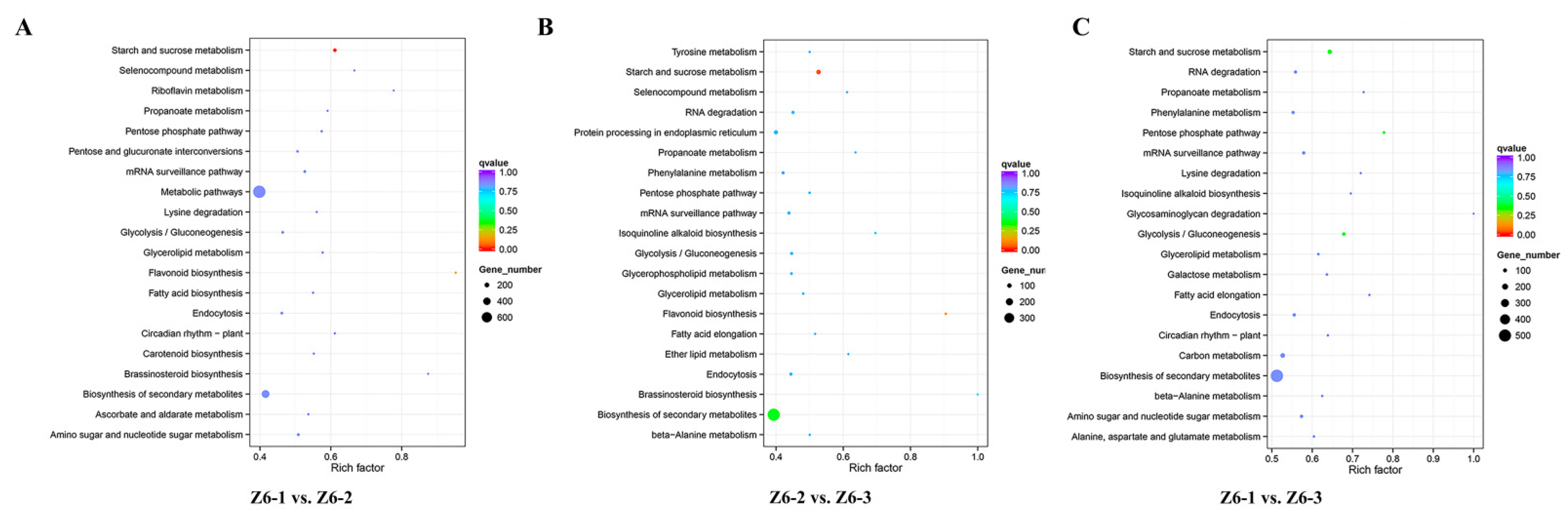
2.2. Transcriptome Analysis of Different Internodes in Lotus
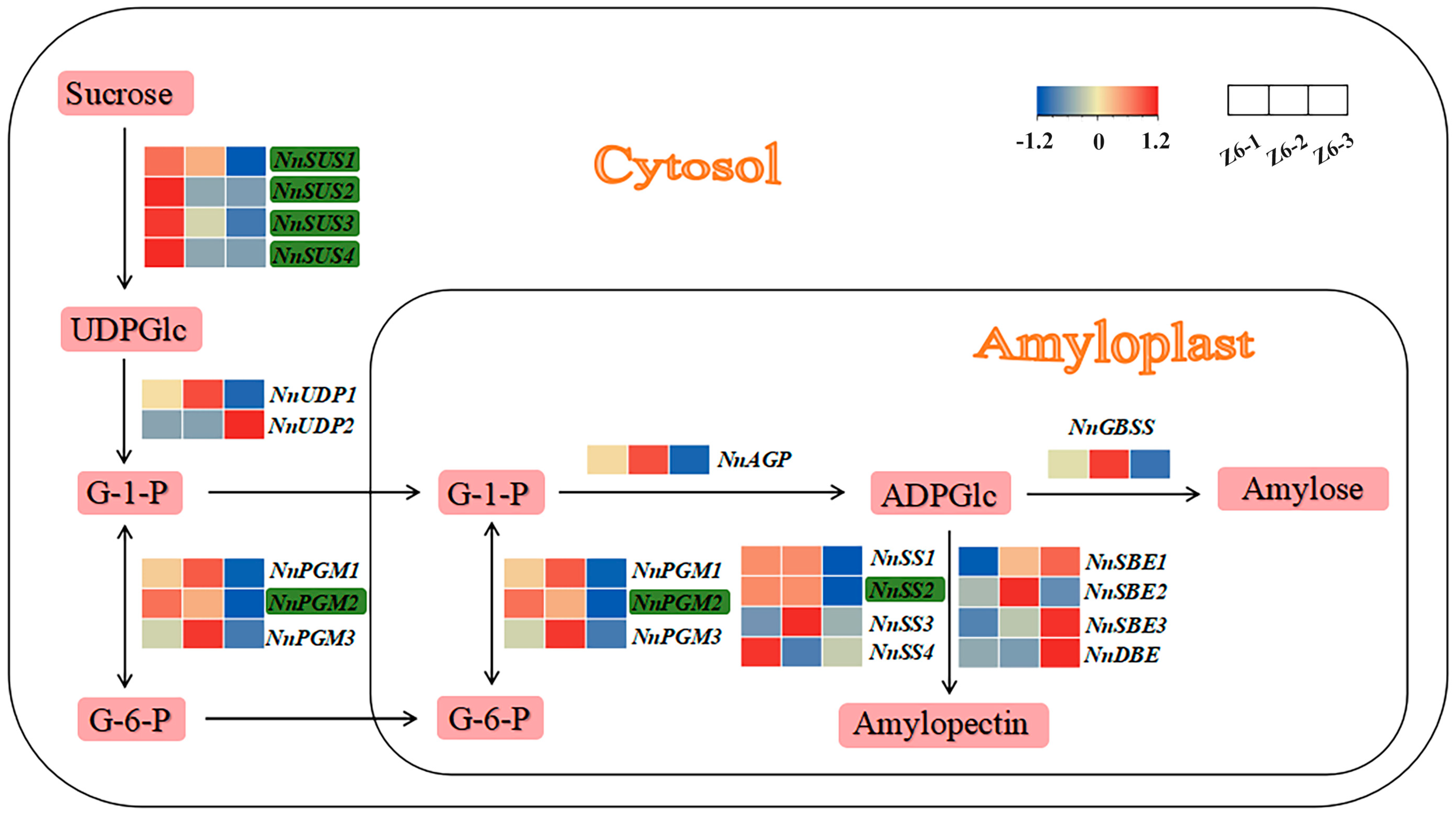
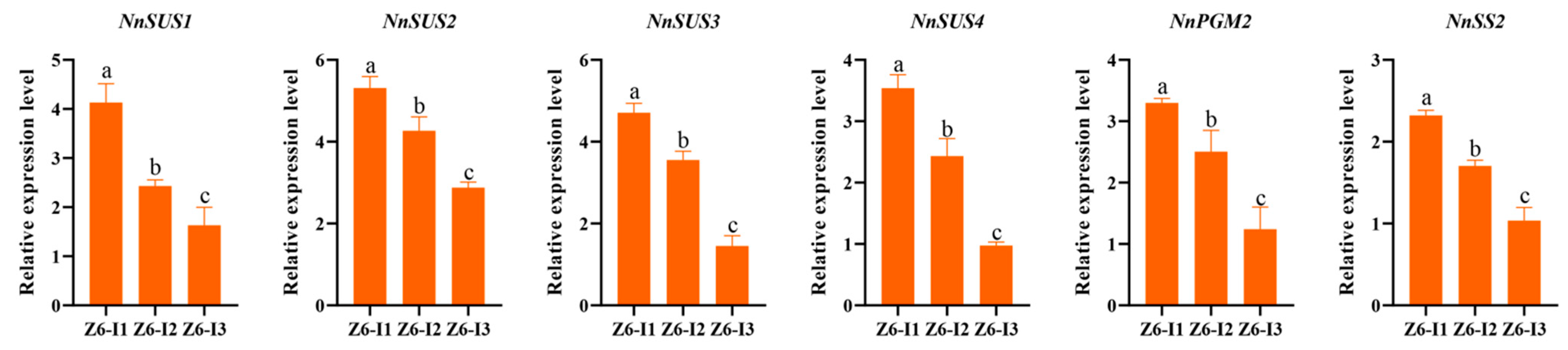
2.3. Expression Profile of Starch Biosynthetic Genes in Different Internodes
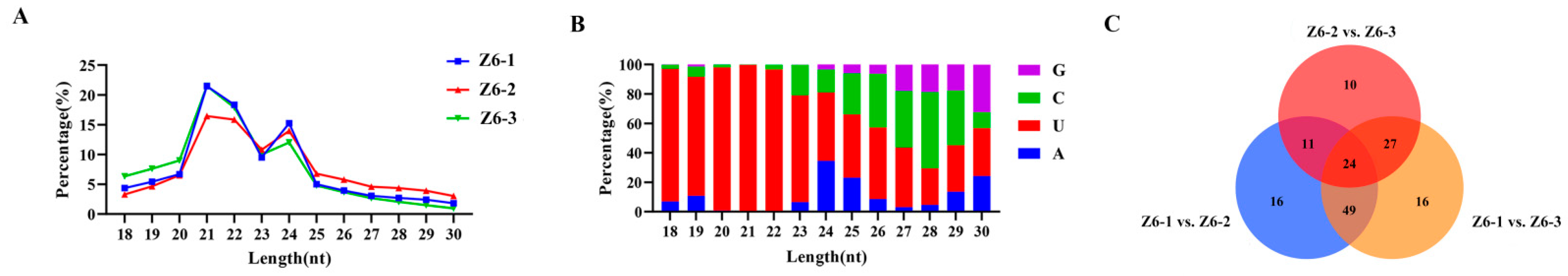
2.4. Sequencing and miRNAs Identification of Lotus Rhizome in Different Internodes
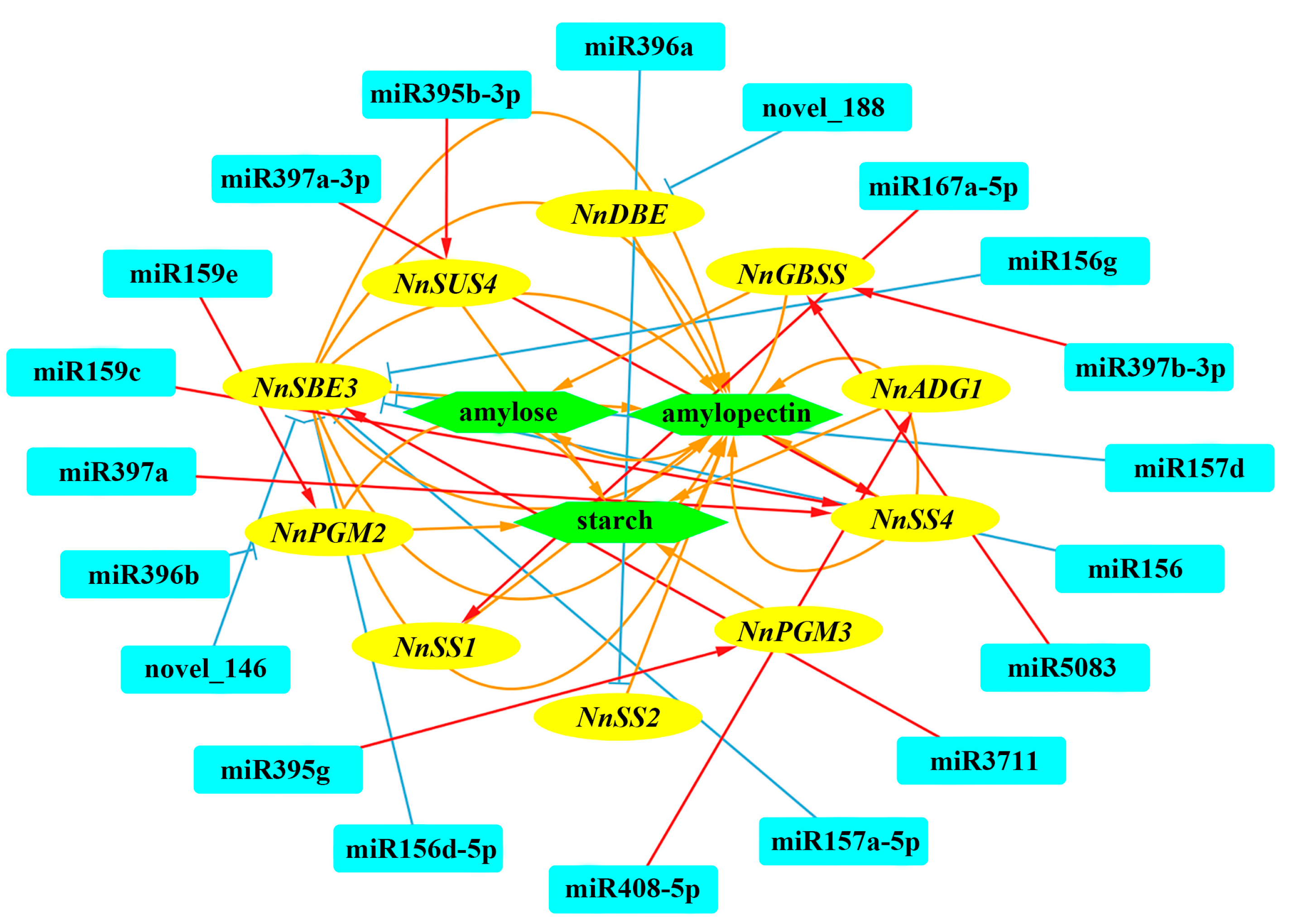
2.5. Targets of Differentially Expressed miRNAs in Different Internodes
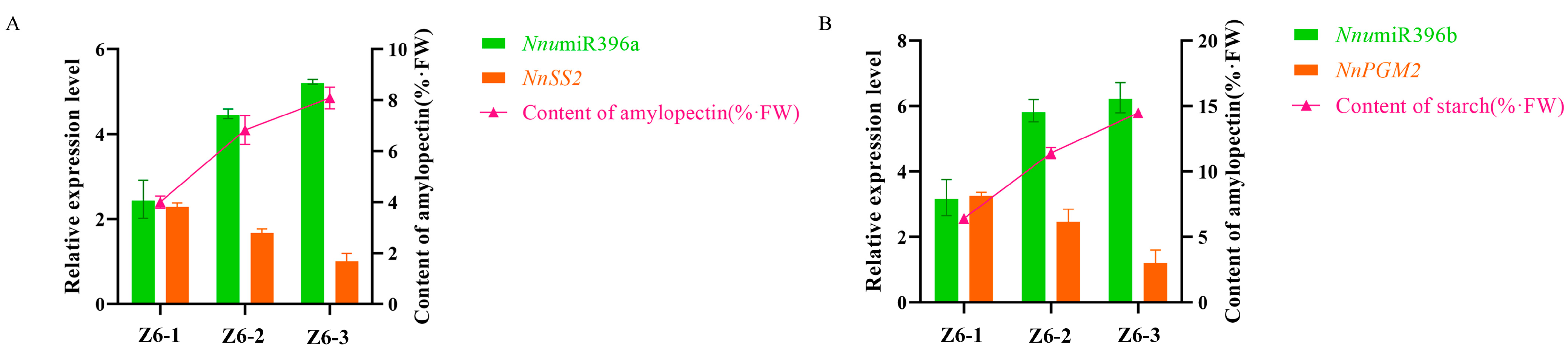
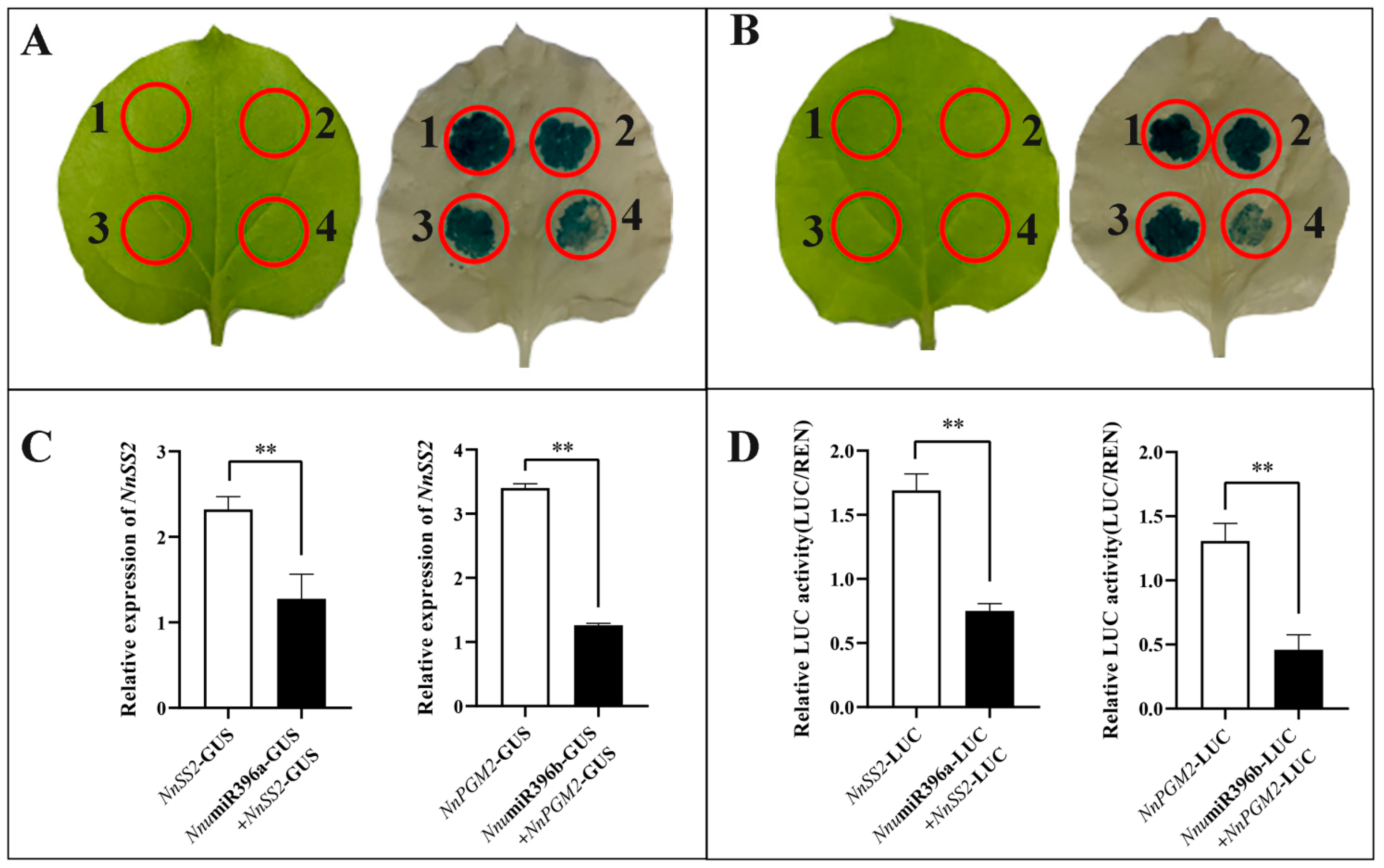
2.6. Validation of the Regulatory Pathways for Starch Biosynthesis Mediated by NnumiR396a-NnSS2 and NnumiR396b-NnPGM2
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Starch Content
4.3. Small RNA and Transcriptome Sequencing
4.4. Bioinformation Analysis of the Data
4.5. QRT-PCR Analysis of mRNAs
4.6. QRT-PCR Analysis of miRNAs
4.7. Transient Expression Analysis
4.8. Transient Dual-Luciferase Detection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shen-Miller, J. Sacred lotus, the long-living fruits of China Antique. Seed Sci. Res. 2002, 12, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Ming, R.; VanBuren, R.; Liu, Y.L.; Yang, M.; Shen-Miller, J. Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biol. 2013, 14, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.B.; Zhang, M.; Wang, S.J. Processing characteristics and flavour of full lotus root powder beverage. J. Sci. Food Agric. 2010, 90, 2482–2489. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.Y.; Li, M.; Cao, D.D.; Yang, P.F. Genetic dissection of rhizome yield-related traits in Nelumbo nucifera through genetic linkage map construction and QTL mapping. Plant Physiol. Biochem. 2021, 160, 155–165. [Google Scholar] [CrossRef]
- Zhao, S.P.; Ruan, F.J.; Shen, W.J.; Deng, K.M.; Jiang, T.; Wu, P.; Feng, K.; Li, L.J. The Effect of nitrogen fertilizer on rhizome quality and starch physicochemical properties in Nelumbo nucifera. Agronomy 2022, 12, 794. [Google Scholar] [CrossRef]
- Zhu, F. Structures, properties, and applications of lotus starches. Food Hydrocolloid. 2017, 63, 332–348. [Google Scholar] [CrossRef]
- Wang, K.; Henry, R.J.; Gilbert, R.G. Causal relations among starch biosynthesis, structure, and properties. Springer Sci. Rev. 2014, 2, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Baroja-Fernandez, E.; Munoz, F.J.; Montero, M.; Etxeberria, E.; Sesma, M.T.; Ovecka, M.; Bahaji, A.; Ezquer, I.; Li, J.; Prat, S.; et al. Enhancing sucrose synthase activity in transgenic potato (Solanum tuberosum L.) tubers results in increased levels of starch, ADPglucose and UDPglucose and total yield. Plant Cell Physiol. 2009, 50, 1651–1662. [Google Scholar] [CrossRef]
- Huang, T.W.; Luo, X.L.; Fan, Z.P.; Yang, Y.N.; Wan, W. Genome-wide identification and analysis of the sucrose synthase gene family in cassava (Manihot esculenta Crantz). Gene 2021, 769, 145191. [Google Scholar] [CrossRef]
- Edurne, B.F.; José, M.F.; Jun, L.; Abdellatif, B.; Goizeder, A.; Manuel, M.; Ed, E.; Maite, H.; Teresa, S.M.; Javier, P.R. Sucrose synthase activity in the sus1/sus2/sus3/sus4 Arabidopsis mutant is sufficient to support normal cellulose and starch production. Proc. Natl. Acad. Sci. USA 2012, 109, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Long, W.; Dong, B.; Wang, Y.; Pan, P.; Wang, Y.; Liu, L.; Chen, X.; Liu, X.; Liu, S.; Tian, Y.; et al. FLOURY ENDOSPERM8, encoding the UDP-glucose pyrophosphorylase 1, affects the synthesis and structure of starch in rice endosperm. J. Plant Biol. 2017, 60, 513–522. [Google Scholar] [CrossRef]
- Uematsu, K.; Suzuki, N.; Iwamae, T.; Inui, M.; Yukawa, H. Alteration of photosynthate partitioning by high-level expression of phosphoglucomutase in tobacco chloroplasts. Biosci. Biotechnol. Biochem. 2012, 76, 1315–1321. [Google Scholar] [CrossRef]
- Uematsu, K.; Suzuki, N.; Iwamae, T.; Inui, M.; Yukawa, H. Expression of Arabidopsis plastidial phosphoglucomutase in tobacco stimulates photosynthetic carbon flow into starch synthesis. J. Plant Physiol. 2012, 169, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Zheng, X.W.; Xie, K.Q.; Liu, J.D.; Zheng, X.F.; Jin, S.R.; Diao, Y.; Hu, Z.L.; Wang, J.X. Development and identification of three functional markers associated with starch content in lotus (Nelumbo nucifera). Sci. Rep. 2020, 10, 4242. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Wang, L.; Zhang, L.; Liu, Y.; Yang, M.; Yuan, Z.; Li, S.; Han, Y. Characterization of genes encoding Granule-Bound Starch Synthase in sacred lotus reveals phylogenetic affinity of Nelumbo to proteales. Plant Mol. Biol. Rep. 2013, 31, 1157–1165. [Google Scholar] [CrossRef]
- Lu, Y.; Li, L.; Zhou, Y.; Gao, Q.; Liang, G.; Chen, X.; Qi, X. Cloning and characterization of the Wx gene encoding a Granule-Bound Starch Synthase in lotus (Nelumbo nucifera Gaertn). Plant Mol. Biol. Rep. 2012, 30, 1210–1217. [Google Scholar] [CrossRef]
- Emmanuel, O.N.; Dodzi, A.K.; Tianyue, A.; Longxin, H.; Xiangru, T. The roles of starch branching enzymes and starch synthase in the biosynthesis of amylose in rice. J. Cereal Sci. 2022, 104, 103393. [Google Scholar] [CrossRef]
- Zhu, F.L.; Cheng, N.; Sun, H.; Diao, Y.; Hu, Z.L. Molecular cloning and characterization of a gene encoding soluble starch synthase III (SSSIII) in lotus (Nelumbo nucifera). Biologia 2020, 75, 279–288. [Google Scholar] [CrossRef]
- Andreas, B.; Katsiaryna, S.; Vanja, T.; Susanne, L.K.; Shahnoor, S.S.; Andersen, H.; Hanne-Grethe, K.; Kare, L.N. Non-GMO potato lines, synthesizing increased amylose and resistant starch, are mainly deficient in isoamylase debranching enzyme. Plant Biotechnol. J. 2020, 18, 2096–2108. [Google Scholar] [CrossRef]
- Nabemoto, M.; Watanabe, R.; Ohsu, M.; Sato, K.; Otani, M.; Nakayachi, O.; Watanabe, M. Molecular characterization of genes encoding isoamylase-type debranching enzyme in tuberous root of sweet potato, Ipomoea batatas (L.). Plant Biotechnol. 2016, 33, 351–359. [Google Scholar] [CrossRef] [Green Version]
- He, M.Y.; Kong, X.J.; Jiang, Y.M.; Qu, H.X.; Zhu, H. MicroRNAs: Emerging regulators in horticultural crops. Trends Plant Sci. 2022, 22, S1360–S1385. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xia, J.; Xia, Z.Q.; Zhang, H.F.; Zeng, C.Y.; Lu, C.; Zhang, W.X.; Wang, W.Q. Potential functions of microRNAs in starch metabolism and development revealed by miRNA transcriptome profiling of cassava cultivars and their wild progenitor. BMC Plant Biol. 2015, 15, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunn, J.E.; MacRae, E. New complexities in the synthesis of sucrose. Curr. Opin. Plant Biol. 2003, 6, 208–214. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, Y.F.; Sunkar, R.; Zhang, W.X. SeqTar: An effective method for identifying microRNA guided cleavage sites from degradome of polyadenylated transcripts in plants. Nucleic Acids Res. 2012, 40, e28. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.L.; Frazier, T.P.; Zhang, B.H. Identification, characterization and expression analysis of MicroRNAs and their targets in the potato (Solanum tuberosum). Gene 2010, 473, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhu, L.P.; Cheng, P.; Xu, L.M.; Liu, Y.L.; Ke, W.D.; Yang, P.F. Transcriptomic analysis of the regulation of rhizome formation in temperate and tropical lotus (Nelumbo nucifera). Sci. Rep. 2015, 5, 13059. [Google Scholar] [CrossRef]
- Cao, D.D.; Damaris, R.N.; Zhang, Y.; Liu, M.H.; Li, M.; Yang, P.F. Proteomic analysis showing the signaling pathways involved in the rhizome enlargement process in Nelumbo nucifera. BMC Genom. 2019, 20, 766. [Google Scholar] [CrossRef]
- Cheng, L.B.; Li, S.Y.; Yin, J.J.; Li, L.J.; Chen, X.H. Genome-Wide analysis of differentially expressed genes relevant to rhizome formation in lotus root (Nelumbo nucifera Gaertn). PLoS ONE 2013, 8, e67116. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z. The major endogenous small RNAs and their roles in plant development. Biotechnol. Bull. 2012, 22, 7–13. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, T.; Xu, Z.N.; Wai, C.M.; Chen, K.; Zhang, X.T.; Wang, S.P.; Ji, B.; Ming, R.; Sunkar, R. Identification of microRNAs, phasiRNAs and their targets in pineapple. Trop Plant Biol. 2016, 9, 176–186. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Ou, L.; Kang, L.; Liu, Y.; Lv, J.; Wei, G.; Yang, B.; Yang, S.; Chen, W.; et al. Identification and characterization of novel microRNAs for fruit development and quality in hot pepper (Capsicum annuum L.). Gene 2017, 608, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Liu, A.L.; Zhang, Y.Y.; Feng, K.; Zhao, S.P.; Li, L.L. NnABI4-Mediated ABA regulation of starch biosynthesis in lotus (Nelumbo nucifera Gaertn). Int. J. Mol. Sci. 2021, 22, 13506. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.L. Sucrose Metabolism: Gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef]
- Mehdi, R.; Lamm, C.E.; Anjanappa, R.B. Symplasmic phloem unloading and radial post-phloem transport via vascular rays in tuberous roots of Manihot esculenta. J. Exp. Bot. 2019, 70, 5559–5573. [Google Scholar] [CrossRef]
- Malinova, I.; Kunz, H.H.; Alseekh, S.; Herbst, K.; Fernie, A.R.; Gierth, M.; Fettke, J. Reduction of the cytosolic phosphoglucomutase in Arabidopsis reveals impact on plant growth, seed and root development, and carbohydrate partitioning. PLoS ONE 2014, 9, e112468. [Google Scholar] [CrossRef] [PubMed]
- Periappuram, C.; Steinhauer, L.; Barton, D.L.; Taylor, D.C.; Chatson, B.; Zou, J.T. The plastidic phosphoglucomutase from Arabidopsis. A reversible enzyme reaction with an important role in metabolic control. Plant Physiol. 2000, 122, 1193–1199. [Google Scholar] [CrossRef] [Green Version]
- He, S.T.; Hao, X.M.; Wang, S.S.; Zhou, W.Z.; Ma, Q.X.; Lu, X.L.; Chen, L.N.; Zhang, P. Starch synthase II plays a crucial role in starch biosynthesis and the formation of multienzyme complexes in cassava storage roots. J. Exp. Bot. 2022, 73, 2540–2557. [Google Scholar] [CrossRef]
- Yu, Y.; Sun, F.Y.; Chen, N.; Sun, G.L.; Wang, C.Y.; Wu, D.X. MiR396 regulatory network and its expression during grain de-velopment in wheat. Protoplasma 2021, 258, 103–113. [Google Scholar] [CrossRef]
- Li, S.C.; Gao, F.Y.; Xie, K.L.; Zeng, X.H.; Cao, Y.; Zeng, J.; He, Z.S.; Ren, Y.; Li, W.; Deng, Q.; et al. The OsmiR396c-OsGRF4-OsGIF1 regulatory module determines grain size and yield in rice. Plant Biotechnol. J. 2016, 14, 2134–2146. [Google Scholar] [CrossRef]
- Hou, G.; Du, C.; Gao, H.; Liu, S.; Sun, W.; Lu, H.; Kang, J.; Xie, Y.; Ma, D.; Wang, C. Identification of microRNAs in developing wheat grain that are potentially involved in regulating grain characteristics and the response to nitrogen levels. BMC Plant Biol. 2020, 20, 87. [Google Scholar] [CrossRef]
- Zhang, K.; Shi, X.; Zhao, X.; Ding, D.; Tang, J.; Niu, J. Investigation of miR396 and growth-regulating factor regulatory network in maize grain filling. Acta Physiol. Plant 2015, 37, 28. [Google Scholar] [CrossRef]
- Ali, M.; Brian, A.W.; Kenneth, M.; Lorian, S.; Barbara, W. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Kerr, S.C.; Gaiti, F.; Tanurdzic, M. De Novo plant transcriptome assembly and annotation using Illumina RNA-Seq reads. Methods Mol. Biol. 2019, 1933, 265–275. [Google Scholar] [CrossRef]
- Zhao, F.Z.; Maren, N.A.; Kosentka, P.Z.; Liao, Y.Y.; Lu, H.Y.; Duduit, J.R.; Huang, D.; Ashrafi, H.; Zhao, T.J.; Huerta, A.I.; et al. An optimized protocol for stepwise optimization of real-time RT-PCR analysis. Hortic. Res. 2021, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Yasuhiro, K.; Yuichi, T. Comparative analysis of various root active promoters by evaluation of GUS expression in transgenic Arabidopsis. Plant Biotechnol. 2021, 38, 443–448. [Google Scholar] [CrossRef]
- Tada, Y. The HKT transporter gene from Arabidopsis, AtHKT1;1, is dominantly expressed in shoot vascular tissue and root tips and is mild salt stress-responsive. Plants 2019, 8, 204. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.B.; Zhu, X.E.; Qi, B.L.; Gao, Z.; Tian, P.; Li, Z.W.; Lin, Z.T.; Zhang, Y.X.; Huang, T.B. SlMIR164A regulates fruit ripening and quality by controlling SlNAM2 and SlNAM3 in tomato. Plant Biotechnol. J. 2022. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Zhao, S.; Deng, K.; Wu, P.; Feng, K.; Li, L. Integrated mRNA and Small RNA Sequencing Reveals a microRNA Regulatory Network Associated with Starch Biosynthesis in Lotus (Nelumbo nucifera Gaertn.) Rhizomes. Int. J. Mol. Sci. 2022, 23, 7605. https://doi.org/10.3390/ijms23147605
Zhu Y, Zhao S, Deng K, Wu P, Feng K, Li L. Integrated mRNA and Small RNA Sequencing Reveals a microRNA Regulatory Network Associated with Starch Biosynthesis in Lotus (Nelumbo nucifera Gaertn.) Rhizomes. International Journal of Molecular Sciences. 2022; 23(14):7605. https://doi.org/10.3390/ijms23147605
Chicago/Turabian StyleZhu, Yamei, Shuping Zhao, Kangming Deng, Peng Wu, Kai Feng, and Liangjun Li. 2022. "Integrated mRNA and Small RNA Sequencing Reveals a microRNA Regulatory Network Associated with Starch Biosynthesis in Lotus (Nelumbo nucifera Gaertn.) Rhizomes" International Journal of Molecular Sciences 23, no. 14: 7605. https://doi.org/10.3390/ijms23147605
APA StyleZhu, Y., Zhao, S., Deng, K., Wu, P., Feng, K., & Li, L. (2022). Integrated mRNA and Small RNA Sequencing Reveals a microRNA Regulatory Network Associated with Starch Biosynthesis in Lotus (Nelumbo nucifera Gaertn.) Rhizomes. International Journal of Molecular Sciences, 23(14), 7605. https://doi.org/10.3390/ijms23147605