
S-Adenosylmethionine Inhibits Colorectal Cancer Cell Migration through Mirna-Mediated Targeting of Notch Signaling Pathway †

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
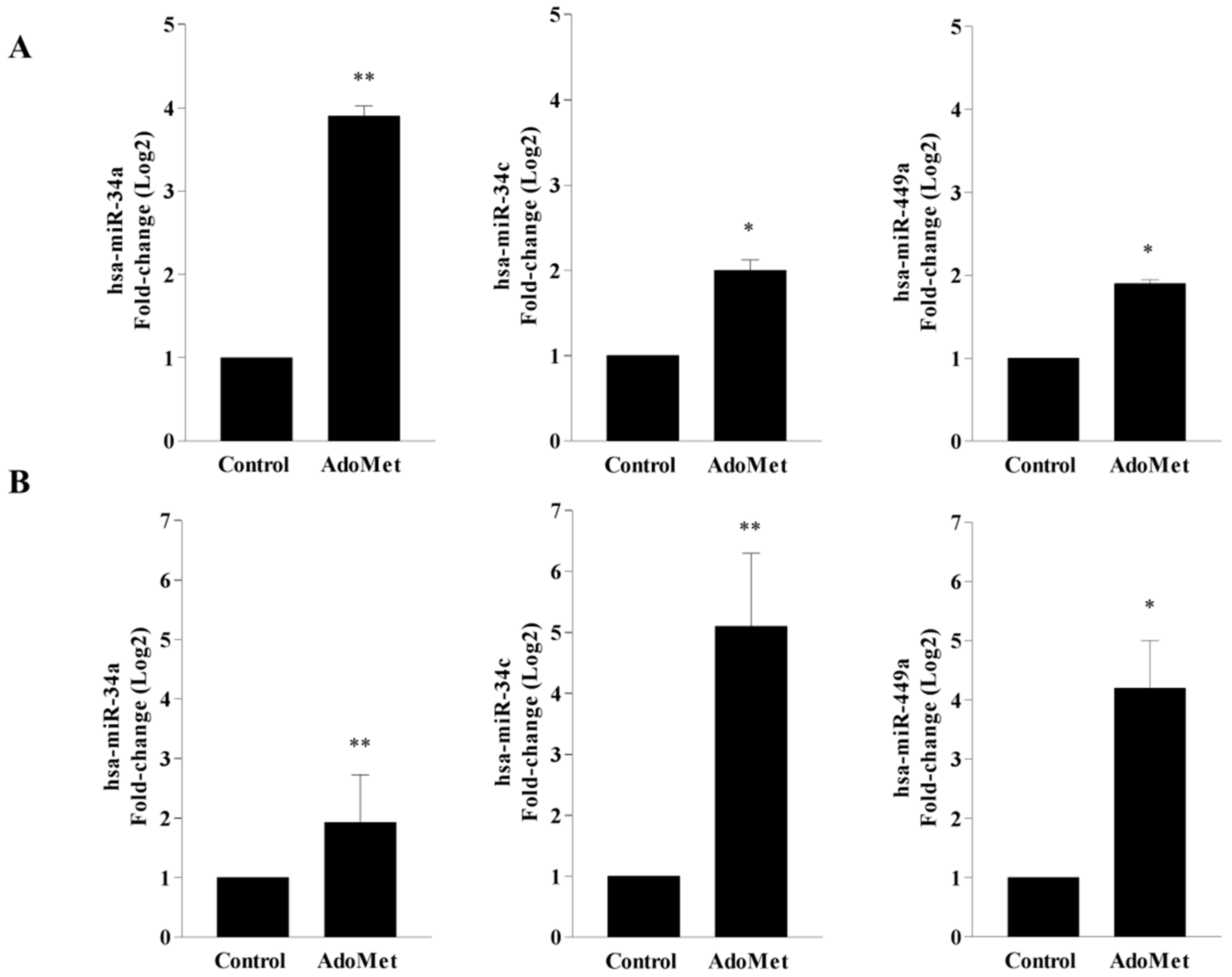
2.1. AdoMet Upregulated miR-34a, miR-34c, and miR-449a Expression in HCT-116 and Caco-2 CRC Cell Lines
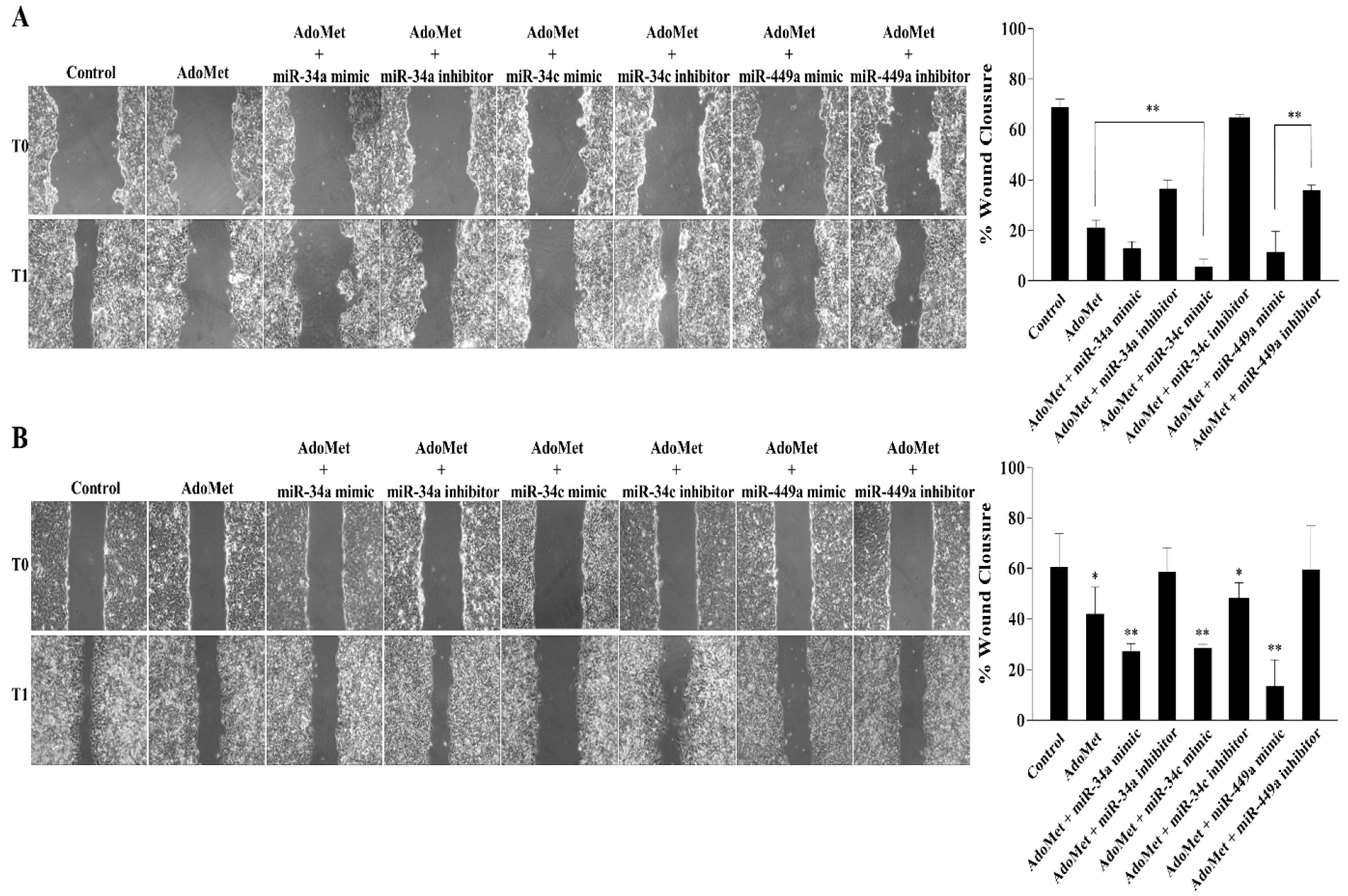
2.2. miR-34a, miR-34c, and miR-449a Mediated the AdoMet-Induced Inhibition of HCT-116 and Caco-2 Cell Migration
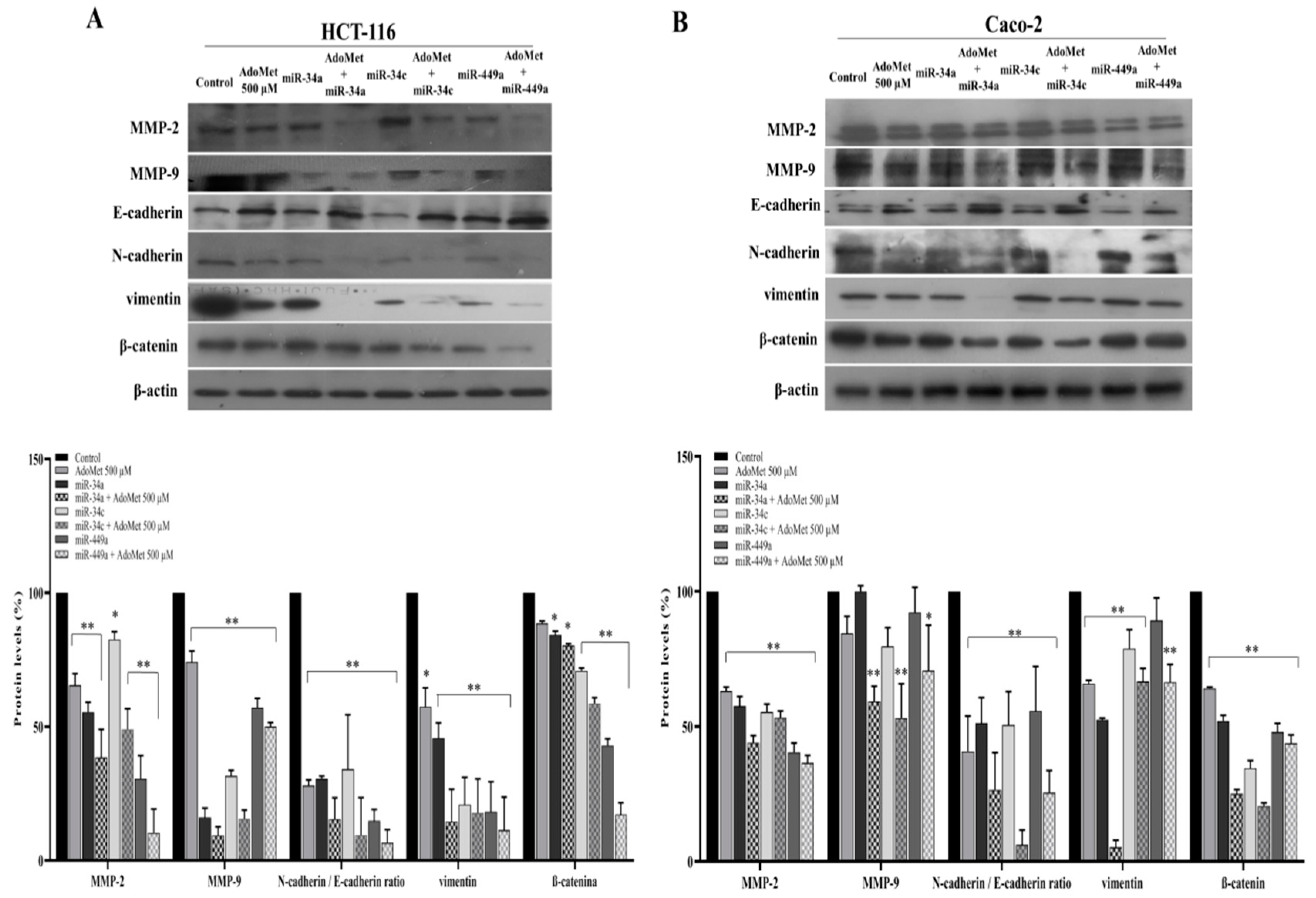
2.3. miR-34a, miR-34c, and miR-449a Mediated AdoMet-Induced Inhibition of Migration- and EMT-Related Protein Expression in HCT-116 and Caco-2 Cells
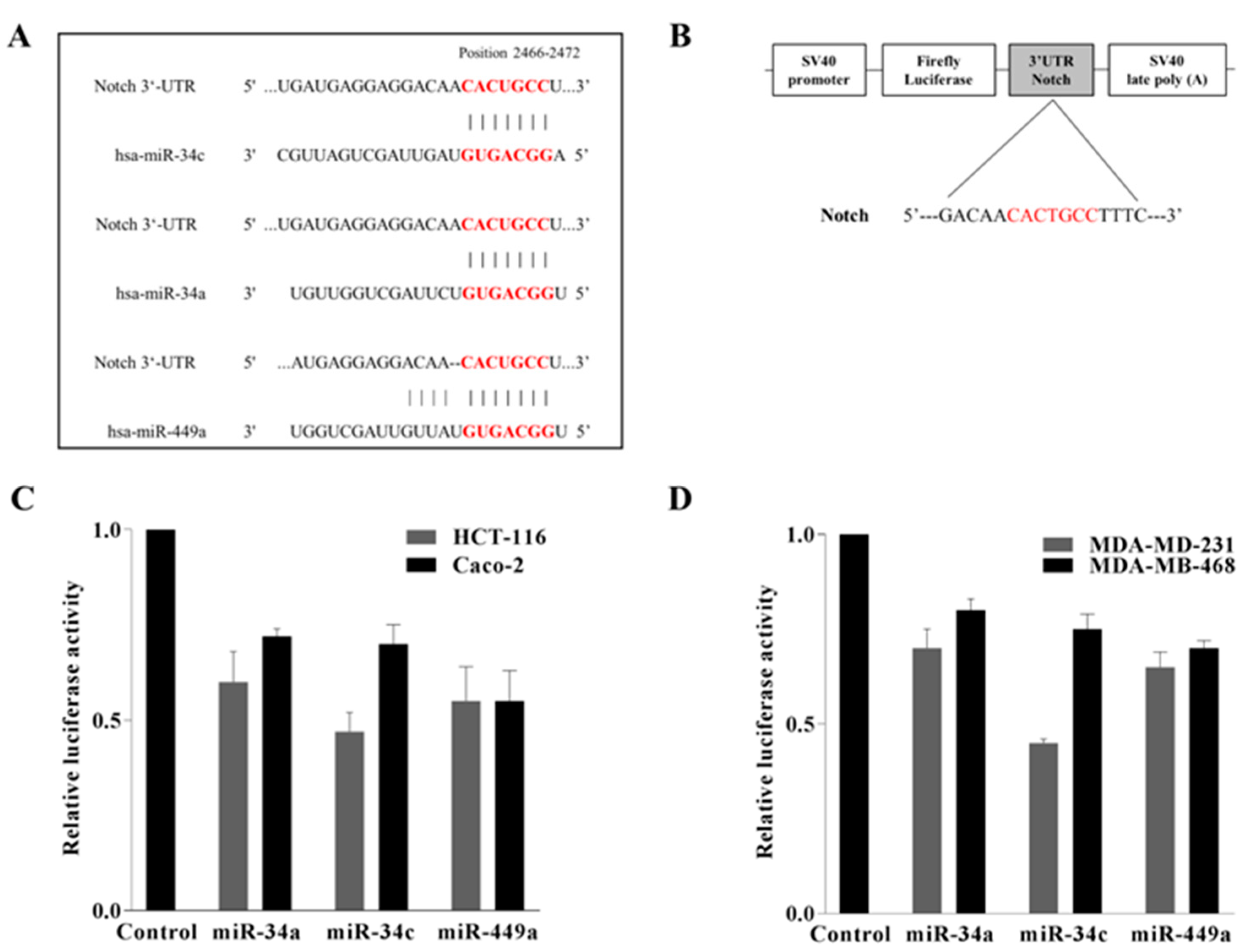
2.4. Notch Was Directly Targeted by miR-34a, miR 34c, and miR 449a in Colorectal HCT-116 and Caco-2 and in Triple Negative MDA-MB-231 and MDA-MB-468 Breast Cancer Cells
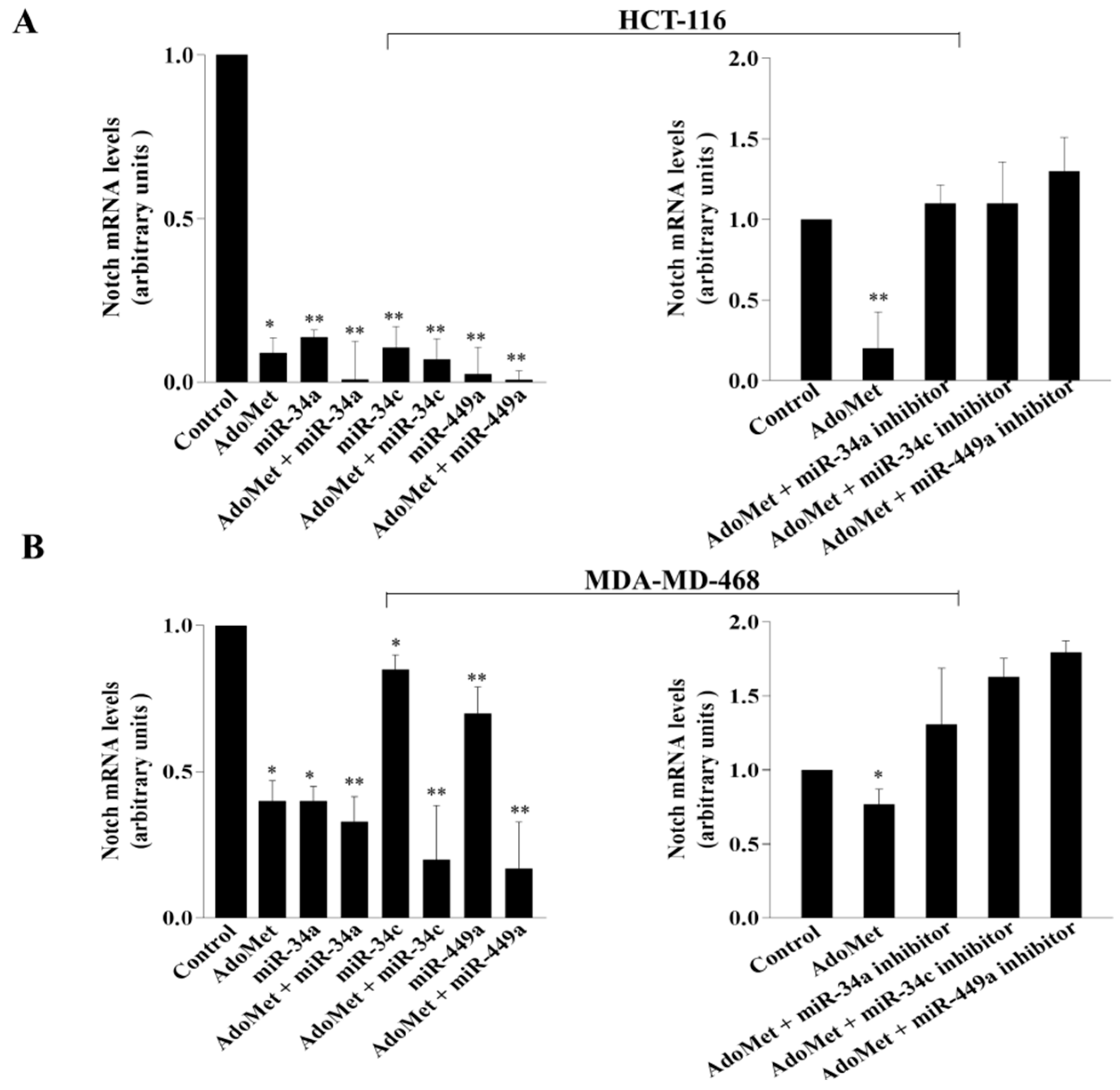
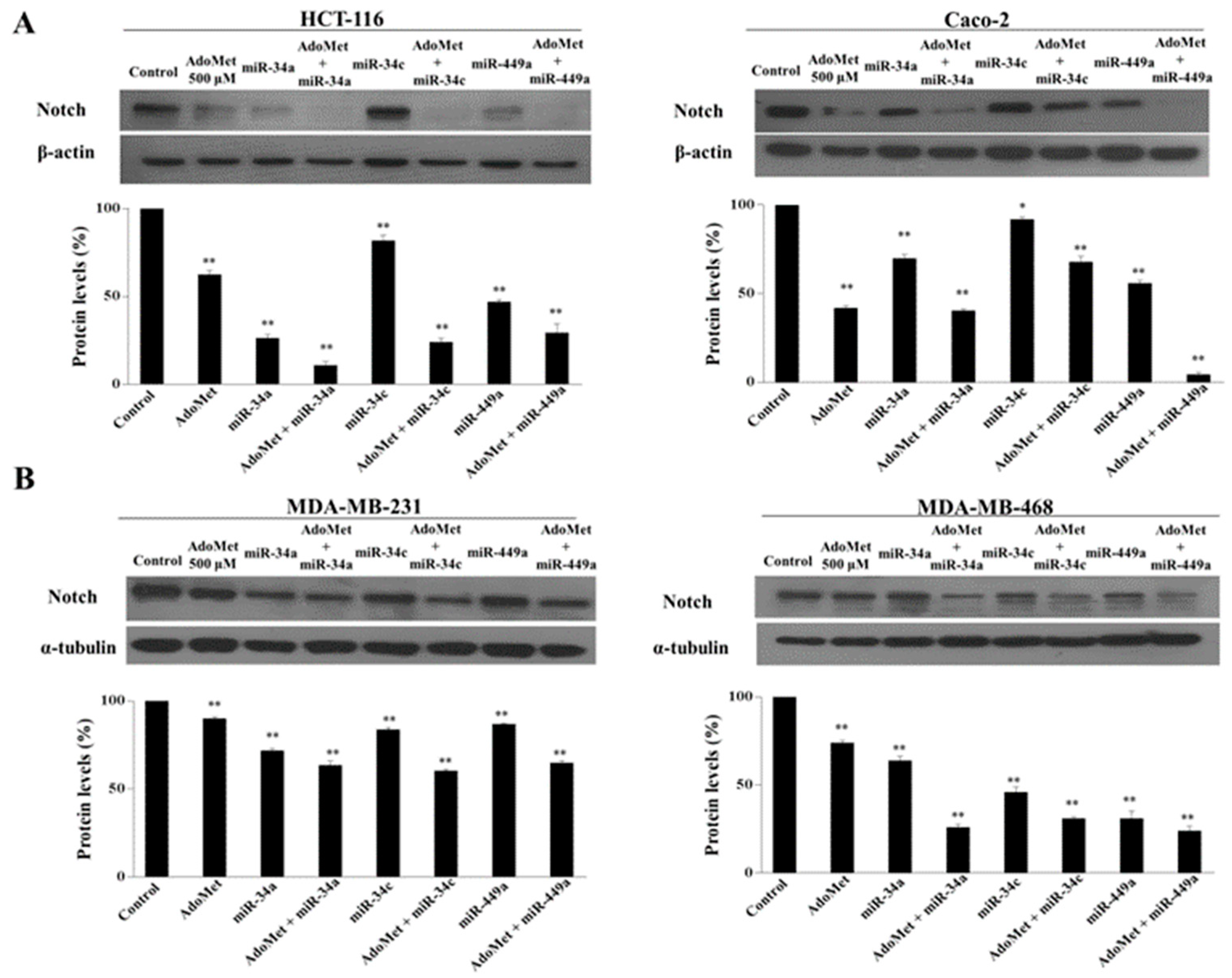
2.5. AdoMet Inhibited Notch Expression in CRC and TNBC Cells through miR-34a, miR-34c, and miR-449a
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Cultures and Treatments
4.3. Cell Transfections
4.4. qRT-PCR for miRNAs Detection and mRNA Expression
4.5. Protein Extraction and Western Blot Analysis
4.6. Migration Process Evaluated by Scratch-Wound Assay
4.7. Luciferase Reporter Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Datta, P.K. Regulation of EMT in colorectal cancer: A culprit in metastasis. Cancers 2017, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.E. Molecular mechanisms of colon cancer progression and metastasis: Recent insights and advancements. Int. J. Mol. Sci. 2020, 22, 130. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Sharma, A.K.; Damodaran, C. A review on Notch signaling and colorectal cancer. Cells 2020, 9, 1549. [Google Scholar] [CrossRef] [PubMed]
- Anusewicz, D.; Orzechowska, M.; Bednarek, A.K. Notch signaling pathway in cancer-review with bioinformatic analysis. Cancers 2021, 13, 768. [Google Scholar] [CrossRef]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The varied roles of Notch in cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaik, J.P.; Alanazi, I.O.; Pathan, A.; Parine, N.R.; Almadi, M.A.; Azzam, N.A.; Aljebreen, A.M.; Alharbi, O.; Alanazi, M.S.; Khan, Z. Frequent activation of Notch signaling pathway in colorectal cancers and its implication in patient survival outcome. J. Oncol. 2020, 2020, 6768942. [Google Scholar] [CrossRef]
- Allen, F.; Maillard, I. Therapeutic targeting of Notch signaling: From cancer to inflammatory disorders. Front. Cell. Dev. Biol. 2021, 9, 649205. [Google Scholar] [CrossRef]
- Vinson, K.E.; George, D.C.; Fender, A.W.; Bertrand, F.E.; Sigounas, G. The Notch pathway in colorectal cancer. Int. J. Cancer 2016, 138, 1835–1842. [Google Scholar] [CrossRef]
- To, K.K.; Tong, C.W.; Wu, M.; Cho, W.C. MicroRNAs in the prognosis and therapy of colorectal cancer: From bench to bedside. World J. Gastroenterol. 2018, 24, 2949–2973. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.T.; Han, C.; Sun, Y.M.; Chen, T.Q.; Chen, Y.Q. Noncoding RNAs in cancer therapy resistance and targeted drug development. J. Hematol. Oncol. 2019, 12, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenetics 2019, 11, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Mollaei, H.; Safaralizadeh, R.; Rostami, Z. MicroRNA replacement therapy in cancer. J. Cell. Physiol. 2019, 234, 12369–12384. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Eberhardt, M.; Schmitz, U.; Vera, J. Systems biology-based investigation of cooperating microRNAs as monotherapy or adjuvant therapy in cancer. Nucleic Acids Res. 2019, 47, 7753–7766. [Google Scholar] [CrossRef]
- Liu, B.; Shyr, Y.; Cai, J.; Liu, Q. Interplay between miRNAs and host genes and their role in cancer. Brief. Funct. Genom. 2018, 18, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Gebert, L.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell. Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Glassy, M.C.; Abak, A.; Hussen, B.M.; Niazi, V.; Taheri, M. The interaction between miRNAs/lncRNAs and Notch pathway in human disorders. Biomed. Pharmacother. 2021, 138, 111496–111508. [Google Scholar] [CrossRef]
- Shen, B. A new golden age of natural products drug discovery. Cell 2015, 163, 1297–1300. [Google Scholar] [CrossRef] [Green Version]
- Alnuqaydan, A.M. Targeting micro-RNAs by natural products: A novel future therapeutic strategy to combat cancer. Am. J. Transl. Res. 2020, 12, 3531–3556. [Google Scholar]
- Mosca, L.; Vitiello, F.; Pagano, M.; Coppola, A.; Veglia Tranchese, R.; Grillo, R.; Cacciapuoti, G.; Porcelli, M. S-adenosylmethionine, a promising antitumor agent in oral and laryngeal cancer. Appl. Sci. 2022, 12, 1746. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Calvisi, D.F.; Feo, C.F.; Feo, F. S-adenosylmethionine: From the discovery of its inhibition of tumorigenesis to its use as a therapeutic agent. Cells 2022, 11, 409. [Google Scholar] [CrossRef] [PubMed]
- Ilisso, C.P.; Sapio, L.; Delle Cave, D.; Illiano, M.; Spina, A.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine affects ERK1/2 and Stat3 pathways and induces apoptosis in osteosarcoma cells. J. Cell. Physiol. 2016, 231, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Cheishvili, D.; Arakelian, A.; Tanvir, I.; Khan, H.A.; Pépin, A.S.; Szyf, M.; Rabbani, S.A. Methyl donor S-adenosylmethionine (SAM) supplementation attenuates breast cancer growth, invasion and metastasis in vivo; therapeutic and chemopreventive applications. Oncotarget 2018, 9, 5169–5183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Liang, X.; Huang, H.; Zhang, G.; Liu, T.; Zhang, J.; Chen, Z.; Zhang, Z.; Chen, Y. S-adenosylmethionine affects cell cycle pathways and suppresses proliferation in liver cells. J. Cancer 2019, 10, 4368–4379. [Google Scholar] [CrossRef] [Green Version]
- Mosca, L.; Minopoli, M.; Pagano, M.; Vitiello, F.; Carriero, M.V.; Cacciapuoti, G.; Porcelli, M. Effects of S-adenosyl-l-methionine on the invasion and migration of head and neck squamous cancer cells and analysis of the underlying mechanisms. Int. J. Oncol. 2020, 56, 1212–1224. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, N.; Arakelian, A.; Muller, W.J.; Szyf, M.; Rabbani, S.A. An enhanced chemopreventive effect of methyl donor S-adenosylmethionine in combination with 25-hydroxyvitamin D in blocking mammary tumor growth and metastasis. Bone Res. 2020, 8, 28–31. [Google Scholar] [CrossRef]
- Mahmood, N.; Arakelian, A.; Cheishvili, D.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine in combination with decitabine shows enhanced anti-cancer effects in repressing breast cancer growth and metastasis. J. Cell. Mol. Med. 2020, 24, 10322–10337. [Google Scholar] [CrossRef]
- Mosca, L.; Pagano, M.; Ilisso, C.P.; Delle Cave, D.; Desiderio, V.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. AdoMet triggers apoptosis in head and neck squamous cancer by inducing ER-stress and potentiates cell sensitivity to cisplatin. J. Cell. Physiol. 2019, 234, 13277–13291. [Google Scholar] [CrossRef]
- Cave, D.D.; Desiderio, V.; Mosca, L.; Ilisso, C.P.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine-mediated apoptosis is potentiated by autophagy inhibition induced by chloroquine in human breast cancer cells. J. Cell. Physiol. 2017, 233, 1370–1383. [Google Scholar] [CrossRef]
- Mosca, L.; Vitiello, F.; Coppola, A.; Borzacchiello, L.; Ilisso, C.P.; Pagano, M.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. Therapeutic potential of the natural compound S-adenosylmethionine as a chemoprotective synergistic agent in breast, and head and neck cancer treatment: Current status of research. Int. J. Mol. Sci. 2020, 21, 8547. [Google Scholar] [CrossRef] [PubMed]
- Ilisso, C.P.; Delle Cave, D.; Mosca, L.; Pagano, M.; Coppola, A.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. Adenosylmethionine regulates apoptosis and autophagy in MCF-7 breast cancer cells through the modulation of specific microRNAs. Cancer Cell Int. 2018, 18, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Coppola, A.; Ilisso, C.P.; Stellavato, A.; Schiraldi, C.; Caraglia, M.; Mosca, L.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine inhibits cell growth and migration of triple negative breast cancer cells through upregulating MiRNA-34c and MiRNA-449a. Int. J. Mol. Sci. 2020, 22, 286. [Google Scholar] [CrossRef]
- Pagano, M.; Mosca, L.; Vitiello, F.; Ilisso, C.P.; Coppola, A.; Borzacchiello, L.; Mele, L.; Caruso, F.P.; Ceccarelli, M.; Caraglia, M.; et al. Mi-RNA-888-5p is involved in S-adenosylmethionine antitumor effects in laryngeal squamous cancer cells. Cancers 2020, 12, 3665. [Google Scholar] [CrossRef]
- Mosca, L.; Vitiello, F.; Borzacchiello, L.; Coppola, A.; Tranchese, R.V.; Pagano, M.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. Mutual correlation between non-coding RNA and S-adenosylmethionine in human cancer: Roles and therapeutic opportunities. Cancers 2021, 13, 3264. [Google Scholar] [CrossRef] [PubMed]
- Mosca, L.; Pagano, M.; Pecoraro, A.; Borzacchiello, L.; Mele, L.; Cacciapuoti, G.; Porcelli, M.; Russo, G.; Russo, A. S-adenosyl-L-methionine overcomes uL3-mediated drug resistance in p53 deleted colon cancercells. Int. J. Mol. Sci. 2020, 22, 103. [Google Scholar] [CrossRef]
- Mosca, L.; Pagano, M.; Borzacchiello, L.; Mele, L.; Russo, A.; Russo, G.; Cacciapuoti, G.; Porcelli, M. S-adenosylmethionine increases the sensitivity of human colorectal cancer cells to 5-fluorouracil by inhibiting P-glycoprotein expression and NF-B activation. Int. J. Mol. Sci. 2021, 22, 9286. [Google Scholar] [CrossRef]
- Zsigrai, S.; Kalmár, A.; Nagy, Z.B.; Barták, B.K.; Valcz, G.; Szigeti, K.A.; Galamb, O.; Dankó, T.; Sebestyén, A.; Barna, G.; et al. S-adenosylmethionine treatment of colorectal cancer cell lines alters DNA methylation, DNA repair and tumor progression-related gene expression. Cells 2020, 9, 1864. [Google Scholar] [CrossRef]
- Zhang, N.; Hu, X.; Du, Y.; Du, J. The role of miRNAs in colorectal cancer progression and chemoradiotherapy. Biomed. Pharmacother. 2021, 134, 111099–111109. [Google Scholar] [CrossRef]
- Niki, M.; Nakajima, K.; Ishikawa, D.; Nishida, J.; Ishifune, C.; Tsukumo, S.I.; Shimada, M.; Nagahiro, S.; Mitamura, Y.; Yasutomo, K. MicroRNA-449a deficiency promotes colon carcinogenesis. Sci. Rep. 2017, 7, 10696–10706. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, D.; Takasu, C.; Kashihara, H.; Nishi, M.; Tokunaga, T.; Higashijima, J.; Yoshikawa, K.; Yasutomo, K.; Shimada, M. The significance of MicroRNA-449a and its potential target HDAC1 in patients with colorectal cancer. Anticancer Res. 2019, 39, 2855–2860. [Google Scholar] [CrossRef] [PubMed]
- Veena, M.S.; Raychaudhuri, S.; Basak, S.K.; Venkatesan, N.; Kumar, P.; Biswas, R.; Chakrabarti, R.; Lu, J.; Su, T.; Gallagher-Jones, M.; et al. Dysregulation of hsa-miR-34a and hsa-miR-449a leads to overexpression of PACS-1 and loss of DNA damage response (DDR) in cervical cancer. J. Biol. Chem. 2020, 295, 17169–17186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. Exp. Clin. Cancer Res. 2019, 38, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Huang, B.; Li, Z.; Li, H.; Sun, L.; Zhang, Q.; Qiu, X.; Wang, E. MicroRNA-449a is downregulated in non-small cell lung cancer and inhibits migration and invasion by targeting c-Met. PLoS ONE 2013, 8, e64759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandbothe, M.; Buurman, R.; Reich, N.; Greiwe, L.; Vajen, B.; Gürlevik, E.; Schäffer, V.; Eilers, M.; Kühnel, F.; Vaquero, A.; et al. The microRNA-449 family inhibits TGF-β-mediated liver cancer cell migration by targeting SOX4. J. Hepatol. 2017, 66, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Salem, N.; Kamal, I.; Al-Maghrabi, J.; Abuzenadah, A.; Peer-Zada, A.A.; Qari, Y.; Al-Ahwal, M.; Al-Qahtani, M.; Buhmeida, A. High expression of matrix metalloproteinases: MMP-2 and MMP-9 predicts poor survival outcome in colorectal carcinoma. Future Oncol. 2016, 12, 323–331. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Strouhalova, K.; Přechová, M.; Gandalovičová, A.; Brábek, J.; Gregor, M.; Rosel, D. Vimentin intermediate filaments as potential target for cancer treatment. Cancers 2020, 12, 184. [Google Scholar] [CrossRef] [Green Version]
- Bian, J.; Dannappel, M.; Wan, C.; Firestein, R. Transcriptional regulation of Wnt/β-catenin pathway in colorectal cancer. Cells 2020, 9, 2125. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.; Bartel, D.P. Predicting effective microRNA target sites in mammalianm RNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Xiu, M.X.; Liu, Y.M. The role of oncogenic Notch2 signaling in cancer: A novel therapeutic target. Am. J. Cancer Res. 2019, 9, 837–854. [Google Scholar] [PubMed]
- Niu, L.; Yang, W.; Duan, L.; Wang, X.; Li, Y.; Xu, C.; Liu, C.; Zhang, Y.; Zhou, W.; Liu, J.; et al. Biological implications and clinical potential of metastasis-related miRNA in colorectal cancer. Mol. Ther.-Nucleic Acids 2020, 23, 42–54. [Google Scholar] [CrossRef]
- Lu, S.C. S-adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in cell growth, apoptosis and liver cancer. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. 1), S73–S77. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Zhang, Z.; Pan, L.; Zhang, Y. MicroRNA-34/449 family and viral infections. Virus Res. 2019, 260, 1–6. [Google Scholar] [CrossRef]
- Li, W.J.; Wang, Y.; Liu, R.; Kasinski, A.L.; Shen, H.; Slack, F.J.; Tang, D.G. MicroRNA-34a: Potent tumor suppressor, cancer stem cell inhibitor, and potential anticancer therapeutic. Front. Cell Dev. Biol. 2021, 9, 640587–640607. [Google Scholar] [CrossRef]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Rokavec, M.; Li, H.; Jiang, L.; Hermeking, H. The p53/miR-34 axis in development and disease. J. Mol. Cell Biol. 2014, 6, 214–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, N.; Lin, C.P.; Ribeiro, M.C.; Biton, A.; Lai, G.; He, X.; Bu, P.; Vogel, H.; Jablons, D.M.; Keller, A.C.; et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014, 28, 438–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyota, M.; Suzuki, H.; Sasaki, Y.; Maruyama, R.; Imai, K.; Shinomura, Y.; Tokino, T. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008, 68, 4123–4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Liu, S.; Chen, P.; Fu, D.; Hou, Y.; Hu, J.; Liu, Z.; Jiang, Y.; Cao, X.; Cheng, C.; et al. miR-449a inhibits colorectal cancer progression by targeting SATB2. Oncotarget 2016, 8, 100975–100988. [Google Scholar] [CrossRef] [Green Version]
- Lan, S.H.; Lin, S.C.; Wang, W.C.; Yang, Y.C.; Lee, J.C.; Lin, P.W.; Chu, M.L.; Lan, K.Y.; Zuchini, R.; Liu, H.S.; et al. Autophagy upregulates miR-449a expression to suppress progression of colorectal cancer. Front. Oncol. 2021, 11, 738144–738156. [Google Scholar] [CrossRef]
- Herszényi, L.; Hritz, I.; Lakatos, G.; Varga, M.Z.; Tulassay, Z. The behavior of matrix metalloproteinases and their inhibitors in colorectal cancer. Int. J. Mol. Sci. 2012, 13, 13240–13263. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, G.; Lin, C.; Lin, P.; Chen, H.; He, R.; Huang, Y.; Yang, S.; Ye, J. Vimentin affects colorectal cancer proliferation, invasion, and migration via regulated by activator protein 1. J. Cell. Phys. 2021, 236, 7591–7604. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Ye, K.; Xu, C.; Hui, T. MiR-34b inhibits the proliferation and promotes apoptosis in colon cancer cells by targeting Wnt/β-catenin signaling pathway. Biosci. Rep. 2019, 39, BSR20191799. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Xu, X.; Chen, D.; Zhao, F.; Wang, W. Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed. Pharmacother. 2019, 110, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.K.; Xu, Z.Y.; Yuan, L.; Mo, S.; Xu, B.; Cheng, X.D.; Qin, J.J. Targeting β-catenin signaling by natural products for cancer prevention and therapy. Front. Pharmacol. 2020, 11, 984. [Google Scholar] [CrossRef] [PubMed]
- Majidinia, M.; Darband, S.G.; Kaviani, M.; Nabavi, S.M.; Jahanban-Esfahlan, R.; Yousefi, B. Cross-regulation between Notch signaling pathway and miRNA machinery in cancer. DNA Repair 2018, 66–67, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Lin, R.; Jin, H.; Si, L.; Jian, W.; Yu, Q.; Yang, S. MicroRNA-34 suppresses proliferation of human ovarian cancer cells by triggering autophagy and apoptosis and inhibits cell invasion by targeting Notch 1. Biochimie 2019, 160, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ai, F.; Li, X.; Tian, L.; Wang, X.; Shen, S.; Liu, F. MicroRNA-34a suppresses colorectal cancer metastasis by regulating Notch signaling. Oncol. Lett. 2017, 14, 2325–2333. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Duan, Q.; Ahmad, A.; Bao, B.; Banerjee, S.; Shi, Y.; Ma, J.; Geng, J.; Chen, Z.; Rahman, K.M.; et al. Genistein inhibits cell growth and induces apoptosis through up-regulation of miR-34a in pancreatic cancer cells. Curr. Drug Targets 2012, 13, 1750–1756. [Google Scholar] [CrossRef]
- Bauer, S.; Ratz, L.; Heckmann-Nötzel, D.; Kaczorowski, A.; Hohenfellner, M.; Kristiansen, G.; Duensing, S.; Altevogt, P.; Klauck, S.M.; Sültmann, H. miR-449a repression leads to enhanced Notch signaling in TMPRSS2: ERG fusion positive prostate cancer cells. Cancers 2021, 13, 964. [Google Scholar] [CrossRef]
- Mercey, O.; Popa, A.; Cavard, A.; Paquet, A.; Chevalier, B.; Pons, N.; Magnone, V.; Zangari, J.; Brest, P.; Zaragosi, L.E.; et al. Characterizing isomiR variants within the microRNA-34/449 family. FEBS Lett. 2017, 591, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Stasio, D.D.; Mosca, L.; Lucchese, A.; Cave, D.D.; Kawasaki, H.; Lombardi, A.; Porcelli, M.; Caraglia, M. Salivary mir-27b expression in oral lichen planus patients: A series of cases and a narrative review of literature. Curr. Top. Med. Chem. 2019, 19, 2816–2823. [Google Scholar] [CrossRef]
- Mele, L.; Del Vecchio, V.; Marampon, F.; Regad, T.; Wagner, S.; Mosca, L.; Bimonte, S.; Giudice, A.; Liccardo, D.; Prisco, C.; et al. β2-AR blockade potentiates MEK1/2 inhibitor effect on HNSCC by regulating the Nrf2-mediated defense mechanism. Cell Death Dis. 2020, 11, 850–863. [Google Scholar] [CrossRef]
- Minici, C.; Mosca, L.; Ilisso, C.P.; Cacciapuoti, G.; Porcelli, M.; Degano, M. Structures of catalytic cycle intermediates of the Pyrococcus furiosus methionine adenosyltransferase demonstrate negative cooperativity in the archaeal orthologues. J. Struct. Biol. 2020, 210, 107462–107472. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Parsons, C.; Slack, F.J. The tumor-suppressive and potential therapeutic functions of miR-34a in epithelial carcinomas. Expert Opin. Ther. Targets 2016, 20, 737–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiesel, V.A.; Stan, S.D. Modulation of Notch signaling pathway by bioactive dietary agents. Int. J. Mol. Sci. 2022, 23, 3532. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borzacchiello, L.; Veglia Tranchese, R.; Grillo, R.; Arpino, R.; Mosca, L.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine Inhibits Colorectal Cancer Cell Migration through Mirna-Mediated Targeting of Notch Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 7673. https://doi.org/10.3390/ijms23147673
Borzacchiello L, Veglia Tranchese R, Grillo R, Arpino R, Mosca L, Cacciapuoti G, Porcelli M. S-Adenosylmethionine Inhibits Colorectal Cancer Cell Migration through Mirna-Mediated Targeting of Notch Signaling Pathway. International Journal of Molecular Sciences. 2022; 23(14):7673. https://doi.org/10.3390/ijms23147673
Chicago/Turabian StyleBorzacchiello, Luigi, Roberta Veglia Tranchese, Roberta Grillo, Roberta Arpino, Laura Mosca, Giovanna Cacciapuoti, and Marina Porcelli. 2022. "S-Adenosylmethionine Inhibits Colorectal Cancer Cell Migration through Mirna-Mediated Targeting of Notch Signaling Pathway" International Journal of Molecular Sciences 23, no. 14: 7673. https://doi.org/10.3390/ijms23147673