
Recent Advances Regarding the Molecular Mechanisms of Triterpenic Acids: A Review (Part I)

 , , ,
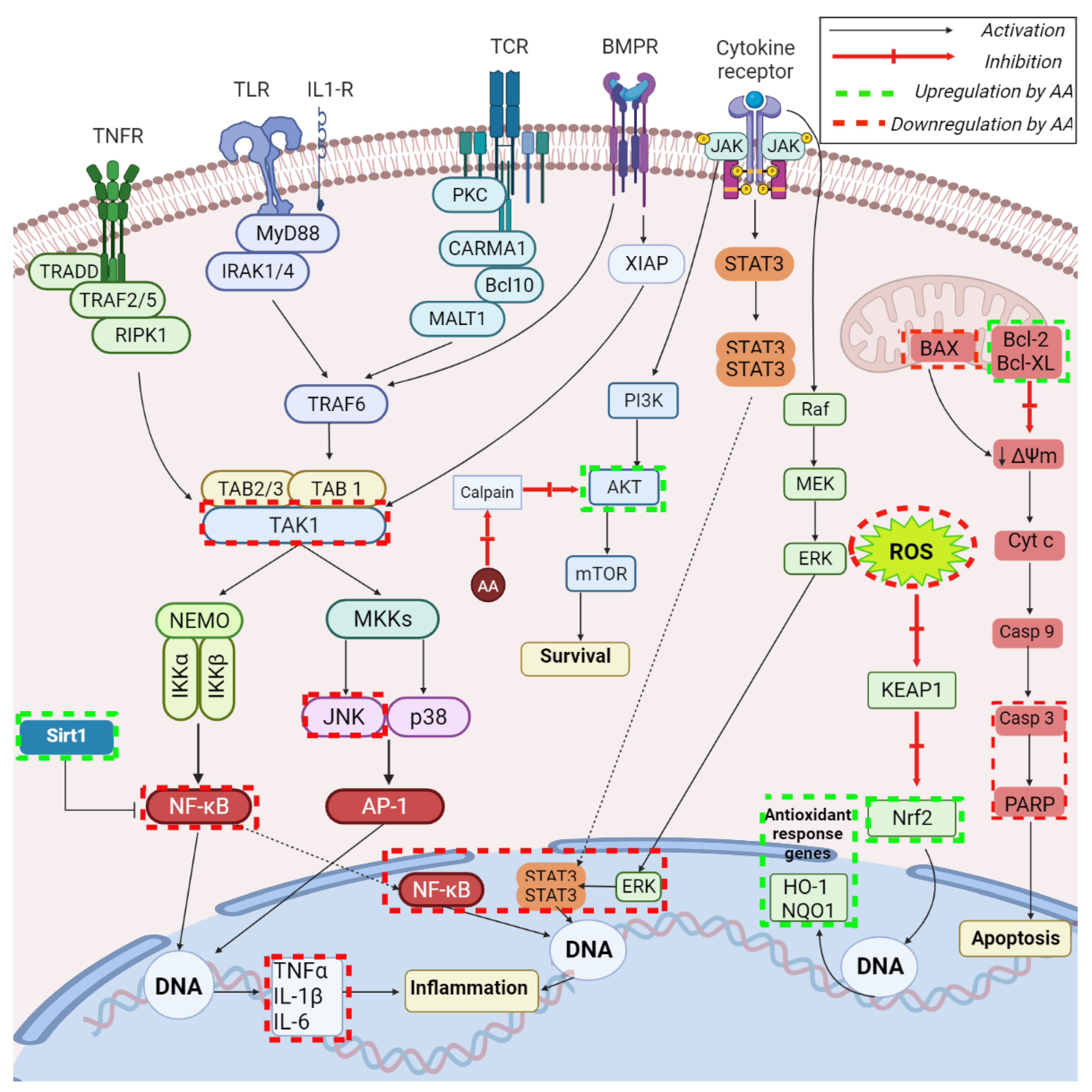
, , , .jpg) ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Asiatic Acid
2.1. Anticancer Activity
2.2. Antidiabetic Activity
2.3. Antiinflammatory Activity
2.4. Neuroprotective Activity
2.5. Cardioprotective Activity
2.6. Hepatoprotective Activity
2.7. Other Biological Activities
3. Oleanolic Acid
3.1. Anticancer Activity
3.2. Antidiabetic Activity
3.3. Antiinfectious Activity
3.4. Lipidemic Activity
3.5. Cardioprotective Activity
3.6. Renal Activity
3.7. Anti-Osteoporotic Activity
3.8. Antiinflammatory Activity
3.9. Neuroprotective Activity
3.10. Other Biological Activities
4. Ursolic Acid
4.1. Anticancer Activity
4.2. Antidiabetic Activity
4.3. Antiinfectious Activity
4.4. Anti-Atherosclerotic Activity
4.5. Neuroprotective Activity
4.6. Antiinflammatory Activity
4.7. Other Biological Activities
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- D’yakonov, V.A.; Dzhemileva, L.U.; Dzhemilev, U.M. Advances in the Chemistry of Natural and Semisynthetic Topoisomerase I/II Inhibitors. Stud. Nat. Prod. Chem. 2017, 54, 21–86. [Google Scholar]
- Ghosh, G.; Subudhi, B.; Mishra, S.K. Antihyperglycemic activity of root bark of Polyalthia longifolia var. pendula and aerial parts of Sida rhombifolia Linn. and its relationship with antioxidant property. Asian J. Chem. 2011, 23, 141–144. [Google Scholar]
- Binaschi, M.; Zunino, F.; Capranico, G. Mechanism of action of DNA topoisomerase inhibitors. Stem Cells 1995, 13, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Aminfar, Z.; Rabiei, B.; Tohidfar, M.; Mirjalili, M.H. Identification of key genes involved in the biosynthesis of triterpenic acids in the mint family. Sci. Rep. 2019, 9, 15826. [Google Scholar] [CrossRef]
- Järvinen, T.A.H.; Liu, E.T. Topoisomerase II α gene (TOP2A) amplification and deletion in cancer—More common than anticipated. Cytopathology 2003, 14, 309–313. [Google Scholar] [CrossRef]
- Burden, D.A.; Osheroff, N. Mechanism of action of eukaryotic topoisomerase II and drugs targeted to the enzyme. Biochim. Biophys. Acta Gene Struct. Expr. 1998, 1400, 139–154. [Google Scholar] [CrossRef]
- Ulici, A.; Milan, A.; Mioc, M.; Ghiulai, R.; Racoviceanu, R.; Șoica, C. Ring-Modified Triterpene Derivatives as Potential Pharmacological Active Compounds. Timis. Med. J. 2021, 2020, 7. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Davletshin, E.V.; Tukhbatullin, A.A.; D’yakonov, V.A.; Yunusbaeva, M.M.; Dzhemileva, L.U.; Dzhemilev, U.M. Pentacyclic triterpene acid conjugated with mitochondria-targeting cation F16: Synthesis and evaluation of cytotoxic activities. Med. Chem. Res. 2021, 30, 940–951. [Google Scholar] [CrossRef]
- Graykowski, D.R.; Wang, Y.-Z.; Upadhyay, A.; Savas, J.N. The Dichotomous Role of Extracellular Vesicles in the Central Nervous System. iScience 2020, 23, 101456. [Google Scholar] [CrossRef]
- Mechanism matters. Nat. Med. 2010, 16, 347. [CrossRef]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Gregori-Puigjané, E.; Setola, V.; Hert, J.; Crews, B.A.; Irwin, J.J.; Lounkine, E.; Marnett, L.; Roth, B.L.; Shoichet, B.K. Identifying mechanism-of-action targets for drugs and probes. Proc. Natl. Acad. Sci. USA 2012, 109, 11178–11183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clegg, L.E.; Mac Gabhann, F. Molecular mechanism matters: Benefits of mechanistic computational models for drug development. Pharmacol. Res. 2015, 99, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najmi, A.; Javed, S.A.; Al Bratty, M.; Alhazmi, H.A. Modern Approaches in the Discovery and Development of Plant-Based Natural Products and Their Analogues as Potential Therapeutic Agents. Molecules 2022, 27, 349. [Google Scholar] [CrossRef]
- Laszczyk, M. Pentacyclic Triterpenes of the Lupane, Oleanane and Ursane Group as Tools in Cancer Therapy. Planta Med. 2009, 75, 1549–1560. [Google Scholar] [CrossRef] [Green Version]
- Soica, C.; Voicu, M.; Ghiulai, R.; Dehelean, C.; Racoviceanu, R.; Trandafirescu, C.; Rosca, O.-J.; Nistor, G.; Mioc, M.; Mioc, A. Natural Compounds in Sex Hormone-Dependent Cancers: The Role of Triterpenes as Therapeutic Agents. Front. Endocrinol. 2021, 11, 612396. [Google Scholar] [CrossRef]
- Lei, P.; Zhang, W.; Men, X. Review on anti-tumor effect of triterpene acid compounds. J. Cancer Res. Ther. 2014, 10, 14. [Google Scholar] [CrossRef]
- Chudzik, M.; Korzonek-Szlacheta, I.; Król, W. Triterpenes as Potentially Cytotoxic Compounds. Molecules 2015, 20, 1610–1625. [Google Scholar] [CrossRef] [Green Version]
- Cháirez-Ramírez, M.H.; Moreno-Jiménez, M.R.; González-Laredo, R.F.; Gallegos-Infante, J.A.; Rocha-Guzmán, N.E. Lupane-type triterpenes and their anti-cancer activities against most common malignant tumors: A review. EXCLI J. 2016, 15, 758–771. [Google Scholar] [CrossRef]
- Singh, B.; Sanjeev, G.; Navgeet, K. Triterpenes in cancer: Significance and their influence. Mol. Biol. Rep. 2016, 43, 881–896. [Google Scholar] [CrossRef]
- Jang, E.; Lee, J.-H. Promising Anticancer Activities of Alismatis rhizome and Its Triterpenes via p38 and PI3K/Akt/mTOR Signaling Pathways. Nutrients 2021, 13, 2455. [Google Scholar] [CrossRef] [PubMed]
- Barreto Vianna, D.R.; Gotardi, J.; Baggio Gnoatto, S.C.; Pilger, D.A. Natural and Semisynthetic Pentacyclic Triterpenes for Chronic Myeloid Leukemia Therapy: Reality, Challenges and Perspectives. ChemMedChem 2021, 16, 1835–1860. [Google Scholar] [CrossRef] [PubMed]
- Ríos, J.-L. Effects of triterpenes on the immune system. J. Ethnopharmacol. 2010, 128, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Renda, G.; Gökkaya, İ.; Şöhretoğlu, D. Immunomodulatory properties of triterpenes. Phytochem. Rev. 2022, 21, 537–563. [Google Scholar] [CrossRef] [PubMed]
- Nazaruk, J.; Borzym-Kluczyk, M. The role of triterpenes in the management of diabetes mellitus and its complications. Phytochem. Rev. 2015, 14, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Sharma, H.; Kumar, P.; Deshmukh, R.R.; Bishayee, A.; Kumar, S. Pentacyclic triterpenes: New tools to fight metabolic syndrome. Phytomedicine 2018, 50, 166–177. [Google Scholar] [CrossRef]
- Castellano, J.M.; Guinda, A.; Delgado, T.; Rada, M.; Cayuela, J.A. Biochemical Basis of the Antidiabetic Activity of Oleanolic Acid and Related Pentacyclic Triterpenes. Diabetes 2013, 62, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Putta, S.; Sastry Yarla, N.; Kumar Kilari, E.; Surekha, C.; Aliev, G.; Basavaraju Divakara, M.; Sridhar Santosh, M.; Ramu, R.; Zameer, F.; Prasad MN, N.; et al. Therapeutic Potentials of Triterpenes in Diabetes and its Associated Complications. Curr. Top. Med. Chem. 2016, 16, 2532–2542. [Google Scholar] [CrossRef]
- Mabhida, S.E.; Dludla, P.V.; Johnson, R.; Ndlovu, M.; Louw, J.; Opoku, A.R.; Mosa, R.A. Protective effect of triterpenes against diabetes-induced β-cell damage: An overview of in vitro and in vivo studies. Pharmacol. Res. 2018, 137, 179–192. [Google Scholar] [CrossRef]
- Oboh, M.; Govender, L.; Siwela, M.; Mkhwanazi, B.N. Anti-Diabetic Potential of Plant-Based Pentacyclic Triterpene Derivatives: Progress Made to Improve Efficacy and Bioavailability. Molecules 2021, 26, 7243. [Google Scholar] [CrossRef]
- De Souza Miranda, R.; da Silva Mascarenhas de Jesus, B.; Silva Luiz, S.R.; Viana, C.B.; Adão Malafaia, C.R.; de Souza Figueiredo, F.; Dos Santos Conceição Carvalho, T.; Silva, M.L.; Londero, V.S.; Costa-Silva, T.A.; et al. Antiinflammatory activity of natural triterpenes—An overview from 2006 to 2021. Phyther. Res. 2022, 36, 1459–1506. [Google Scholar] [CrossRef]
- Razali, N.N.M.; Ng, C.T.; Fong, L.Y. Cardiovascular Protective Effects of Centella asiatica and Its Triterpenes: A Review. Planta Med. 2019, 85, 1203–1215. [Google Scholar] [CrossRef] [Green Version]
- Lobine, D.; Ahmed, S.; Aschner, M.; Khan, H.; Mirzaei, H.; Mahomoodally, M.F. Antiurolithiatic effects of pentacyclic triterpenes: The distance traveled from therapeutic aspects. Drug Dev. Res. 2020, 81, 671–684. [Google Scholar] [CrossRef]
- Paduch, R.; Kandefer-Szerszen, M. Antitumor and Antiviral Activity of Pentacyclic Triterpenes. Mini. Rev. Org. Chem. 2014, 11, 262–268. [Google Scholar] [CrossRef]
- Agra, L.C.; Ferro, J.N.S.; Barbosa, F.T.; Barreto, E. Triterpenes with healing activity: A systematic review. J. Dermatol. Treat. 2015, 26, 465–470. [Google Scholar] [CrossRef]
- Ghiulai, R.; Roşca, O.J.; Antal, D.S.; Mioc, M.; Mioc, A.; Racoviceanu, R.; Macaşoi, I.; Olariu, T.; Dehelean, C.; Creţu, O.M.; et al. Tetracyclic and pentacyclic triterpenes with high therapeutic efficiency in wound healing approaches. Molecules 2020, 25, 5557. [Google Scholar] [CrossRef]
- Gohil, K.; Patel, J.; Gajjar, A. Pharmacological review on Centella asiatica: A potential herbal cure-all. Indian J. Pharm. Sci. 2010, 72, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.C.; Bhattamisra, S.K.; Chellappan, D.K.; Candasamy, M. Actions and Therapeutic Potential of Madecassoside and Other Major Constituents of Centella asiatica: A Review. Appl. Sci. 2021, 11, 8475. [Google Scholar] [CrossRef]
- Meeran, M.F.N.; Goyal, S.N.; Suchal, K.; Sharma, C.; Patil, C.R.; Ojha, S.K. Pharmacological properties, molecular mechanisms, and pharmaceutical development of asiatic acid: A pentacyclic triterpenoid of therapeutic promise. Front. Pharmacol. 2018, 9, 892. [Google Scholar] [CrossRef]
- Lv, J.; Sharma, A.; Zhang, T.; Wu, Y.; Ding, X. Pharmacological Review on Asiatic Acid and Its Derivatives: A Potential Compound. SLAS Technol. 2018, 23, 111–127. [Google Scholar] [CrossRef] [Green Version]
- Gou, X.; Bai, H.; Liu, L.; Chen, H.; Shi, Q.; Chang, L.; Ding, M.; Shi, Q.; Zhou, M.; Chen, W.; et al. Asiatic Acid Interferes with Invasion and Proliferation of Breast Cancer Cells by Inhibiting WAVE3 Activation through PI3K/AKT Signaling Pathway. BioMed Res. Int. 2020, 2020, 1874387. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wang, S.L.; Wang, Y.C.; Wu, Y.N.; Yu, X.; Zhao, W.Z.; Wang, J.H. High WAVE3 expression correlates with proliferation, migration and invasion in human ovarian cancer. Oncotarget 2017, 8, 41189–41201. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.; Chen, K.; Huang, J.; Chu, D.; Li, J.; Huang, K.; Ma, C. Asiatic acid inhibits angiogenesis and vascular permeability through the VEGF/VEGFR2 signaling pathway to inhibit the growth and metastasis of breast cancer in mice. Phyther. Res. 2021, 35, 6389–6400. [Google Scholar] [CrossRef]
- Zhu, Z.; Cui, L.; Yang, J.; Vong, C.T.; Hu, Y.; Xiao, J.; Chan, G.; He, Z.; Zhong, Z. Anticancer effects of asiatic acid against doxorubicin-resistant breast cancer cells via an AMPK-dependent pathway in vitro. Phytomedicine 2021, 92, 153737. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Liu, Y.-T.; Chuang, Y.-C.; Lo, Y.-S.; Lin, C.-C.; Hsi, Y.-T.; Hsieh, M.-J.; Chen, M.-K. Asiatic Acid, Extracted from Centella asiatica and Induces Apoptosis Pathway through the Phosphorylation p38 Mitogen-Activated Protein Kinase in Cisplatin-Resistant Nasopharyngeal Carcinoma Cells. Biomolecules 2020, 10, 184. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Geng, J.; Guo, W.; Gao, J.; Zhu, X. Asiatic acid inhibits lung cancer cell growth in vitro and in vivo by destroying mitochondria. Acta Pharm. Sin. B 2017, 7, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Kuma, A.; Matsui, M.; Mizushima, N. LC3, an Autophagosome Marker, Can be Incorporated into Protein Aggregates Independent of Autophagy: Caution in the Interpretation of LC3 Localization. Autophagy 2007, 3, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Ren, J.; Zhou, Q.; Yang, Q.; Li, B. Effect of asiatic acid on epithelial-mesenchymal transition of human alveolar epithelium A549 cells induced by TGF-β1. Oncol. Lett. 2019, 17, 4285–4292. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhou, B.P. Snail. Cell Adh. Migr. 2010, 4, 199–203. [Google Scholar] [CrossRef]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Lian, G.-Y.; Wang, Q.-M.; Tang, P.M.-K.; Zhou, S.; Huang, X.-R.; Lan, H.-Y. Combination of Asiatic Acid and Naringenin Modulates NK Cell Anti-cancer Immunity by Rebalancing Smad3/Smad7 Signaling. Mol. Ther. 2018, 26, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Lian, G.-Y.; Wang, Q.-M.; Mak, T.S.-K.; Huang, X.-R.; Yu, X.-Q.; Lan, H.-Y. Inhibition of tumor invasion and metastasis by targeting TGF-β-Smad-MMP2 pathway with Asiatic acid and Naringenin. Mol. Ther.-Oncolytics 2021, 20, 277–289. [Google Scholar] [CrossRef]
- Cheng, Q.; Liao, M.; Hu, H.; Li, H.; Wu, L. Asiatic Acid (AA) Sensitizes Multidrug-Resistant Human Lung Adenocarcinoma A549/DDP Cells to Cisplatin (DDP) via Downregulation of P-Glycoprotein (MDR1) and Its Targets. Cell. Physiol. Biochem. 2018, 47, 279–292. [Google Scholar] [CrossRef]
- Eliseeva, I.A.; Kim, E.R.; Guryanov, S.G.; Ovchinnikov, L.P.; Lyabin, D.N. Y-box-binding protein 1 (YB-1) and its functions. Biochem. 2011, 76, 1402–1433. [Google Scholar] [CrossRef]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Swargiary, G.; Mani, S. Molecular docking and simulation studies of phytocompounds derived from Centella asiatica and Andrographis paniculata against hexokinase II as mitocan agents. Mitochondrion 2021, 61, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Huang, J.; Ma, Y.; Chen, W.; Fan, Q.; Sun, X.; Shao, M.; Cai, H. Asiatic acid inhibits proliferation, migration and induces apoptosis by regulating Pdcd4 via the PI3K/Akt/mTOR/p70S6K signaling pathway in human colon carcinoma cells. Oncol. Lett. 2018, 15, 8223–8230. [Google Scholar] [CrossRef]
- Li, J.; Chen, K.; Huang, J.; Chu, D.; Tian, M.; Huang, K.; Ma, C. Asiatic Acid Induces Endoplasmic Reticulum Stress and Activates the Grp78/IRE1α/JNK and Calpain Pathways to Inhibit Tongue Cancer Growth. Front. Pharmacol. 2021, 12, 690612. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-X.; Yu, K.; Dong, J.; Zhao, L.; Liu, Z.; Zhang, Q.; Li, S.; Du, Y.; Cheng, H. Precise Prediction of Calpain Cleavage Sites and Their Aberrance Caused by Mutations in Cancer. Front. Genet. 2019, 10, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Yao, C.; Tang, J.; Liu, Y.; Huang, C.; Yu, S.; Wei, H.; Han, Y.; Chen, G. Enhanced GRP78 protein expression via the IRE1α/ASK1/p38 MAPK pathway during As2O3-induced endoplasmic reticulum stress in BEAS-2B cells. Toxicology 2021, 462, 152962. [Google Scholar] [CrossRef]
- Masola, B.; Oguntibeju, O.O.; Oyenihi, A.B. Centella asiatica ameliorates diabetes-induced stress in rat tissues via influences on antioxidants and inflammatory cytokines. Biomed. Pharmacother. 2018, 101, 447–457. [Google Scholar] [CrossRef]
- Smriti, K.; Pai, K.M.; Ravindranath, V.; Pentapati, K.C. Role of salivary malondialdehyde in assessment of oxidative stress among diabetics. J. Oral Biol. Craniofacial Res. 2016, 6, 42–45. [Google Scholar] [CrossRef] [Green Version]
- Swaroop, J.; Naidu, J.; Rajarajeswari, D. Association of TNF-α with insulin resistance in type 2 diabetes mellitus. Indian J. Med. Res. 2012, 135, 127. [Google Scholar] [CrossRef]
- Kartika, R.; Purnamasari, D.; Pradipta, S.; Larasati, R.A.; Wibowo, H. Impact of Low Interferon-γ and IL-10 Levels on TNF-α and IL-6 Production by PHA-Induced PBMCs in Type 2 Diabetes Mellitus. J. Inflamm. Res. 2020, 13, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Bankaitis, K.V.; Fingleton, B. Targeting IL4/IL4R for the treatment of epithelial cancer metastasis. Clin. Exp. Metastasis 2015, 32, 847–856. [Google Scholar] [CrossRef]
- Sun, W.; Xu, G.; Guo, X.; Luo, G.; Wu, L.; Hou, Y.; Guo, X.; Zhou, J.; Xu, T.; Qin, L.; et al. Protective effects of asiatic acid in a spontaneous type 2 diabetic mouse model. Mol. Med. Rep. 2017, 16, 1333–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Meng, F.; Chen, L.; Li, X.; Cen, L.-J.; Wen, Y.; Zhang, H.; Li, C. Inhibition of Methylglyoxal-Induced AGEs/RAGE Expression Contributes to Dermal Protection by N-Acetyl-L-Cysteine. Cell. Physiol. Biochem. 2017, 41, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Song, D.; Wang, M.; Chen, K.; Zhang, T. α7 nicotinic acetylcholine receptor agonist attenuates the cerebral injury in a rat model of cardiopulmonary bypass by activating the Akt/GSK3β pathway. Mol. Med. Rep. 2017, 16, 7979–7986. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-N.; Wu, C.-G.; Shi, B.-M.; Ke, Q.; Ding, Y. The protective effect of asiatic acid on podocytes in the kidney of diabetic rats. Am. J. Transl. Res. 2018, 10, 3733–3741. [Google Scholar] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Fu, P.; Zhang, Q.; Ma, X.; Jiang, P. Protective effects of Asiatic acid against pelvic inflammatory disease in rats. Exp. Ther. Med. 2019, 17, 4687–4692. [Google Scholar] [CrossRef] [Green Version]
- Yuyun, X.; Xi, C.; Qing, Y.; Lin, X.; Ke, R.; Bingwei, S. Asiatic acid attenuates lipopolysaccharide-induced injury by suppressing activation of the Notch signaling pathway. Oncotarget 2018, 9, 15036–15046. [Google Scholar] [CrossRef]
- Dong, S.-H.; Liu, Y.-W.; Wei, F.; Tan, H.-Z.; Han, Z.-D. Asiatic acid ameliorates pulmonary fibrosis induced by bleomycin (BLM) via suppressing pro-fibrotic and inflammatory signaling pathways. Biomed. Pharmacother. 2017, 89, 1297–1309. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Xia, X.; Dai, C.; Yu, H.; Huang, X.; Chen, A.; Tan, Y.; Wang, L. Asiatic acid prevents the development of interstitial lung disease in a hypochlorous acid-induced mouse model of scleroderma. Oncol. Lett. 2018, 15, 8711–8716. [Google Scholar] [CrossRef]
- Liu, N.; Fu, D.; Yang, J.; Liu, P.; Song, X.; Wang, X.; Li, R.; Fu, Z.; Chen, J.; Gong, X.; et al. Asiatic acid attenuates hypertrophic and fibrotic differentiation of articular chondrocytes via AMPK/PI3K/AKT signaling pathway. Arthritis Res. Ther. 2020, 22, 112. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, G.; Hu, Z.; Tang, S.; Xu, J.; Shang, P.; Tang, Q.; Liu, H. Asiatic acid ameliorates obesity-related osteoarthritis by inhibiting myeloid differentiation protein-2. Food Funct. 2020, 11, 5513–5524. [Google Scholar] [CrossRef]
- Park, J.-H.; Seo, Y.H.; Jang, J.-H.; Jeong, C.-H.; Lee, S.; Park, B. Asiatic acid attenuates methamphetamine-induced neuroinflammation and neurotoxicity through blocking of NF-kB/STAT3/ERK and mitochondria-mediated apoptosis pathway. J. Neuroinflamm. 2017, 14, 240. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.-Y.; Sun, J.; Cong, Q.-J.; Chen, W.-X.; Ahsan, H.M.; Gao, J.; Qian, J.-J. Asiatic Acid Protects Dopaminergic Neurons from Neuroinflammation by Suppressing Mitochondrial ROS Production. Biomol. Ther. 2019, 27, 442–449. [Google Scholar] [CrossRef]
- Qian, Y.; Xin, Z.; Lv, Y.; Wang, Z.; Zuo, L.; Huang, X.; Li, Y.; Xin, H.-B. Asiatic acid suppresses neuroinflammation in BV2 microglia via modulation of the Sirt1/NF-κB signaling pathway. Food Funct. 2018, 9, 1048–1057. [Google Scholar] [CrossRef]
- Ding, H.; Xiong, Y.; Sun, J.; Chen, C.; Gao, J.; Xu, H. Asiatic Acid Prevents Oxidative Stress and Apoptosis by Inhibiting the Translocation of α-Synuclein Into Mitochondria. Front. Neurosci. 2018, 12, 431. [Google Scholar] [CrossRef]
- Lu, C.W.; Lin, T.Y.; Wang, S.J.; Huang, S.K. Asiatic acid, an active substance of Centella asiatica, presynaptically depresses glutamate release in the rat hippocampus. Eur. J. Pharmacol. 2019, 865, 172781. [Google Scholar] [CrossRef]
- Lu, C.-W.; Lin, T.-Y.; Pan, T.-L.; Wang, P.-W.; Chiu, K.-M.; Lee, M.-Y.; Wang, S.-J. Asiatic Acid Prevents Cognitive Deficits by Inhibiting Calpain Activation and Preserving Synaptic and Mitochondrial Function in Rats with Kainic Acid-Induced Seizure. Biomedicines 2021, 9, 284. [Google Scholar] [CrossRef]
- Welbat, J.; Chaisawang, P.; Pannangrong, W.; Wigmore, P. Neuroprotective Properties of Asiatic Acid against 5-Fluorouracil Chemotherapy in the Hippocampus in an Adult Rat Model. Nutrients 2018, 10, 1053. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Mong, M.; Yang, Y.; Yin, M. Asiatic acid and maslinic acid attenuated kainic acid-induced seizure through decreasing hippocampal inflammatory and oxidative stress. Epilepsy Res. 2018, 139, 28–34. [Google Scholar] [CrossRef]
- Wong, J.H.; Reza, F.; Muthuraju, S.; Chuang, H.G.; Zhang, J.; Senik, M.H.; Yusof, S.R.; Mohamad, H.; Muhammad, T.S.T.; Ismail, N.H.; et al. Acute application of Centella asiatica extract enhanced AMPAR-mediated postsynaptic currents in rat entorhinal cortex. J. Integr. Neurosci. 2020, 19, 217–227. [Google Scholar] [CrossRef]
- Arora, R.; Kumar, R.; Agarwal, A.; Reeta, K.H.; Gupta, Y.K. Comparison of three different extracts of Centella asiatica for anti-amnesic, antioxidant and anticholinergic activities: In vitro and in vivo study. Biomed. Pharmacother. 2018, 105, 1344–1352. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Zhao, P.; Cheng, J.; Gong, W.; Zhang, J. Asiatic acid exerts neuroprotective effect against hypoxicischemic brain injury in neonatal rats via inhibition of oxidative damage. Trop. J. Pharm. Res. 2021, 20, 1903–1908. [Google Scholar] [CrossRef]
- Han, F.; Yan, N.; Huo, J.; Chen, X.; Fei, Z.; Li, X. Asiatic acid attenuates traumatic brain injury via upregulating Nrf2 and HO-1 expression. Int. J. Clin. Exp. Med. 2018, 11, 360–366. [Google Scholar]
- Huang, W.; Gao, F.; Hu, F.; Huang, J.; Wang, M.; Xu, P.; Zhang, R.; Chen, J.; Sun, X.; Zhang, S.; et al. Asiatic Acid Prevents Retinal Ganglion Cell Apoptosis in a Rat Model of Glaucoma. Front. Neurosci. 2018, 12, 489. [Google Scholar] [CrossRef] [Green Version]
- Yi, C.; Si, L.; Xu, J.; Yang, J.; Wang, Q.; Wang, X. Effect and mechanism of asiatic acid on autophagy in myocardial ischemia-reperfusion injury in vivo and in vitro. Exp. Ther. Med. 2020, 20, 1. [Google Scholar] [CrossRef]
- Yi, C.; Song, M.; Sun, L.; Si, L.; Yu, D.; Li, B.; Lu, P.; Wang, W.; Wang, X. Asiatic Acid Alleviates Myocardial Ischemia-Reperfusion Injury by Inhibiting the ROS-Mediated Mitochondria-Dependent Apoptosis Pathway. Oxid. Med. Cell Longev. 2022, 2022, 1–16. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, Z.; Quan, M.; Lv, Y.; Li, Y.; Xin, H.-B.; Qian, Y. Asiatic acid protests against myocardial ischemia/reperfusion injury via modulation of glycometabolism in rat cardiomyocyte. Drug Des. Devel. Ther. 2018, 12, 3573–3582. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Tian, X.; Ruan, Y.; Xing, J.; Meng, Z. Asiatic acid alleviates Ang-II induced cardiac hypertrophy and fibrosis via miR-126/PIK3R2 signaling. Nutr. Metab. 2021, 18, 71. [Google Scholar] [CrossRef]
- Meng, Z.; Li, H.; Si, C.; Liu, Y.; Teng, S. Asiatic acid inhibits cardiac fibrosis throughNrf2/HO-1 and TGF-β1/Smads signaling pathways in spontaneous hypertension rats. Int. Immunopharmacol. 2019, 74, 105712. [Google Scholar] [CrossRef]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights Into Pathogenesis and Treatment. Front. Pharmacol. 2021, 12, 787541. [Google Scholar] [CrossRef]
- Fong, L.Y.; Ng, C.T.; Yong, Y.K.; Hakim, M.N.; Ahmad, Z. Asiatic acid stabilizes cytoskeletal proteins and prevents TNF-α-induced disorganization of cell-cell junctions in human aortic endothelial cells. Vascul. Pharmacol. 2019, 117, 15–26. [Google Scholar] [CrossRef]
- Maneesai, P.; Bunbupha, S.; Kukongviriyapan, U.; Senggunprai, L.; Kukongviriyapan, V.; Prachaney, P.; Pakdeechote, P. Effect of asiatic acid on the Ang II-AT1R-NADPH oxidase-NF-κB pathway in renovascular hypertensive rats. Naunyn-Schmiedebergs Arch. Pharmacol. 2017, 390, 1073–1083. [Google Scholar] [CrossRef]
- Lu, Y.; Kan, H.; Wang, Y.; Wang, D.; Wang, X.; Gao, J.; Zhu, L. Asiatic acid ameliorates hepatic ischemia/reperfusion injury in rats via mitochondria-targeted protective mechanism. Toxicol. Appl. Pharmacol. 2018, 338, 214–223. [Google Scholar] [CrossRef]
- Qi, Z.; Ci, X.; Huang, J.; Liu, Q.; Yu, Q.; Zhou, J.; Deng, X. Asiatic acid enhances Nrf2 signaling to protect HepG2 cells from oxidative damage through Akt and ERK activation. Biomed. Pharmacother. 2017, 88, 252–259. [Google Scholar] [CrossRef]
- Xu, Y.; Yao, J.; Zou, C.; Zhang, H.; Zhang, S.; Liu, J.; Ma, G.; Jiang, P.; Zhang, W. Asiatic acid protects against hepatic ischemia/reperfusion injury by inactivation of Kupffer cells via PPARγ/NLRP3 inflammasome signaling pathway. Oncotarget 2017, 8, 86339–86355. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Qi, Z.; Wang, S.; Feng, H.; Deng, X.; Ci, X. Asiatic Acid Exhibits Anti-inflammatory and Antioxidant Activities against Lipopolysaccharide and d-Galactosamine-Induced Fulminant Hepatic Failure. Front. Immunol. 2017, 8, 785. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Qiao, Q.; Vonglorkham, S.; Feng, Z.; Pang, L.; Chen, S.; Wang, D.; Lao, L.; Lin, X.; Wei, J. Asiatic acid ameliorates acute hepatic injury by reducing endoplasmic reticulum stress and triggering hepatocyte autophagy. Biomed. Pharmacother. 2020, 129, 110375. [Google Scholar] [CrossRef]
- Wang, D.; Lao, L.; Pang, X.; Qiao, Q.; Pang, L.; Feng, Z.; Bai, F.; Sun, X.; Lin, X.; Wei, J. Asiatic acid from Potentilla chinensis alleviates non-alcoholic fatty liver by regulating endoplasmic reticulum stress and lipid metabolism. Int. Immunopharmacol. 2018, 65, 256–267. [Google Scholar] [CrossRef]
- Wei, L.; Chen, Q.; Guo, A.; Fan, J.; Wang, R.; Zhang, H. Asiatic acid attenuates CCl 4 -induced liver fibrosis in rats by regulating the PI3K/AKT/mTOR and Bcl-2/Bax signaling pathways. Int. Immunopharmacol. 2018, 60, 1–8. [Google Scholar] [CrossRef]
- Fan, J.; Chen, Q.; Wei, L.; Zhou, X.; Wang, R.; Zhang, H. Asiatic acid ameliorates CC l4-induced liver fibrosis in rats: Involvement of Nrf2/ARE, NF-κB/IκBα, and JAK1/STAT3 signaling pathways. Drug Des. Dev. Ther. 2018, 12, 3595–3605. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-W.; Tu, L.-L.; Zhang, Y.; Pan, J.-C.; Zheng, G.-L.; Yin, L.-N. Liver-targeted delivery of asiatic acid nanostructured lipid carrier for the treatment of liver fibrosis. Drug Deliv. 2021, 28, 2534–2547. [Google Scholar] [CrossRef]
- Gaudio, A.; Xourafa, A.; Rapisarda, R.; Zanoli, L.; Signorelli, S.S.; Castellino, P. Hematological Diseases and Osteoporosis. Int. J. Mol. Sci. 2020, 21, 3538. [Google Scholar] [CrossRef]
- Hong, G.; Zhou, L.; Han, X.; Sun, P.; Chen, Z.; He, W.; Tickner, J.; Chen, L.; Shi, X.; Xu, J. Asiatic Acid Inhibits OVX-Induced Osteoporosis and Osteoclastogenesis Via Regulating RANKL-Mediated NF-κb and Nfatc1 Signaling Pathways. Front. Pharmacol. 2020, 11, 331. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; He, J.; Zhao, Y.; Chen, H.; Tan, N. Asiatic acid prevents renal fibrosis in UUO rats via promoting the production of 15d-PGJ2, an endogenous ligand of PPAR-γ. Acta Pharmacol. Sin. 2020, 41, 373–382. [Google Scholar] [CrossRef]
- Wróbel, A.; Zapała, Ł.; Kluz, T.; Rogowski, A.; Misiek, M.; Juszczak, K.; Sieńko, J.; Gold, D.; Stangel-Wójcikiewicz, K.; Poleszak, E.; et al. The Potential of Asiatic Acid in the Reversion of Cyclophosphamide-Induced Hemorrhagic Cystitis in Rats. Int. J. Mol. Sci. 2021, 22, 5853. [Google Scholar] [CrossRef]
- Yang, C.; Guo, Y.; Huang, T.; Zhao, J.; Huang, X.-J.; Tang, H.; An, N.; Pan, Q.; Xu, Y.; Liu, H. Asiatic acid protects against cisplatin-induced acute kidney injury via anti-apoptosis and anti-inflammation. Biomed. Pharmacother. 2018, 107, 1354–1362. [Google Scholar] [CrossRef]
- Pollier, J.; Goossens, A. Oleanolic acid. Phytochemistry 2012, 77, 10–15. [Google Scholar] [CrossRef]
- Ghiulai, R.; Avram, S.; Stoian, D.; Pavel, I.Z.; Coricovac, D.; Oprean, C.; Vlase, L.; Farcas, C.; Mioc, M.; Minda, D.; et al. Lemon Balm Extracts Prevent Breast Cancer Progression In Vitro and In Ovo on Chorioallantoic Membrane Assay. Evid.-Based Complement. Altern. Med. 2020, 2020, 6489159. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Wen, X.; Sun, H. Oleanolic acid derivatives for pharmaceutical use: A patent review. Expert Opin. Ther. Pat. 2016, 26, 643–655. [Google Scholar] [CrossRef]
- Žiberna, L.; Šamec, D.; Mocan, A.; Nabavi, S.; Bishayee, A.; Farooqi, A.; Sureda, A.; Nabavi, S. Oleanolic Acid Alters Multiple Cell Signaling Pathways: Implication in Cancer Prevention and Therapy. Int. J. Mol. Sci. 2017, 18, 643. [Google Scholar] [CrossRef] [Green Version]
- Castellano, J.M.; Ramos-Romero, S.; Perona, J.S. Oleanolic Acid: Extraction, Characterization and Biological Activity. Nutrients 2022, 14, 623. [Google Scholar] [CrossRef]
- Missaoui, N.; Chouaibi, S.; Limam, S.; Mhamdi, N.; Zahmoul, T.; Hamchi, H.; Mokni, M.; Hmissa, S. Signification of forkhead box A1 (FOXA1) expression in thyroid cancers. J. Egypt. Natl. Cancer Inst. 2019, 31, 11. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Yang, Z.; Jiang, X.; Zhang, J.; Guo, X. Oleanolic acid inhibits cell proliferation migration and invasion and induces SW579 thyroid cancer cell line apoptosis by targeting forkhead transcription factor A. Anticancer Drugs 2019, 30, 812–820. [Google Scholar] [CrossRef]
- Guo, Y.; Han, B.; Luo, K.; Ren, Z.; Cai, L.; Sun, L. NOX2-ROS-HIF-1α signaling is critical for the inhibitory effect of oleanolic acid on rectal cancer cell proliferation. Biomed. Pharmacother. 2017, 85, 733–739. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Cao, Y.; Li, P.; Tang, X.; Yang, M.; Gu, S.; Xiong, K.; Li, T.; Xiao, T. Oleanolic Acid Induces Autophagy and Apoptosis via the AMPK-mTOR Signaling Pathway in Colon Cancer. J. Oncol. 2021, 2021, 8281718. [Google Scholar] [CrossRef]
- Medina-O’Donnell, M.; Rivas, F.; Reyes-Zurita, F.J.; Cano-Muñoz, M.; Martinez, A.; Lupiañez, J.A.; Parra, A. Oleanolic Acid Derivatives as Potential Inhibitors of HIV-1 Protease. J. Nat. Prod. 2019, 82, 2886–2896. [Google Scholar] [CrossRef]
- Reyes-Zurita, F.J.; Medina-O’Donnell, M.; Ferrer-Martin, R.M.; Rufino-Palomares, E.E.; Martin-Fonseca, S.; Rivas, F.; Martínez, A.; García-Granados, A.; Pérez-Jiménez, A.; García-Salguero, L.; et al. The oleanolic acid derivative, 3-O-succinyl-28-O-benzyl oleanolate, induces apoptosis in B16–F10 melanoma cells via the mitochondrial apoptotic pathway. RSC Adv. 2016, 6, 93590–93601. [Google Scholar] [CrossRef] [Green Version]
- Medina-O’Donnell, M.; Rivas, F.; Reyes-Zurita, F.J.; Martinez, A.; Galisteo-González, F.; Lupiañez, J.A.; Parra, A. Synthesis and in vitro antiproliferative evaluation of PEGylated triterpene acids. Fitoterapia 2017, 120, 25–40. [Google Scholar] [CrossRef]
- Medina-O’Donnell, M.; Rivas, F.; Reyes-Zurita, F.J.; Martinez, A.; Lupiañez, J.A.; Parra, A. Diamine and PEGylated-diamine conjugates of triterpenic acids as potential anticancer agents. Eur. J. Med. Chem. 2018, 148, 325–336. [Google Scholar] [CrossRef]
- Jannus, F.; Medina-O’Donnell, M.; Rivas, F.; Díaz-Ruiz, L.; Rufino-Palomares, E.E.; Lupiáñez, J.A.; Parra, A.; Reyes-Zurita, F.J. A Diamine-PEGylated Oleanolic Acid Derivative Induced Efficient Apoptosis through a Death Receptor and Mitochondrial Apoptotic Pathway in HepG2 Human Hepatoma Cells. Biomolecules 2020, 10, 1375. [Google Scholar] [CrossRef]
- Xu, Y.; Shu, B.; Tian, Y.; Wang, G.; Wang, Y.; Wang, J.; Dong, Y. Oleanolic acid induces osteosarcoma cell apoptosis by inhibition of Notch signaling. Mol. Carcinog. 2018, 57, 896–902. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, Y.; Zweidler-McKay, P.A.; Hughes, D.P.M. Critical Role of Notch Signaling in Osteosarcoma Invasion and Metastasis. Clin. Cancer Res. 2008, 14, 2962–2969. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Yin, J.; Cheng, X.; Lu, X.; Ni, L.; Xi, Y.; Yin, G.; Lu, G.; Sun, W.; Wei, M. Oleanolic acid attenuates TGF-β1-induced epithelial-mesenchymal transition in NRK-52E cells. BMC Complement. Altern. Med. 2018, 18, 205. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, W.; Zhao, J.; Zhang, H.; Zhang, Q.; Liang, Y.; Chen, S.; Liu, H.; Zong, S.; Tian, Y.; et al. Oleanolic Acid Inhibits Epithelial–Mesenchymal Transition of Hepatocellular Carcinoma by Promoting iNOS Dimerization. Mol. Cancer Ther. 2019, 18, 62–74. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Liu, X.; Huang, M.; Wei, Z.; Zhang, M.; He, M.; Zheng, Z.; Dong, H.; Liu, D. Oleanolic acid inhibits the migration and invasion of hepatocellular carcinoma cells by promoting microRNA-122 expression. Pharmazie 2021, 76, 422–427. [Google Scholar] [CrossRef]
- Tran, T.H.; Montano, M.A. MicroRNAs. In Translating MicroRNAs to the Clinic; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–15. [Google Scholar]
- Hosny, S.; Sahyon, H.; Youssef, M.; Negm, A. Oleanolic Acid Suppressed DMBA-Induced Liver Carcinogenesis through Induction of Mitochondrial-Mediated Apoptosis and Autophagy. Nutr. Cancer 2021, 73, 968–982. [Google Scholar] [CrossRef]
- Zhou, W.; Zeng, X.; Wu, X. Effect of oleanolic acid on apoptosis and autophagy of SMMC-7721 hepatoma cells. Med. Sci. Monit. 2020, 26, e921606-1. [Google Scholar] [CrossRef]
- Peng, F.; Li, G.; Xie, Y.; Yin, H.; Li, X.; Yang, Y. Compositional characterization of Pyrus ussuriensis Maxim and their antioxidant activities and induction of apoptosis in Bel-7402 cell. J. Food Biochem. 2020, 44, e13222. [Google Scholar] [CrossRef]
- Wang, W.; Wu, L.; Li, J.; Ji, J.; Chen, K.; Yu, Q.; Li, S.; Feng, J.; Liu, T.; Zhang, J.; et al. Alleviation of Hepatic Ischemia Reperfusion Injury by Oleanolic Acid Pretreating via Reducing HMGB1 Release and Inhibiting Apoptosis and Autophagy. Mediat. Inflamm. 2019, 2019, 3240713. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Cao, Y.; Li, W.; Wang, Y.; Xu, J.; Xu, G. Molecular evidence for better efficacy of hypocrellin A and oleanolic acid combination in suppression of HCC growth. Eur. J. Pharmacol. 2019, 842, 281–290. [Google Scholar] [CrossRef]
- Xu, G.; Wang, J.; Wu, F.; Wang, N.; Zhou, W.; Wang, Q.; Pan, W.; Ao, G.; Yang, J. YAP and 14-3-3γ are involved in HS-OA-induced growth inhibition of hepatocellular carcinoma cells: A novel mechanism for hydrogen sulfide releasing oleanolic acid. Oncotarget 2016, 7, 52150–52165. [Google Scholar] [CrossRef]
- Liese, J.; Hinrichs, T.M.; Lange, M.; Fulda, S. Cotreatment with sorafenib and oleanolic acid induces reactive oxygen species-dependent and mitochondrial-mediated apoptotic cell death in hepatocellular carcinoma cells. Anticancer Drugs 2019, 30, 209–217. [Google Scholar] [CrossRef]
- Khan, M.W.; Zhao, P.; Khan, A.; Raza, F.; Raza, S.M.; Sarfraz, M.; Chen, Y.; Li, M.; Yang, T.; Ma, X.; et al. Synergism of cisplatin-oleanolic acid co-loaded calcium carbonate nanoparticles on hepatocellular carcinoma cells for enhanced apoptosis and reduced hepatotoxicity. Int. J. Nanomed. 2019, 14, 3753–3771. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.-J.; Jo, H.-J.; Lee, K.-J.; Choi, J.W.; An, J.H. Oleanolic acid induces p53-dependent apoptosis via the ERK/JNK/AKT pathway in cancer cell lines in prostatic cancer xenografts in mice. Oncotarget 2018, 9, 26370–26386. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.-F.; Peng, H.-P.; Lu, X.-R.; Hu, Y.; Xu, Z.-H.; Xu, J.-K. Oleanolic acid regulates the Treg/Th17 imbalance in gastric cancer by targeting IL-6 with miR-98-5p. Cytokine 2021, 148, 155656. [Google Scholar] [CrossRef]
- Gao, Z.; Deng, G.; Li, Y.; Huang, H.; Sun, X.; Shi, H.; Yao, X.; Gao, L.; Ju, Y.; Luo, M. Actinidia chinensis Planch prevents proliferation and migration of gastric cancer associated with apoptosis, ferroptosis activation and mesenchymal phenotype suppression. Biomed. Pharmacother. 2020, 126, 110092. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front. Immunol. 2016, 7, 279. [Google Scholar] [CrossRef] [Green Version]
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Baltierra-Uribe, S.L.; Castillo-Cruz, J.; Campillo-Navarro, M.; Hernández-Pérez, A.D.; Luna-Angulo, A.B.; Chacón-Salinas, R.; Coral-Vázquez, R.M.; Estrada-García, I.; et al. Ursolic and Oleanolic Acids Induce Mitophagy in A549 Human Lung Cancer Cells. Molecules 2019, 24, 3444. [Google Scholar] [CrossRef] [Green Version]
- Edathara, P.M.; Chintalapally, S.; Makani, V.K.K.; Pant, C.; Yerramsetty, S.; Rao, M.D.; Bhadra, M.P. Inhibitory role of oleanolic acid and esculetin in HeLa cells involve multiple signaling pathways. Gene 2021, 771, 145370. [Google Scholar] [CrossRef]
- Gu, Y.; Mohammad, I.; Liu, Z. Overview of the STAT-3 signaling pathway in cancer and the development of specific inhibitors (Review). Oncol. Lett. 2020, 19, 2585–2594. [Google Scholar] [CrossRef]
- An, Q.; Hu, Q.; Wang, B.; Cui, W.; Wu, F.; Ding, Y. Oleanolic acid alleviates diabetic rat carotid artery injury through the inhibition of NLRP3 inflammasome signaling pathways. Mol. Med. Rep. 2017, 16, 8413–8419. [Google Scholar] [CrossRef]
- Gamede, M.; Mabuza, L.; Ngubane, P.; Khathi, A. Plant-derived oleanolic acid ameliorates markers of subclinical inflammation and innate immunity activation in diet-induced pre-diabetic rats. Ther. Adv. Endocrinol. Metab. 2020, 11, 204201882093577. [Google Scholar] [CrossRef]
- Gamede, M.; Mabuza, L.; Ngubane, P.; Khathi, A. Preventing the onset of diabetes-induced chronic kidney disease during prediabetes: The effects of oleanolic acid on selected markers of chronic kidney disease in a diet-induced prediabetic rat model. Biomed. Pharmacother. 2021, 139, 111570. [Google Scholar] [CrossRef]
- Iskender, H.; Dokumacioglu, E.; Terim Kapakin, K.A.; Yenice, G.; Mohtare, B.; Bolat, I.; Hayirli, A. Effects of oleanolic acid on inflammation and metabolism in diabetic rats. Biotech. Histochem. 2022, 97, 269–276. [Google Scholar] [CrossRef]
- Li, W.; Zeng, H.; Xu, M.; Huang, C.; Tao, L.; Li, J.; Zhang, T.; Chen, H.; Xia, J.; Li, C.; et al. Oleanolic Acid Improves Obesity-Related Inflammation and Insulin Resistance by Regulating Macrophages Activation. Front. Pharmacol. 2021, 12, 697483. [Google Scholar] [CrossRef]
- Li, W.-F.; Wang, P.; Li, H.; Li, T.-Y.; Feng, M.; Chen, S.-F. Oleanolic acid protects against diabetic cardiomyopathy via modulation of the nuclear factor erythroid 2 and insulin signaling pathways. Exp. Ther. Med. 2017, 14, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Wu, G.; Cheng, X.; Fan, J.; Peng, J.; Su, H.; Xu, Z.; Cao, M.; Long, Z.; Hao, Y.; et al. Oleanolic acid attenuates PCBs-induced adiposity and insulin resistance via HNF1b-mediated regulation of redox and PPARγ signaling. Free Radic. Biol. Med. 2018, 124, 122–134. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Jiang, M.; Xie, X.; Yang, H.; Wang, X.; Xiao, L.; Wang, N. Oleanolic acid ameliorates high glucose-induced endothelial dysfunction via PPARδ activation. Sci. Rep. 2017, 7, 40237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.-K.; Hao, H.; Bian, Y.; Ge, Y.-X.; Lu, S.; Xie, H.-X.; Wang, K.-M.; Tao, H.; Yuan, C.; Zhang, J.; et al. Discovery of New α-Glucosidase Inhibitors: Structure-Based Virtual Screening and Biological Evaluation. Front. Chem. 2021, 9, 639279. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Hu, X.; Xu, X.; Zhang, G.; Gong, D. Inhibitory mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α-glucosidase. Int. J. Biol. Macromol. 2018, 107, 1844–1855. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Dong, S.; Wu, Y.; Zhao, H.; Li, X.; Gao, W. Oleanolic acid and ursolic acid as potential inhibitors of human salivary α-amylase: Insights from in vitro assays and in silico simulations. J. Mol. Model. 2017, 23, 248. [Google Scholar] [CrossRef]
- Shan, T.; Ye, J.; Jia, J.; Wang, Z.; Jiang, Y.; Wang, Y.; Wang, Y.; Zheng, K.; Ren, Z. Viral UL8 Is Involved in the Antiviral Activity of Oleanolic Acid Against HSV-1 Infection. Front. Microbiol. 2021, 12, 2046. [Google Scholar] [CrossRef]
- Catteau, L.; Reichmann, N.; Olson, J.; Pinho, M.; Nizet, V.; Van Bambeke, F.; Quetin-Leclercq, J. Synergy between Ursolic and Oleanolic Acids from Vitellaria paradoxa Leaf Extract and β-Lactams against Methicillin-Resistant Staphylococcus aureus: In Vitro and In Vivo Activity and Underlying Mechanisms. Molecules 2017, 22, 2245. [Google Scholar] [CrossRef] [Green Version]
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Verstraeten, S.; Catteau, L.; Boukricha, L.; Quetin-Leclercq, J.; Mingeot-Leclercq, M.-P. Effect of Ursolic and Oleanolic Acids on Lipid Membranes: Studies on MRSA and Models of Membranes. Antibiotics 2021, 10, 1381. [Google Scholar] [CrossRef]
- Lin, Y.-N.; Chang, H.-Y.; Wang, C.C.N.; Chu, F.-Y.; Shen, H.-Y.; Chen, C.-J.; Lim, Y.-P. Oleanolic Acid Inhibits Liver X Receptor Alpha and Pregnane X Receptor to Attenuate Ligand-Induced Lipogenesis. J. Agric. Food Chem. 2018, 66, 10964–10976. [Google Scholar] [CrossRef]
- Luo, H.; Liu, J.; Ouyang, Q.; Xuan, C.; Wang, L.; Li, T.; Liu, J. The effects of oleanolic acid on atherosclerosis in different animal models. Acta Biochim. Biophys. Sin. 2017, 49, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, H.; Fujii, Y.; Okada-Iwabu, M.; Iwabu, M.; Kano, K.; Kawana, H.; Hato, M.; Nakamura, Y.; Terada, T.; Kimura-Someya, T.; et al. Human adiponectin receptor AdipoR1 assumes closed and open structures. Commun. Biol. 2020, 3, 446. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.-Q.; Shen, J.; Chen, C.-P.; Ma, X.; Lin, C.; Ouyang, Q.; Xuan, C.-X.; Liu, J.; Sun, H.-B.; Liu, J. Lipid-lowering effects of oleanolic acid in hyperlipidemic patients. Chin. J. Nat. Med. 2018, 16, 339–346. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Tian, Z.; Qi, D.; Li, Y.; Jiang, H. Antihypertensive activity of oleanolic acid is mediated via downregulation of secretory phospholipase A2 and fatty acid synthase in spontaneously hypertensive rats. Int. J. Mol. Med. 2020, 46, 2019–2034. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.M.; Choi, Y.H.; Yoon, J.J.; Lee, Y.J.; Cho, K.W.; Kang, D.G.; Lee, H.S. Oleanolic acid modulates the renin-angiotensin system and cardiac natriuretic hormone concomitantly with volume and pressure balance in rats. Eur. J. Pharmacol. 2017, 809, 231–241. [Google Scholar] [CrossRef]
- Pan, Y.; Zhou, F.; Song, Z.; Huang, H.; Chen, Y.; Shen, Y.; Jia, Y.; Chen, J. Oleanolic acid protects against pathogenesis of atherosclerosis, possibly via FXR-mediated angiotensin (Ang)-(1–7) upregulation. Biomed. Pharmacother. 2018, 97, 1694–1700. [Google Scholar] [CrossRef]
- Cheon, S.-Y.; Jin, B.-R.; Kim, H.-J.; An, H.-J. Oleanolic Acid Ameliorates Benign Prostatic Hyperplasia by Regulating PCNA-Dependent Cell Cycle Progression In Vivo and In Vitro. J. Nat. Prod. 2020, 83, 1183–1189. [Google Scholar] [CrossRef]
- Yang, J.; Li, X.; Yang, H.; Long, C. Oleanolic Acid Improves the Symptom of Renal Ischemia Reperfusion Injury via the PI3K/AKT Pathway. Urol. Int. 2020, 105, 215–220. [Google Scholar] [CrossRef]
- Zhao, D.; Luan, Z. Oleanolic Acid Attenuates Renal Fibrosis through TGF- β/Smad Pathway in a Rat Model of Unilateral Ureteral Obstruction. Evid.-Based Complement. Altern. Med. 2020, 2020, 2085303. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Liu, J.; Song, L.; Liu, Z.; Han, G.; Yuan, D.; Wang, T.; Dun, Y.; Zhou, Z.; Liu, Z.; et al. Oleanolic acid rejuvenates testicular function through attenuating germ cell DNA damage and apoptosis via deactivation of NF-κB, p53 and p38 signalling pathways. J. Pharm. Pharmacol. 2017, 69, 295–304. [Google Scholar] [CrossRef]
- Shu, B.; Zhao, Y.; Wang, Y.; Wang, G.; Shang, X.; Britt, M.; Olmedo, M.; Chelly, M.; Morandi, M.M.; Barton, S.; et al. Oleanolic Acid Enhances Mesenchymal Stromal Cell Osteogenic Potential by Inhibition of Notch Signaling. Sci. Rep. 2017, 7, 7002. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Gao, L.-J.; Fan, Y.-S.; Chen, Y.; Li, Q. Network Pharmacology-Based Analysis on the Action Mechanism of Oleanolic Acid to Alleviate Osteoporosis. ACS Omega 2021, 6, 28410–28420. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Li, X.; Zhao, Y.; Qiao, P.; Tang, D.; Chen, Y.; Xue, C.; Li, C.; Liu, S.; Wang, J.; et al. Oleanolic acid exerts bone protective effects in ovariectomized mice by inhibiting osteoclastogenesis. J. Pharmacol. Sci. 2018, 137, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Shu, B.; Wang, C.; Zhao, Y.; Cheng, W.; Sha, N.; Li, C.; Wang, Q.; Lu, S.; Wang, Y. Oleanolic acid exerts inhibitory effects on the late stage of osteoclastogenesis and prevents bone loss in osteoprotegerin knockout mice. J. Cell. Biochem. 2020, 121, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Shi, L.; Li, J.; Zeng, Y.; Liu, W.; Tang, S.; Jia, L.; Zhang, J.; Gan, G. Oleanolic acid inhibits RANKL-induced osteoclastogenesis via ER alpha/miR-503/RANK signaling pathway in RAW264.7 cells. Biomed. Pharmacother. 2019, 117, 109045. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Yan, W.; Xu, K.; Chen, M.; Chen, Z.; Ran, J.; Xiong, Y.; Wu, L.; Seth, R.K. Oleanolic Acid Decreases IL-1β-Induced Activation of Fibroblast-Like Synoviocytes via the SIRT3-NF-κB Axis in Osteoarthritis. Oxid. Med. Cell. Longev. 2020, 2020, 7517219. [Google Scholar] [CrossRef]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Nie, J.; Jiang, P. Oleanolic acid mitigates interleukin-1β-induced chondrocyte dysfunction by regulating miR-148-3p-modulated FGF2 expression. J. Gene Med. 2020, 22, e3169. [Google Scholar] [CrossRef]
- Kang, Y.-M.; Lee, M.; An, H.-J. Oleanolic acid protects against mast cell-mediated allergic responses by suppressing Akt/NF-κB and STAT1 activation. Phytomedicine 2021, 80, 153340. [Google Scholar] [CrossRef]
- Hartenstein, B. Th2 cell-specific cytokine expression and allergen-induced airway inflammation depend on JunB. EMBO J. 2002, 21, 6321–6329. [Google Scholar] [CrossRef]
- Kang, Y.-M.; Kim, H.-M.; Lee, M.; An, H.-J. Oleanolic Acid Alleviates Atopic Dermatitis-like Responses In Vivo and In Vitro. Int. J. Mol. Sci. 2021, 22, 12000. [Google Scholar] [CrossRef]
- Yin, Y.; Zhang, Y.; Li, H.; Zhao, Y.; Cai, E.; Zhu, H.; Li, P.; Liu, J. Triterpenoids from fruits of Sorbus pohuashanensis inhibit acetaminophen-induced acute liver injury in mice. Biomed. Pharmacother. 2019, 109, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, K.; Xia, Y.; Mo, W.; Wang, F.; Dai, W.; Niu, P. The Hepatoprotection by Oleanolic Acid Preconditioning: Focusing on PPAR α Activation. PPAR Res. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, N.; Xue, C.; Zhang, L.; Zhang, T.; Wang, C.; Bi, C.; Shan, A. Oleanolic acid enhances tight junctions and ameliorates inflammation in Salmonella typhimurium-induced diarrhea in mice via the TLR4/NF-κB and MAPK pathway. Food Funct. 2020, 11, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Phull, A.-R.; Eo, S.-H.; Kim, S.J. Oleanolic acid (OA) regulates inflammation and cellular dedifferentiation of chondrocytes via MAPK signaling pathways. Cell. Mol. Biol. 2017, 63, 12. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.-B.; Wang, R.-X.; Deng, H.-J.; Wang, Y.-H.; Tang, J.-D.; Cao, F.-Y.; Wang, J.-H. Protective effects of oleanolic acid on oxidative stress and the expression of cytokines and collagen by the AKT/NF-κB pathway in silicotic rats. Mol. Med. Rep. 2017, 15, 3121–3128. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.-P.; Li, X.-H.; Li, Y.; Huang, X.-T.; Luo, Z.-Q. The protective effect of oleanolic acid on NMDA-induced MLE-12 cells apoptosis and lung injury in mice by activating SIRT1 and reducing NF-κB acetylation. Int. Immunopharmacol. 2019, 70, 520–529. [Google Scholar] [CrossRef]
- Wang, J.-L.; Ren, C.-H.; Feng, J.; Ou, C.-H.; Liu, L. Oleanolic acid inhibits mouse spinal cord injury through suppressing inflammation and apoptosis via the blockage of p38 and JNK MAPKs. Biomed. Pharmacother. 2020, 123, 109752. [Google Scholar] [CrossRef]
- Han, Y.; Tong, Z.; Wang, C.; Li, X.; Liang, G. Oleanolic acid exerts neuroprotective effects in subarachnoid hemorrhage rats through SIRT1-mediated HMGB1 deacetylation. Eur. J. Pharmacol. 2021, 893, 173811. [Google Scholar] [CrossRef]
- Guo, Q.; He, J.; Zhang, H.; Yao, L.; Li, H. Oleanolic acid alleviates oxidative stress in Alzheimer’s disease by regulating stanniocalcin-1 and uncoupling protein-2 signalling. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1263–1271. [Google Scholar] [CrossRef]
- Wang, K.; Sun, W.; Zhang, L.; Guo, W.; Xu, J.; Liu, S.; Zhou, Z.; Zhang, Y. Oleanolic Acid Ameliorates Aβ25-35 Injection-induced Memory Deficit in Alzheimer’s Disease Model Rats by Maintaining Synaptic Plasticity. CNS Neurol. Disord.—Drug Targets 2018, 17, 389–399. [Google Scholar] [CrossRef]
- Msibi, Z.N.P.; Mabandla, M.V. Oleanolic Acid Mitigates 6-Hydroxydopamine Neurotoxicity by Attenuating Intracellular ROS in PC12 Cells and Striatal Microglial Activation in Rat Brains. Front. Physiol. 2019, 10, 1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Sun, L.; Ji, X.; Shi, R.; Xu, F.; Gu, J. Neuroprotective effects of oleanolic acid against cerebral ischemia-reperfusion injury in mice. Exp. Neurol. 2021, 343, 113785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Lin, K.L.; Law, C.Y.; Liu, B.; Fu, X.Q.; Tse, W.S.; Wong, S.S.M.; Sze, S.C.W.; Yung, K.K.L. Oleanolic acid enhances neural stem cell migration, proliferation and differentiation in vitro by inhibiting GSK3β activity. Cell Death Discov. 2021, 7, 303. [Google Scholar] [CrossRef] [PubMed]
- Soares, I.C.R.; Santos, S.A.A.R.; Coelho, R.F.; Alves, Y.A.; Vieira-Neto, A.E.; Tavares, K.C.S.; Magalhaes, F.E.A.; Campos, A.R. Oleanolic acid promotes orofacial antinociception in adult zebrafish (Danio rerio) through TRPV1 receptors. Chem. Biol. Interact. 2019, 299, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, Y.-F.; Wu, Q.; Xu, S.-F.; Shi, F.-G.; Klaassen, C.D. Oleanolic acid reprograms the liver to protect against hepatotoxicants, but is hepatotoxic at high doses. Liver Int. 2019, 39, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-S.; Sung, H.-Y.; Kim, M.S.; Kim, J.-L.; Kang, M.-K.; Gong, J.-H.; Park, H.-S.; Kang, Y.-H. Oleanolic acid suppresses resistin induction in adipocytes by modulating Tyk-STAT signaling. Nutr. Res. 2013, 33, 144–153. [Google Scholar] [CrossRef]
- Cao, C.Y.; Liu, Y.L.; Ding, Q.; Deng, Y.Y.; Wu, Q.F.; Xu, Y.X.; Liu, J.J.; Fan, W.X. Effect of oleanolic acid on 5α-reductase activity, DPCs proliferation and gene expression correlated with androgenetic alopecia in vitro. Acta Med. Mediterr. 2019, 35, 2159–2165. [Google Scholar] [CrossRef]
- Seo, D.Y.; Lee, S.R.; Heo, J.-W.; No, M.-H.; Rhee, B.D.; Ko, K.S.; Kwak, H.-B.; Han, J. Ursolic acid in health and disease. Korean J. Physiol. Pharmacol. 2018, 22, 235. [Google Scholar] [CrossRef] [Green Version]
- Hussain, H.; Green, I.R.; Ali, I.; Khan, I.A.; Ali, Z.; Al-Sadi, A.M.; Ahmed, I. Ursolic acid derivatives for pharmaceutical use: A patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 1061–1072. [Google Scholar] [CrossRef]
- Kashyap, D.; Tuli, H.S.; Sharma, A.K. Ursolic acid (UA): A metabolite with promising therapeutic potential. Life Sci. 2016, 146, 201–213. [Google Scholar] [CrossRef]
- Bergamin, L.S.; Figueiró, F.; Dietrich, F.; de Mattos Manica, F.; Filippi-Chiela, E.C.; Mendes, F.B.; Jandrey, E.H.F.; Lopes, D.V.; Oliveira, F.H.; Nascimento, I.C.; et al. Interference of ursolic acid treatment with glioma growth: An in vitro and in vivo study. Eur. J. Pharmacol. 2017, 811, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Conway, G.E.; Zizyte, D.; Mondala, J.R.M.; He, Z.; Lynam, L.; Lecourt, M.; Barcia, C.; Howe, O.; Curtin, J.F. Ursolic Acid Inhibits Collective Cell Migration and Promotes JNK-Dependent Lysosomal Associated Cell Death in Glioblastoma Multiforme Cells. Pharmaceuticals 2021, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Lin, J.; Zhang, L.; Lin, J.; Wang, L.; Chen, D.; Peng, J. Comparative proteomics—Network analysis of proteins responsible for ursolic acid–induced cytotoxicity in colorectal cancer cells. Tumor Biol. 2017, 39, 101042831769501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Chen, Y.; Wei, L.; Hong, Z.; Sferra, T.J.; Peng, J. Ursolic acid inhibits colorectal cancer angiogenesis through suppression of multiple signaling pathways. Int. J. Oncol. 2013, 43, 1666–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Shin, E.; Jung, J.; Park, J.; Kim, D.; Shim, B.; Kim, S.-H. Ursolic Acid Induces Apoptosis in Colorectal Cancer Cells Partially via Upregulation of MicroRNA-4500 and Inhibition of JAK2/STAT3 Phosphorylation. Int. J. Mol. Sci. 2018, 20, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cai, Q.; Liu, J.; Peng, J.; Chen, Y.; Sferra, T.; Lin, J. Ursolic acid suppresses the invasive potential of colorectal cancer cells by regulating the TGF-β1/ZEB1/miR-200c signaling pathway. Oncol. Lett. 2019, 18, 3274–3282. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.Y.; Sp, N.; Lee, J.-M.; Jang, K.-J. Antitumor Effects of Ursolic Acid through Mediating the Inhibition of STAT3/PD-L1 Signaling in Non-Small Cell Lung Cancer Cells. Biomedicines 2021, 9, 297. [Google Scholar] [CrossRef]
- Chen, H.; Wu, X.; Duan, Y.; Zhi, D.; Zou, M.; Zhao, Z.; Zhang, X.; Yang, X.; Zhang, J. Ursolic acid isolated from Isodonexcisoides induces apoptosis and inhibits invasion of GBC-SD gallbladder carcinoma cells. Oncol. Lett. 2019, 18, 1467–1474. [Google Scholar] [CrossRef] [Green Version]
- Mu, D.; Zhou, G.; Li, J.; Su, B.; Guo, H. Ursolic acid activates the apoptosis of prostate cancer via ROCK/PTEN mediated mitochondrial translocation of cofilin-1. Oncol. Lett. 2017, 17, 3202–3206. [Google Scholar] [CrossRef] [Green Version]
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Deregowska, A.; Wnuk, M. Ursolic acid-mediated changes in glycolytic pathway promote cytotoxic autophagy and apoptosis in phenotypically different breast cancer cells. Apoptosis 2017, 22, 800–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Xu, B.; Wang, X.; Zheng, B.; Du, J.; Liu, S. The Analysis of the Anti-Tumor Mechanism of Ursolic Acid Using Connectively Map Approach in Breast Cancer Cells Line MCF-7. Cancer Manag. Res. 2020, 12, 3469–3476. [Google Scholar] [CrossRef] [PubMed]
- de Cárcer, G.; Venkateswaran, S.V.; Salgueiro, L.; El Bakkali, A.; Somogyi, K.; Rowald, K.; Montañés, P.; Sanclemente, M.; Escobar, B.; de Martino, A.; et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun. 2018, 9, 3012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Hu, Y.-L.; Wang, H. Ursolic acid inhibits breast cancer growth by inhibiting proliferation, inducing autophagy and apoptosis, and suppressing inflammatory responses via the PI3K/AKT and NF-κB signaling pathways in vitro. Exp. Ther. Med. 2017, 14, 3623–3631. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, T.; Gong, E.S.; Liu, R.H. Antiproliferative Activity of Ursolic Acid in MDA-MB-231 Human Breast Cancer Cells through Nrf2 Pathway Regulation. J. Agric. Food Chem. 2020, 68, 7404–7415. [Google Scholar] [CrossRef]
- Xiang, F.; Fan, Y.; Ni, Z.; Liu, Q.; Zhu, Z.; Chen, Z.; Hao, W.; Yue, H.; Wu, R.; Kang, X. Ursolic Acid Reverses the Chemoresistance of Breast Cancer Cells to Paclitaxel by Targeting MiRNA-149-5p/MyD88. Front. Oncol. 2019, 9, 501. [Google Scholar] [CrossRef] [Green Version]
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Emerging Role of MitomiRs in the Pathophysiology of Human Disease. In microRNA: Medical Evidence; Springer: Cham, Switzerland, 2015; pp. 123–154. [Google Scholar]
- Lin, J.; Chen, S.; Lu, C.; Lin, J.; Yen, G. Ursolic acid promotes apoptosis, autophagy, and chemosensitivity in gemcitabine-resistant human pancreatic cancer cells. Phyther. Res. 2020, 34, 2053–2066. [Google Scholar] [CrossRef]
- Kim, S.-H.; Jin, H.; Meng, R.Y.; Kim, D.-Y.; Liu, Y.C.; Chai, O.H.; Park, B.H.; Kim, S.M. Activating Hippo Pathway via Rassf1 by Ursolic Acid Suppresses the Tumorigenesis of Gastric Cancer. Int. J. Mol. Sci. 2019, 20, 4709. [Google Scholar] [CrossRef] [Green Version]
- Junco, J.J.; Cho, J.; Mancha, A.; Malik, G.; Wei, S.-J.; Kim, D.J.; Liang, H.; DiGiovanni, J.; Slaga, T.J. Role of AMPK and PPARα in the anti-skin cancer effects of ursolic acid. Mol. Carcinog. 2018, 57, 1698–1706. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, R.; Wu, R.; Ramirez, C.N.; Sargsyan, D.; Li, S.; Wang, L.; Cheng, D.; Wang, C.; Hudlikar, R.; et al. DNA methylome and transcriptome alterations and cancer prevention by triterpenoid ursolic acid in UVB-induced skin tumor in mice. Mol. Carcinog. 2019, 58, 1738–1753. [Google Scholar] [CrossRef]
- Wang, S.; Meng, X.; Dong, Y. Ursolic acid nanoparticles inhibit cervical cancer growth in vitro and in vivo via apoptosis induction. Int. J. Oncol. 2017, 50, 1330–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Xia, P. Cellular Inhibitor of Apoptosis Protein-1 (cIAP1) Plays a Critical Role in β-Cell Survival under Endoplasmic Reticulum Stress. J. Biol. Chem. 2012, 287, 32236–32245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Chen, Y.; Zhou, J.; Ma, L.; Shan, Y.; Cheng, X.; Wang, Y.; Zhang, Z.; Ji, X.; Chen, L.; et al. Ursolic acid promotes apoptosis and mediates transcriptional suppression of CT45A2 gene expression in non-small-cell lung carcinoma harbouring EGFR T790M mutations. Br. J. Pharmacol. 2019, 176, 4609–4624. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.-R.; Meng, R.Y.; Rah, S.-Y.; Jin, H.; Ray, N.; Kim, S.-H.; Park, B.H.; Kim, S.M. Reactive Oxygen Species-Mediated Autophagy by Ursolic Acid Inhibits Growth and Metastasis of Esophageal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9409. [Google Scholar] [CrossRef]
- Kim, G.-H.; Kan, S.-Y.; Kang, H.; Lee, S.; Ko, H.M.; Kim, J.H.; Lim, J.-H. Ursolic Acid Suppresses Cholesterol Biosynthesis and Exerts Anti-Cancer Effects in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 4767. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Meng, X.; Cai, X.; Yuan, C.; Zhao, Z.; Zhong, L.; Shi, Y.; Yin, F. Therapeutic effect of ursolic acid on fetal development in pregnant rats with gestational diabetes mellitus via AGEs-RAGE signaling pathway. J. Food Biochem. 2021, 45, e13651. [Google Scholar] [CrossRef]
- Kay, A.M.; Simpson, C.L.; Stewart, J.A. The Role of AGE/RAGE Signaling in Diabetes-Mediated Vascular Calcification. J. Diabetes Res. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, J.; Yan, Y.; Liu, D.; Wang, C.; Wang, H. Inhibition of glycosidase by ursolic acid: In vitro, in vivo and in silico study. J. Sci. Food Agric. 2020, 100, 986–994. [Google Scholar] [CrossRef]
- Wang, X.; Gong, Y.; Zhou, B.; Yang, J.; Cheng, Y.; Zhao, J.; Qi, M. Ursolic acid ameliorates oxidative stress, inflammation and fibrosis in diabetic cardiomyopathy rats. Biomed. Pharmacother. 2018, 97, 1461–1467. [Google Scholar] [CrossRef]
- Choi, W.H.; Lee, I.A. The Mechanism of Action of Ursolic Acid as a Potential Anti-Toxoplasmosis Agent, and Its Immunomodulatory Effects. Pathogens 2019, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Jesus, J.A.; Fragoso, T.N.; Yamamoto, E.S.; Laurenti, M.D.; Silva, M.S.; Ferreira, A.F.; Lago, J.H.G.; Gomes, G.S.; Passero, L.F.D. Therapeutic effect of ursolic acid in experimental visceral leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the Brain: A Cytokine To Remember. J. Immunol. 2012, 189, 4213–4219. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Wang, W.; Zhang, J.; Wang, T.; Liu, M.; Yang, M.; Sun, Z.; Li, X.; Li, Y. Antimicrobial and antibiofilm activities of ursolic acid against carbapenem-resistant Klebsiella pneumoniae. J. Antibiot. 2020, 73, 382–391. [Google Scholar] [CrossRef]
- Shu, C.; Zhao, H.; Jiao, W.; Liu, B.; Cao, J.; Jiang, W. Antifungal efficacy of ursolic acid in control of Alternaria alternata causing black spot rot on apple fruit and possible mechanisms involved. Sci. Hortic. 2019, 256, 108636. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Z.; Li, X.; Wei, Q.; Li, H.; Li, C.; Chen, H.; Liu, C.; He, K. Ursolic Acid Improves Monocrotaline-Induced Right Ventricular Remodeling by Regulating Metabolism. J. Cardiovasc. Pharmacol. 2020, 75, 545–555. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, W.; Zeng, X.; Hao, Z. Ursolic Acid Attenuates Atherosclerosis in ApoE−/− Mice: Role of LOX-1 Mediated by ROS/NF-κB Pathway. Molecules 2018, 23, 1101. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Kang, Y.; Su, G.; Sun, J.; Zhang, Y. Activation of the TLR4/MyD88 signaling pathway contributes to the development of human hepatocellular carcinoma via upregulation of IL-23 and IL-17A. Oncol. Lett. 2018, 15, 9647–9654. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zheng, H.; Sui, Z.; Jing, F.; Quan, X.; Zhao, W.; Liu, G. Ursolic acid exhibits anti-inflammatory effects through blocking TLR4-MyD88 pathway mediated by autophagy. Cytokine 2019, 123, 154726. [Google Scholar] [CrossRef]
- Yu, Y.-M.; Tsai, C.-C.; Tzeng, Y.-W.; Chang, W.-C.; Chiang, S.-Y.; Lee, M.-F. Ursolic acid suppresses leptin-induced cell proliferation in rat vascular smooth muscle cells. Can. J. Physiol. Pharmacol. 2017, 95, 811–818. [Google Scholar] [CrossRef]
- Ding, H.; Wang, H.; Zhu, L.; Wei, W. Ursolic Acid Ameliorates Early Brain Injury After Experimental Traumatic Brain Injury in Mice by Activating the Nrf2 Pathway. Neurochem. Res. 2017, 42, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.; Li, R.; Kadeyala, P.K.; Liu, S.; Schachner, M. The human natural killer-1 (HNK-1) glycan mimetic ursolic acid promotes functional recovery after spinal cord injury in mouse. J. Nutr. Biochem. 2018, 55, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Deng, S.; Liu, F.; He, Z. Ursolic Acid Ameliorates Inflammation in Cerebral Ischemia and Reperfusion Injury Possibly via High Mobility Group Box 1/Toll-Like Receptor 4/NFκB Pathway. Front. Neurol. 2018, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Mou, L.; Liao, L.; Zhang, Y.; Ming, D.; Jiang, J. Ursolic acid ameliorates Nthy-ori 3-1 cells injury induced by IL-1β through limiting MALAT1/miR-206/PTGS1 ceRNA network and NF-κB signaling pathway. Psychopharmacology 2021, 238, 1141–1156. [Google Scholar] [CrossRef]
- Lin, L.; Yin, Y.; Hou, G.; Han, D.; Kang, J.; Wang, Q. Ursolic acid attenuates cigarette smoke-induced emphysema in rats by regulating PERK and Nrf2 pathways. Pulm. Pharmacol. Ther. 2017, 44, 111–121. [Google Scholar] [CrossRef]
- Lin, L.; Hou, G.; Han, D.; Yin, Y.; Kang, J.; Wang, Q. Ursolic acid alleviates airway-vessel remodeling and muscle consumption in cigarette smoke-induced emphysema rats. BMC Pulm. Med. 2019, 19, 103. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhang, Y.; Wang, Z.; Shen, Y.; Zhang, M.; Nie, Q.; Hou, Y.; Bai, G. Ursolic Acid, a Natural Nutraceutical Agent, Targets Caspase3 and Alleviates Inflammation-Associated Downstream Signal Transduction. Mol. Nutr. Food Res. 2017, 61, 1700332. [Google Scholar] [CrossRef]
- Samivel, R.; Nagarajan, R.P.; Subramanian, U.; Khan, A.A.; Masmali, A.; Almubrad, T.; Akhtar, S. Inhibitory Effect of Ursolic Acid on Ultraviolet B Radiation-Induced Oxidative Stress and Proinflammatory Response-Mediated Senescence in Human Skin Dermal Fibroblasts. Oxid. Med. Cell. Longev. 2020, 2020, 1–17. [Google Scholar] [CrossRef]
- Yu, R.; Chen, J.; Xu, J.; Cao, J.; Wang, Y.; Thomas, S.S.; Hu, Z. Suppression of muscle wasting by the plant-derived compound ursolic acid in a model of chronic kidney disease. J. Cachexia Sarcopenia Muscle 2017, 8, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Rathor, R.; Agrawal, A.; Kumar, R.; Suryakumar, G.; Singh, S.N. Ursolic acid ameliorates hypobaric hypoxia-induced skeletal muscle protein loss via upreagulating Akt pathway: An experimental study using rat model. IUBMB Life 2021, 73, 375–389. [Google Scholar] [CrossRef]
- Kim, E.Y.; Sudini, K.; Singh, A.K.; Haque, M.; Leaman, D.; Khuder, S.; Ahmed, S. Ursolic acid facilitates apoptosis in rheumatoid arthritis synovial fibroblasts by inducing SP1-mediated Noxa expression and proteasomal degradation of Mcl-1. FASEB J. 2018, 32, 6174–6185. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-N.; Wang, C.C.N.; Chang, H.-Y.; Chu, F.-Y.; Hsu, Y.-A.; Cheng, W.-K.; Ma, W.-C.; Chen, C.-J.; Wan, L.; Lim, Y.-P. Ursolic Acid, a Novel Liver X Receptor α (LXRα) Antagonist Inhibiting Ligand-Induced Nonalcoholic Fatty Liver and Drug-Induced Lipogenesis. J. Agric. Food Chem. 2018, 66, 11647–11662. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, W.; Zheng, L.; Zhang, B.; Yang, X.; Zhang, Q.; Wang, N.; Wang, Y.; Yang, J.; Sha, J.; et al. Ameliorative effect of ursolic acid on ochratoxin A-induced renal cytotoxicity mediated by Lonp1/Aco2/Hsp75. Toxicon 2019, 168, 141–146. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mioc, M.; Milan, A.; Malița, D.; Mioc, A.; Prodea, A.; Racoviceanu, R.; Ghiulai, R.; Cristea, A.; Căruntu, F.; Șoica, C. Recent Advances Regarding the Molecular Mechanisms of Triterpenic Acids: A Review (Part I). Int. J. Mol. Sci. 2022, 23, 7740. https://doi.org/10.3390/ijms23147740
Mioc M, Milan A, Malița D, Mioc A, Prodea A, Racoviceanu R, Ghiulai R, Cristea A, Căruntu F, Șoica C. Recent Advances Regarding the Molecular Mechanisms of Triterpenic Acids: A Review (Part I). International Journal of Molecular Sciences. 2022; 23(14):7740. https://doi.org/10.3390/ijms23147740
Chicago/Turabian StyleMioc, Marius, Andreea Milan, Daniel Malița, Alexandra Mioc, Alexandra Prodea, Roxana Racoviceanu, Roxana Ghiulai, Andreea Cristea, Florina Căruntu, and Codruța Șoica. 2022. "Recent Advances Regarding the Molecular Mechanisms of Triterpenic Acids: A Review (Part I)" International Journal of Molecular Sciences 23, no. 14: 7740. https://doi.org/10.3390/ijms23147740
APA StyleMioc, M., Milan, A., Malița, D., Mioc, A., Prodea, A., Racoviceanu, R., Ghiulai, R., Cristea, A., Căruntu, F., & Șoica, C. (2022). Recent Advances Regarding the Molecular Mechanisms of Triterpenic Acids: A Review (Part I). International Journal of Molecular Sciences, 23(14), 7740. https://doi.org/10.3390/ijms23147740