
Specific Targeting of Antiapoptotic Bcl-2 Proteins as a Radiosensitizing Approach in Solid Tumors

 ,
, 
 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
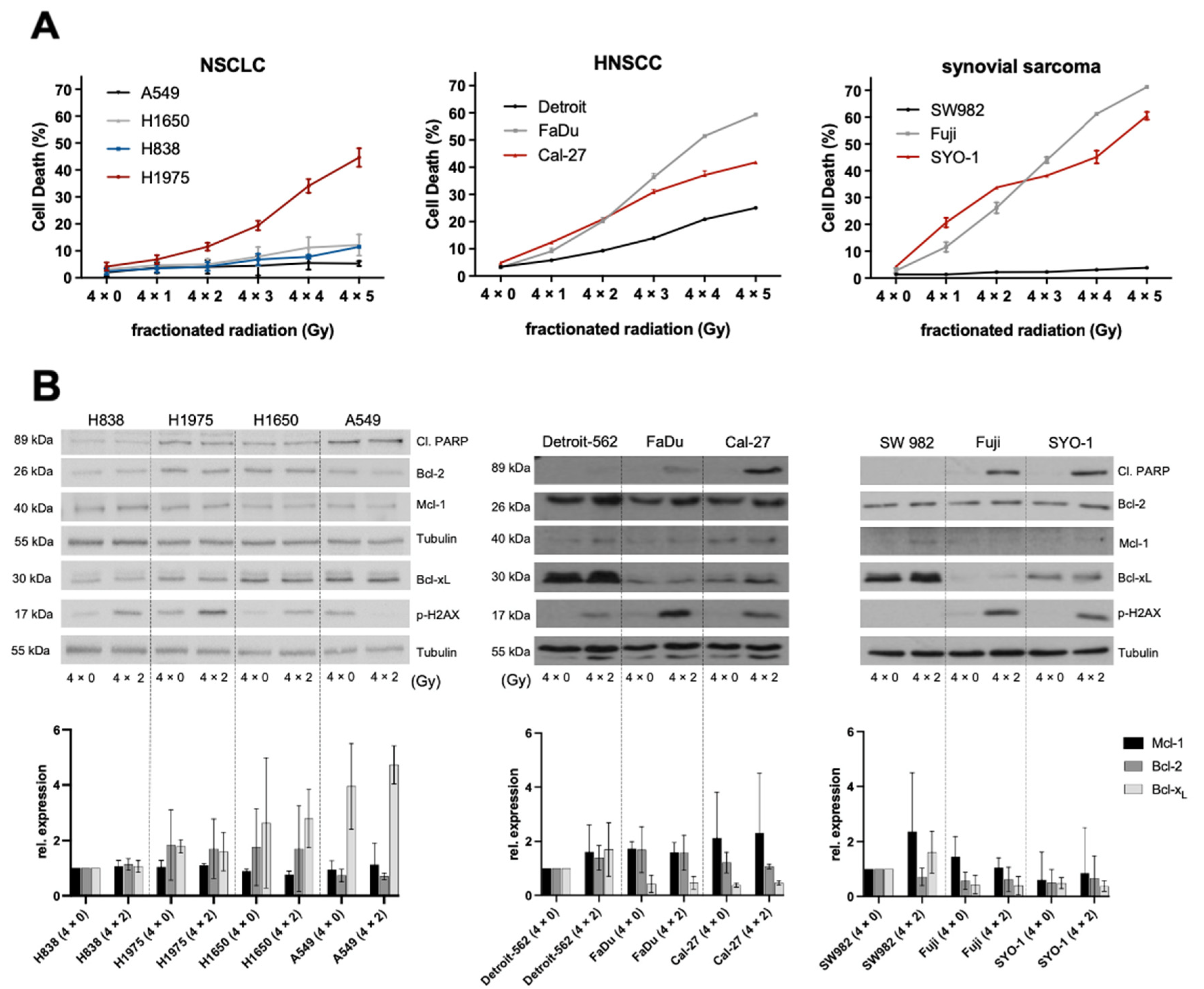
2.1. Ionizing Radiation and Its Effects on NSCLC, HNSCC and Synovial Sarcoma
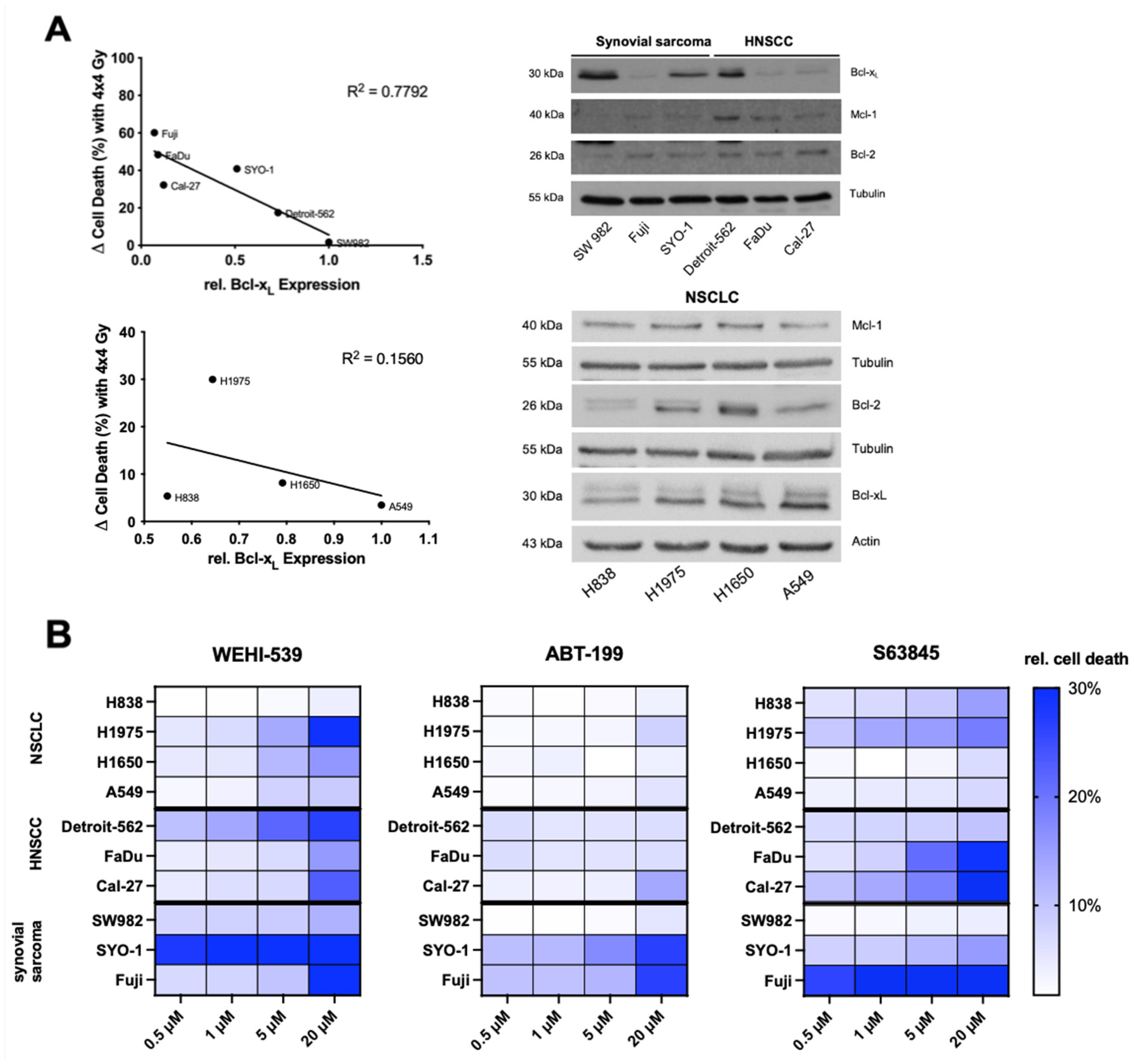
2.2. Higher Expression Levels of Basal Bcl-xL Negatively Correlate with Radiation-Induced Cell Death in HNSCC and Synovial Sarcoma Cells
2.3. Selective Inhibition of the Bcl-2 Protein Family Induces Apoptosis in NSCLC, HNSCC and Synovial Sarcoma Cell Lines
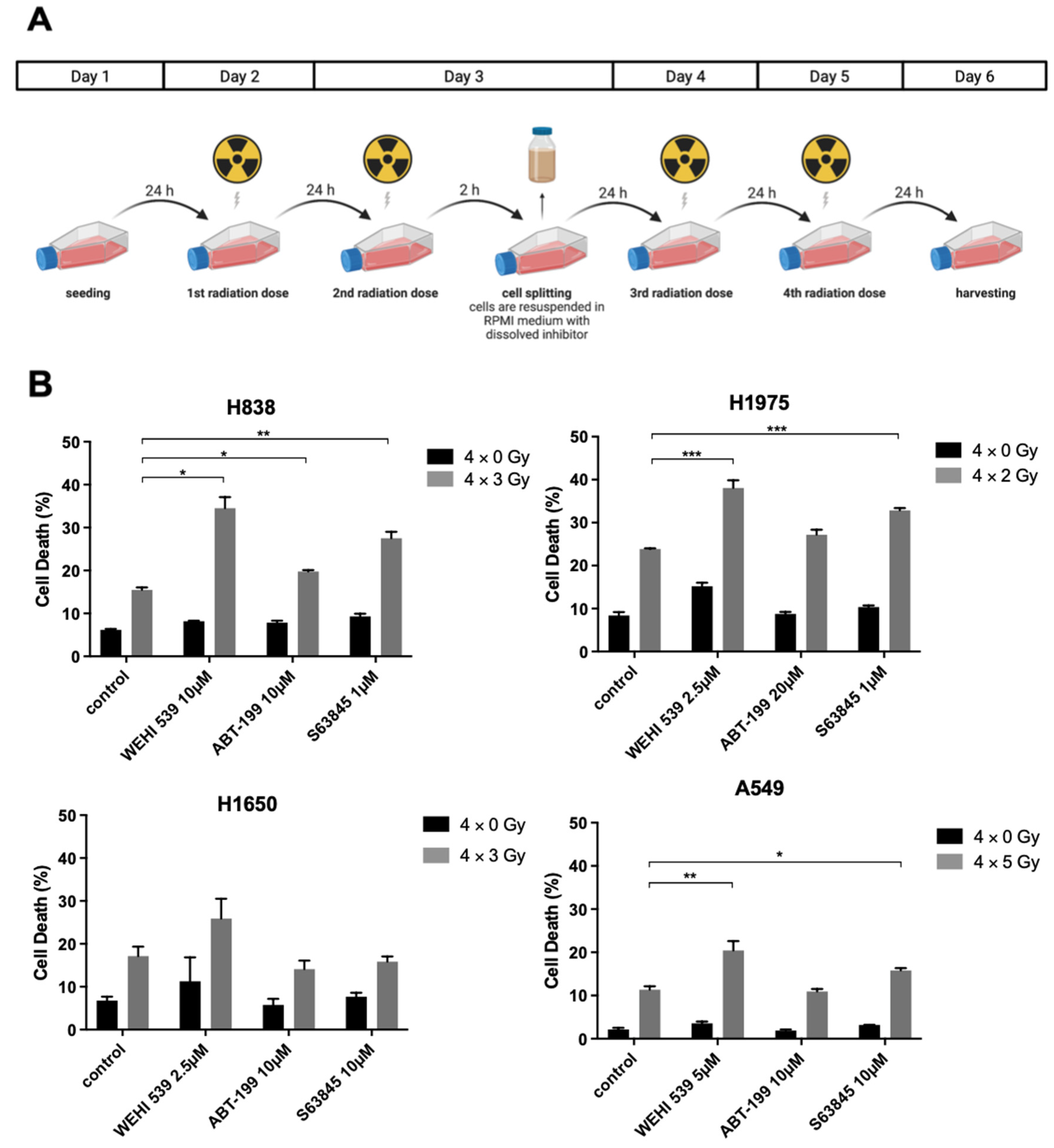
2.4. Mcl-1 and Bcl-xL Inhibition Can Lead to Higher Radiation-Induced Cytotoxicity among NSCLC Cell Lines
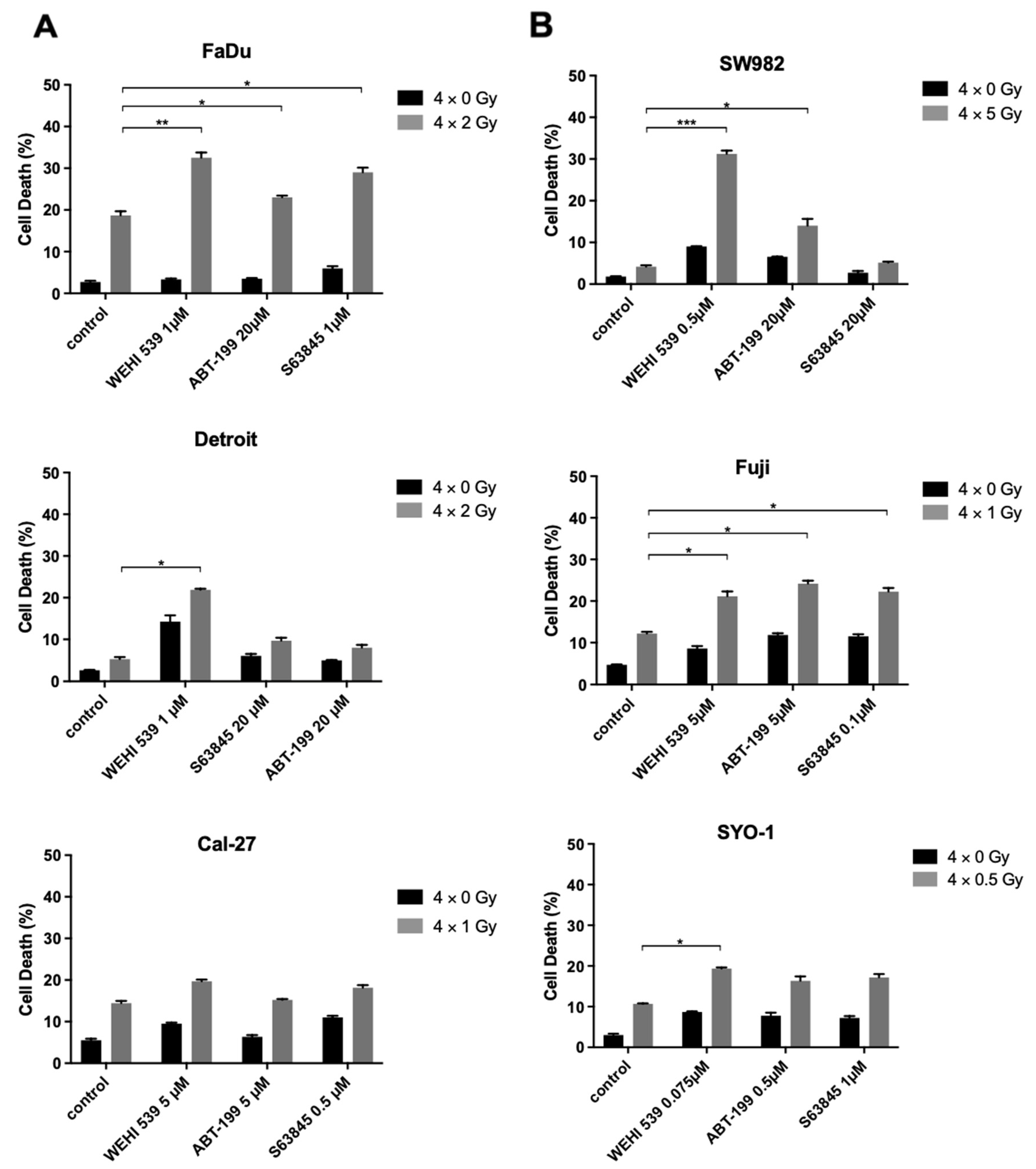
2.5. Bcl-xL Inhibition Plays a Key Role in Radiosensitization among the Resistant HNSCC and Synovial Sarcoma Cell Lines
3. Discussion
4. Material and Methods
4.1. Cell Lines and Reagents
4.2. Protein Isolation, SDS-Page, Densitometry and Western Blotting
4.3. Cell-Death Analysis by Flow Cytometry
4.4. Photon-Beam Radiotherapy for In Vitro Experiments
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.; Edwards, H.; Taub, J.W.; Ge, Y. Evaluating venetoclax and its potential in treatment-naive acute myeloid leukemia. Cancer Mana. Res. 2019, 11, 3197–3213. [Google Scholar]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Lessene, G.; Czabotar, P.E.; Sleebs, B.E.; Zobel, K.; Lowes, K.N.; Adams, J.M.; Baell, J.B.; Colman, P.M.; Deshayes, K.; Fairbrother, W.J.; et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [Google Scholar] [CrossRef]
- Rohner, L.; Reinhart, R.; Iype, J.; Bachmann, S.; Kaufmann, T.; Fux, M. Impact of BH3-mimetics on Human and Mouse Blood Leukocytes: A Comparative Study. Sci. Rep. 2020, 10, 222. [Google Scholar] [CrossRef]
- Orth, M.; Lauber, K.; Niyazi, M.; Friedl, A.; Li, M.; Maihöfer, C.; Schüttrumpf, L.; Ernst, A.; Niemöller, O.M.; Belka, C. Current concepts in clinical radiation oncology. Radiat. Environ. Biophys. 2014, 53, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Ikushima, H. Radiation therapy: State of the art and the future. J. Med. Investig. 2010, 57, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.M.; Hong, Y.; Lee, S.; Liu, P.; Lim, J.H.; Lee, Y.H.; Lee, T.H.; Chang, K.T.; Hong, Y. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 26880–26913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portugal, J.; Mansilla, S.; Bataller, M. Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr. Pharm. Des. 2010, 16, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Ritter, V.; Krautter, F.; Klein, D.; Jendrossek, V.; Rudner, J. Bcl-2/Bcl-xL inhibitor ABT-263 overcomes hypoxia-driven radioresistence and improves radiotherapy. Cell Death Dis. 2021, 12, 694. [Google Scholar] [CrossRef]
- Rudner, J.; Lepple-Wienhues, A.; Budach, W.; Berschauer, J.; Friedrich, B.; Wesselborg, S.; Schulze-Osthoff, K.; Belka, C. Wild-type, mitochondrial and ER-restricted Bcl-2 inhibit DNA damage-induced apoptosis but do not affect death receptor-induced apoptosis. J. Cell Sci. 2001, 114, 4161–4172. [Google Scholar] [CrossRef]
- Wolfsperger, F.; A Hogh-Binder, S.; Schittenhelm, J.; Psaras, T.; Ritter, V.; Bornes, L.; Huber, S.M.; Jendrossek, V.; Rudner, J. Deubiquitylating enzyme USP9x regulates radiosensitivity in glioblastoma cells by Mcl-1-dependent and -independent mechanisms. Cell Death Dis. 2016, 7, e2039. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.; De Salvo, G.L.; Brennan, B.; van Noesel, M.M.; De Paoli, A.; Casanova, M.; Francotte, N.; Kelsey, A.; Alaggio, R.; Oberlin, O.; et al. Synovial sarcoma in children and adolescents: The European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann. Oncol. 2015, 26, 567–572. [Google Scholar] [CrossRef]
- Aupérin, A.; Le Péchoux, C.; Rolland, E.; Curran, W.J.; Furuse, K.; Fournel, P.; Belderbos, J.; Clamon, G.; Ulutin, H.C.; Paulus, R.; et al. Meta-analysis of concomitant versus sequential radiochemotherapy in locally advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 2181–2190. [Google Scholar] [CrossRef]
- Strojan, P.; Hutcheson, K.A.; Eisbruch, A.; Beitler, J.J.; Langendijk, J.A.; Lee, A.W.; Corry, J.; Mendenhall, W.M.; Smee, R.; Rinaldo, A.; et al. Treatment of late sequelae after radiotherapy for head and neck cancer. Cancer Treat. Rev. 2017, 59, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Liu, Z.; Myers, J.N. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J. Cell. Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Bian, D.; Zhang, X.; Zhang, H.; Zhu, Z. Inhibition of Bcl-2 and Bcl-xL overcomes the resistance to the third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer. Mol. Med. Rep. 2021, 23, 48. [Google Scholar] [CrossRef] [PubMed]
- Laudanski, J.; Niklinska, W.; Burzykowski, T.; Chyczewski, L.; Niklinski, J. Prognostic significance of p53 and bcl-2 abnormalities in operable nonsmall cell lung cancer. Eur. Respir. J. 2001, 17, 660–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.S.; Pajak, T.F.; Forastiere, A.A.; Jacobs, J.; Campbell, B.H.; Saxman, S.B.; Kish, J.A.; Kim, H.E.; Cmelak, A.J.; Rotman, M.; et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2004, 350, 1937–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, W.E.E.; Poettgen, C.; Gauler, T.C.; Friedel, G.; Veit, S.; Heinrich, V.; Welter, S.; Budach, W.; Spengler, W.; Kimmich, M.; et al. Phase III Study of Surgery Versus Definitive Concurrent Chemoradiotherapy Boost in Patients With Resectable Stage IIIA(N2) and Selected IIIB Non-Small-Cell Lung Cancer After Induction Chemotherapy and Concurrent Chemoradiotherapy (ESPATUE). J. Clin. Oncol. 2015, 33, 4194–4201. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Cordell, K.G.; Lee, J.S.; Worden, F.P.; Prince, M.E.; Tran, H.H.; Wolf, G.T.; Urba, S.G.; Chepeha, D.B.; Teknos, T.N.; et al. EGFR, p16, HPV Titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J. Clin. Oncol. 2008, 26, 3128–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.A.; Kumar, B.; Cordell, K.G.; Prince, M.E.; Tran, H.H.; Wolf, G.T.; Chepeha, D.B.; Teknos, T.N.; Wang, S.; Eisbruch, A.; et al. Targeting apoptosis to overcome cisplatin resistance: A translational study in head and neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, S106–S108. [Google Scholar] [CrossRef] [Green Version]
- Ow, T.J.; Fulcher, C.D.; Thomas, C.; Broin, P.Ó.; López, A.; Reyna, D.E.; Smith, R.V.; Sarta, C.; Prystowsky, M.B.; Schlecht, N.F.; et al. Optimal targeting of BCL-family proteins in head and neck squamous cell carcinoma requires inhibition of both BCL-xL and MCL-1. Oncotarget 2019, 10, 494–510. [Google Scholar] [CrossRef]
- Fairchild, C.K., Jr.; Floros, K.; Jacob, S.; Coon, C.; Puchalapalli, M.; Hu, B.; Harada, H.; Dozmorov, M.; Koblinski, J.; Smith, S.; et al. Unmasking BCL-2 Addiction in Synovial Sarcoma by Overcoming Low NOXA. Cancers 2021, 13, 2310. [Google Scholar] [CrossRef]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, J.-B.; Espenel, S.; Louati, S.; Gauthier, A.; Garcia, M.-A.; Vial, N.; Malésys, C.; Ardail, D.; Alphonse, G.; Wozny, A.-S.; et al. Combining radiation to EGFR and Bcl-2 blockade: A new approach to target cancer stem cells in head and neck squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2021, 147, 1905–1916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Guttikonda, S.; Roberts, L.; Uziel, T.; Semizarov, D.; Elmore, S.W.; Leverson, J.D.; Lam, L.T. Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene 2011, 30, 1963–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Coppola, D.; Livingston, S.; Cress, W.D.; Haura, E.B. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol. Ther. 2005, 4, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamil, S.; Stoica, C.; Hackett, T.L.; Duronio, V. MCL-1 localizes to sites of DNA damage and regulates DNA damage response. Cell Cycle 2010, 9, 2843–2855. [Google Scholar] [CrossRef] [Green Version]
- Mattoo, A.R.; Pandita, R.K.; Chakraborty, S.; Charaka, V.; Mujoo, K.; Hunt, C.R.; Pandita, T.K. MCL-1 Depletion Impairs DNA Double-Strand Break Repair and Reinitiation of Stalled DNA Replication Forks. Mol. Cell. Biol. 2017, 37, e00535-e16. [Google Scholar] [CrossRef] [Green Version]
- Munkhbaatar, E.; Dietzen, M.; Agrawal, D.; Anton, M.; Jesinghaus, M.; Boxberg, M.; Pfarr, N.; Bidola, P.; Uhrig, S.; Höckendorf, U.; et al. MCL-1 gains occur with high frequency in lung adenocarcinoma and can be targeted therapeutically. Nat. Commun. 2020, 11, 4527. [Google Scholar] [CrossRef]
- Barrott, J.J.; Zhu, J.-F.; Smith-Fry, K.; Susko, A.M.; Nollner, D.; Burrell, L.D.; Pozner, A.; Capecchi, M.R.; Yap, J.T.; Cannon-Albright, L.A.; et al. The Influential Role of BCL2 Family Members in Synovial Sarcomagenesis. Mol. Cancer Res. 2017, 15, 1733–1740. [Google Scholar] [CrossRef] [Green Version]
- de Vos, S.; Leonard, J.P.; Friedberg, J.W.; Zain, J.; Dunleavy, K.; Humerickhouse, R.; Hayslip, J.; Pesko, J.; Wilson, W.H. Safety and efficacy of navitoclax, a BCL-2 and BCL-X(L) inhibitor, in patients with relapsed or refractory lymphoid malignancies: Results from a phase 2a study. Leuk. Lymphoma 2021, 62, 810–818. [Google Scholar] [CrossRef]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.-L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra240. [Google Scholar] [CrossRef] [PubMed]
- Scherr, A.-L.; Mock, A.; Gdynia, G.; Schmitt, N.; Heilig, C.E.; Korell, F.; Rhadakrishnan, P.; Hoffmeister, P.; Metzeler, K.H.; Schulze-Osthoff, K.; et al. Identification of BCL-XL as highly active survival factor and promising therapeutic target in colorectal cancer. Cell Death Dis. 2020, 11, 875. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Chou, H.C.; Fu, Y.N.; Yeh, C.L.; Cheng, H.W.; Chang, I.C.; Liu, K.J.; Chang, G.C.; Tsai, T.F.; Tsai, S.F.; et al. EGFR over-expression in non-small cell lung cancers harboring EGFR mutations is associated with marked down-regulation of CD82. Biochim. Biophys. Acta. 2015, 1852, 1540–1549. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.K.; Kim, H.P.; Han, S.W.; Oh, D.Y.; Im, S.-A.; Bang, Y.-J.; Kim, T.-Y. KRAS mutant lung cancer cells are differentially responsive to MEK inhibitor due to AKT or STAT3 activation: Implication for combinatorial approach. Mol. Carcinog. 2010, 49, 353–362. [Google Scholar] [CrossRef]
- Teicher, B.A.; Polley, E.C.; Kunkel, M.; Evans, D.A.; Silvers, T.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Selby, M.; et al. Sarcoma Cell Line Screen of Oncology Drugs and Investigational Agents Identifies Patterns Associated with Gene and microRNA Expression. Mol. Cancer Ther. 2015, 14, 2452–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalu, N.N.; Mazumdar, T.; Peng, S.; Shen, L.; Sambandam, V.; Rao, X.; Xi, Y.; Li, L.; Qi, Y.; Gleber-Netto, F.O.; et al. Genomic characterization of human papillomavirus-positive and -negative human squamous cell cancer cell lines. Oncotarget 2017, 8, 86369–86383. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobol, B.; Azzam Nieto, O.; Eberlein, E.L.; Scherr, A.-L.; Ismail, L.; Kessler, A.; Nader, L.; Schwab, M.; Hoffmeister, P.; Schmitt, N.; et al. Specific Targeting of Antiapoptotic Bcl-2 Proteins as a Radiosensitizing Approach in Solid Tumors. Int. J. Mol. Sci. 2022, 23, 7850. https://doi.org/10.3390/ijms23147850
Sobol B, Azzam Nieto O, Eberlein EL, Scherr A-L, Ismail L, Kessler A, Nader L, Schwab M, Hoffmeister P, Schmitt N, et al. Specific Targeting of Antiapoptotic Bcl-2 Proteins as a Radiosensitizing Approach in Solid Tumors. International Journal of Molecular Sciences. 2022; 23(14):7850. https://doi.org/10.3390/ijms23147850
Chicago/Turabian StyleSobol, Benjamin, Osama Azzam Nieto, Emily Lara Eberlein, Anna-Lena Scherr, Lars Ismail, Annika Kessler, Luisa Nader, Maximilian Schwab, Paula Hoffmeister, Nathalie Schmitt, and et al. 2022. "Specific Targeting of Antiapoptotic Bcl-2 Proteins as a Radiosensitizing Approach in Solid Tumors" International Journal of Molecular Sciences 23, no. 14: 7850. https://doi.org/10.3390/ijms23147850
APA StyleSobol, B., Azzam Nieto, O., Eberlein, E. L., Scherr, A.-L., Ismail, L., Kessler, A., Nader, L., Schwab, M., Hoffmeister, P., Schmitt, N., Jäger, D., Welte, S., Seidensaal, K., Christopoulos, P., Heilig, C., Kriegsmann, K., Fröhling, S., Kriegsmann, M., Hess, J., & Köhler, B. C. (2022). Specific Targeting of Antiapoptotic Bcl-2 Proteins as a Radiosensitizing Approach in Solid Tumors. International Journal of Molecular Sciences, 23(14), 7850. https://doi.org/10.3390/ijms23147850