
Analysis of Binding Determinants for Different Classes of Competitive and Noncompetitive Inhibitors of Glycine Transporters


Abstract
:1. Introduction
2. Results and Discussion
2.1. Model Building
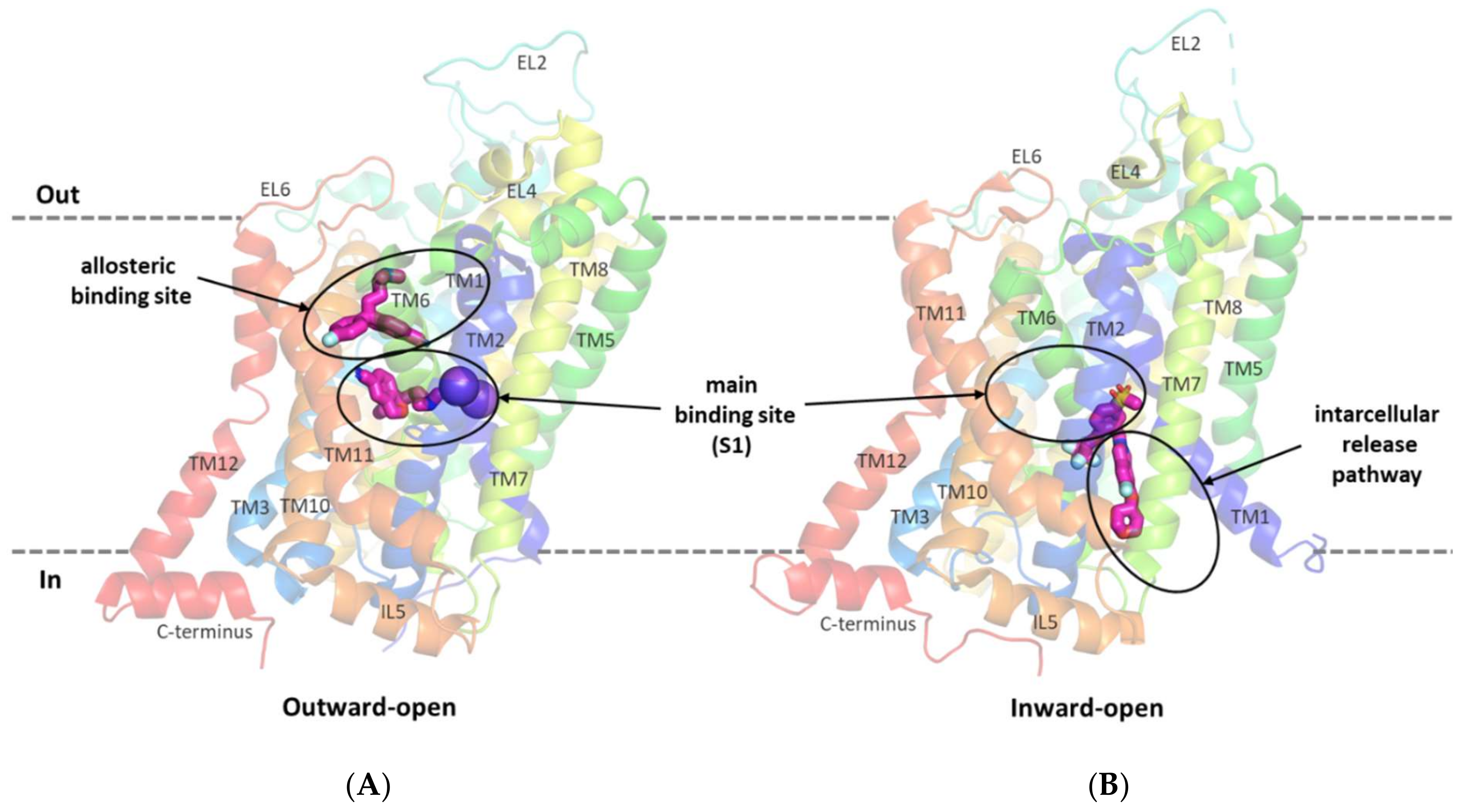
2.2. Structure of GlyT-1 and GlyT-2 Binding Sites
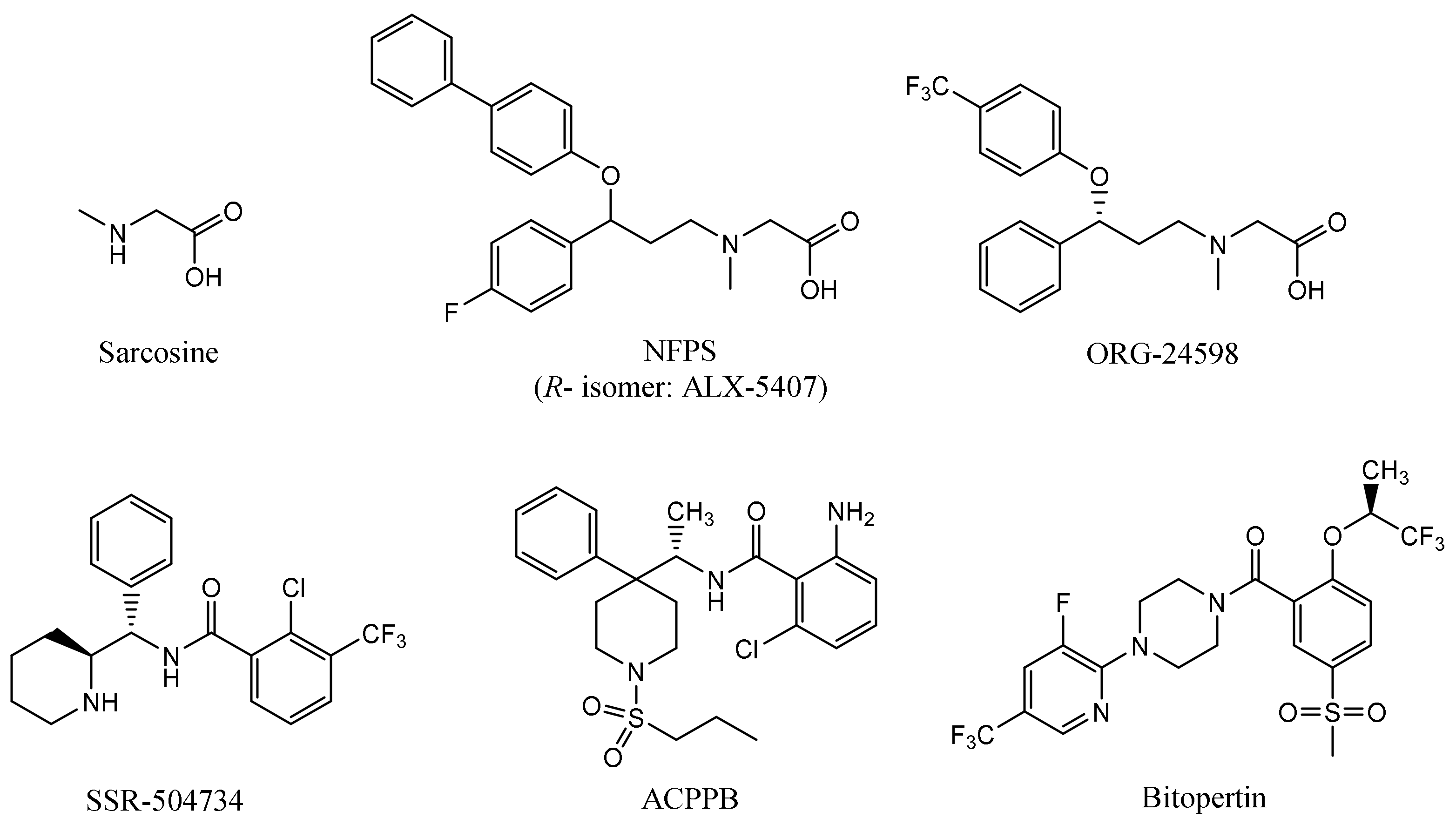
2.3. Ligand Binding Studies
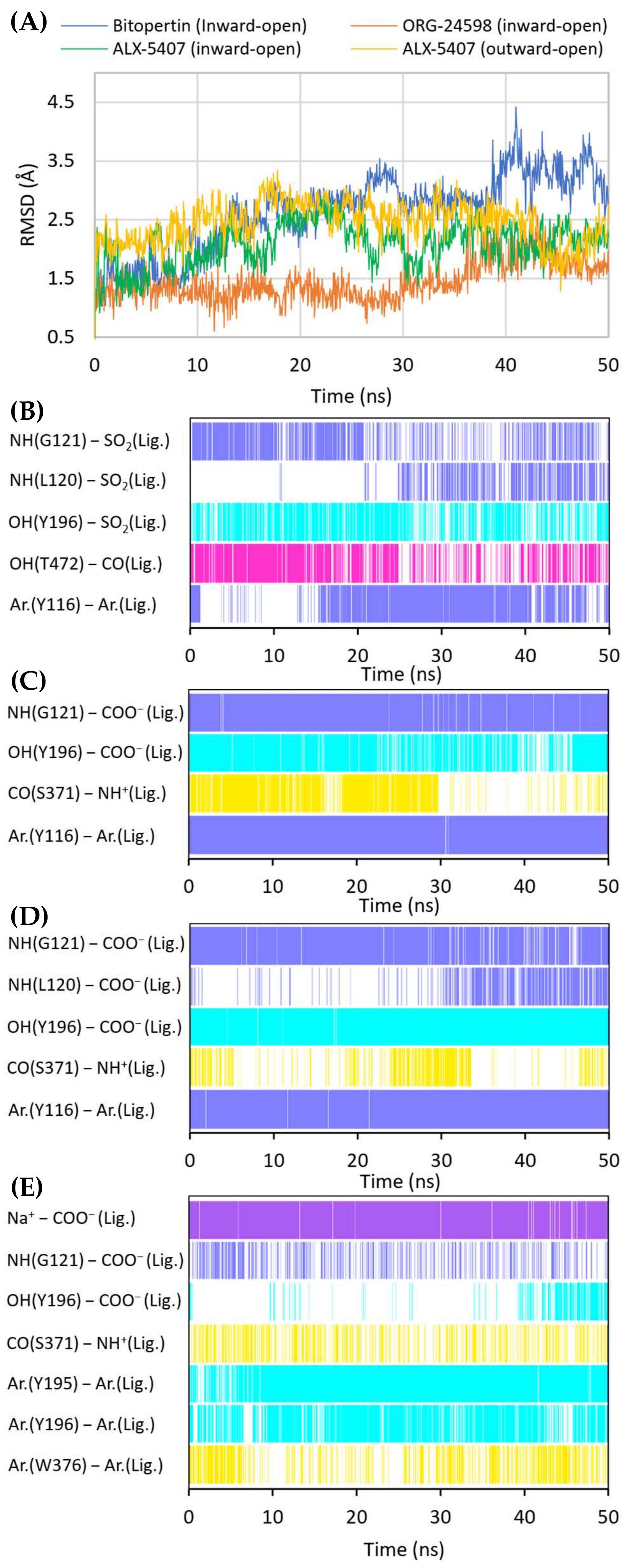
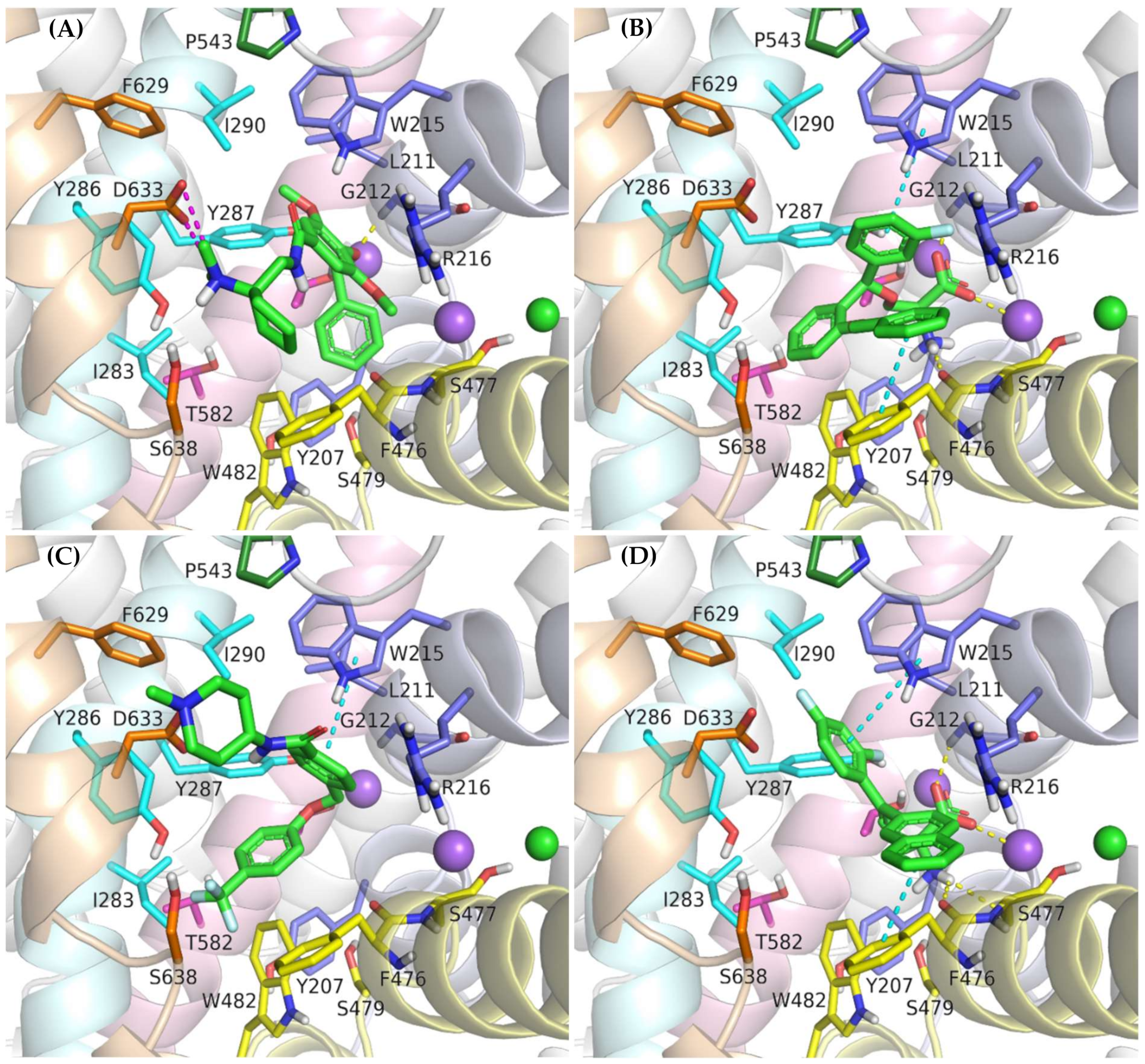
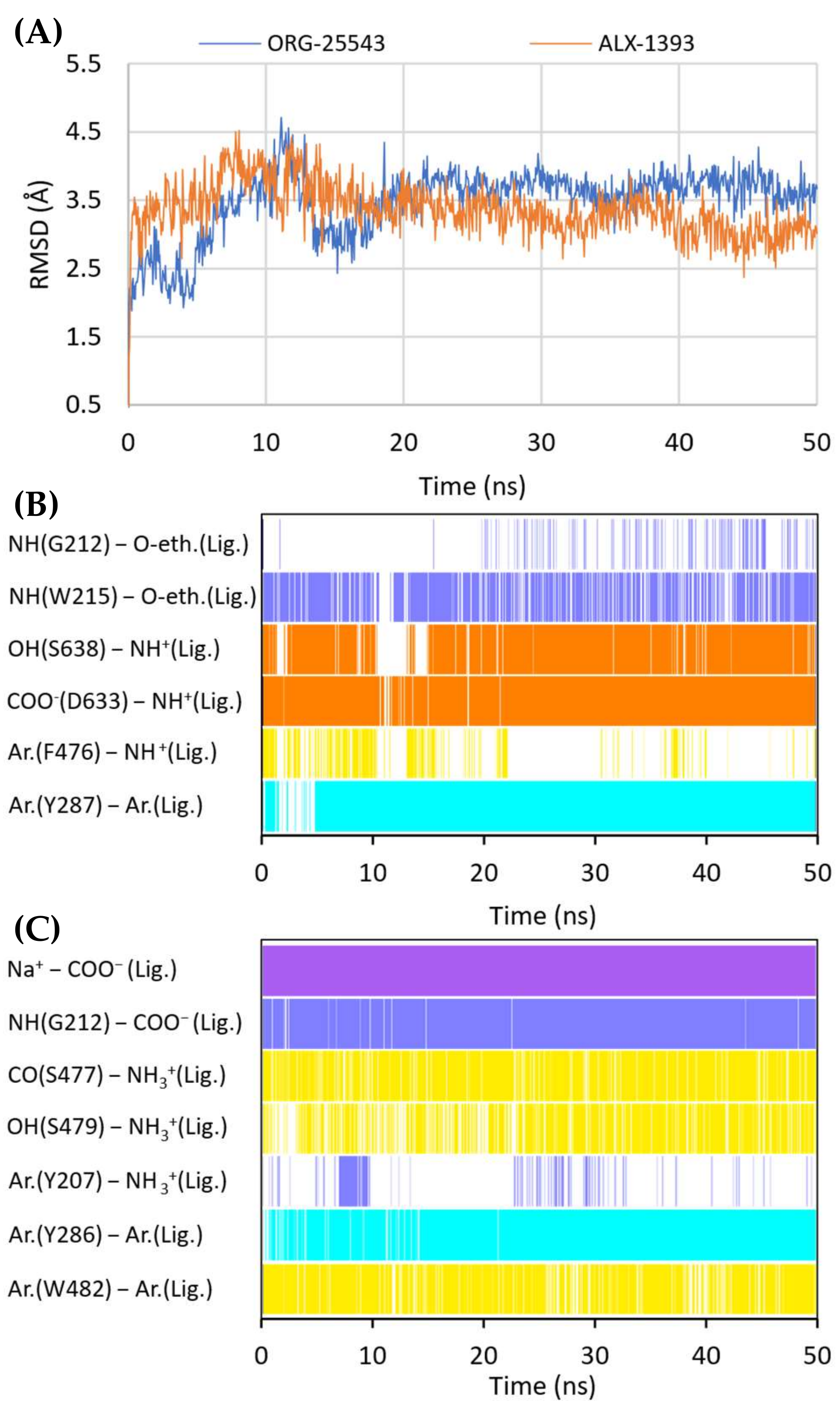
2.4. Binding Mode of Noncompetitive GlyT-1 Inhibitors
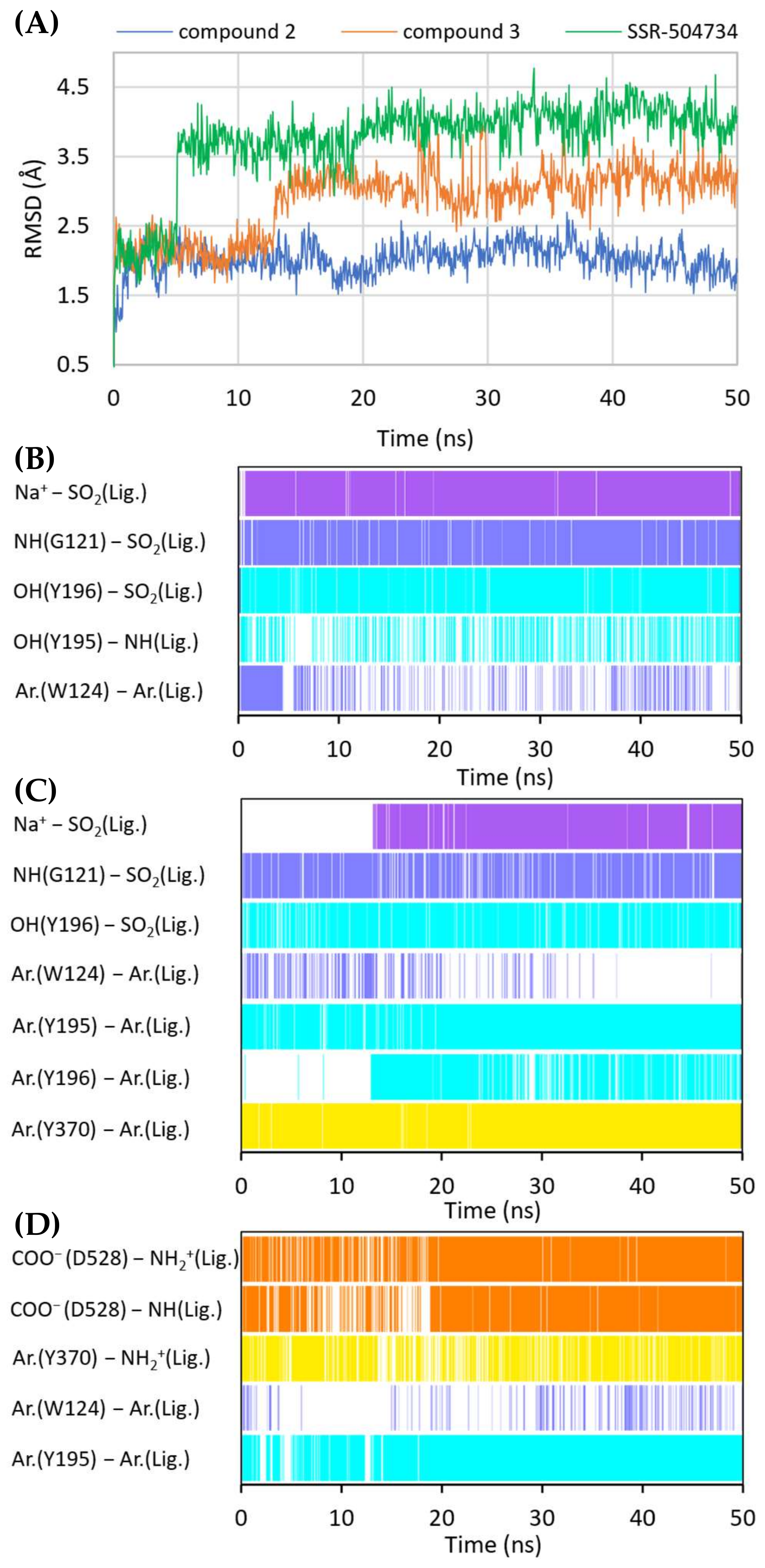
2.5. Binding Mode of Competitive GlyT-1 Inhibitors
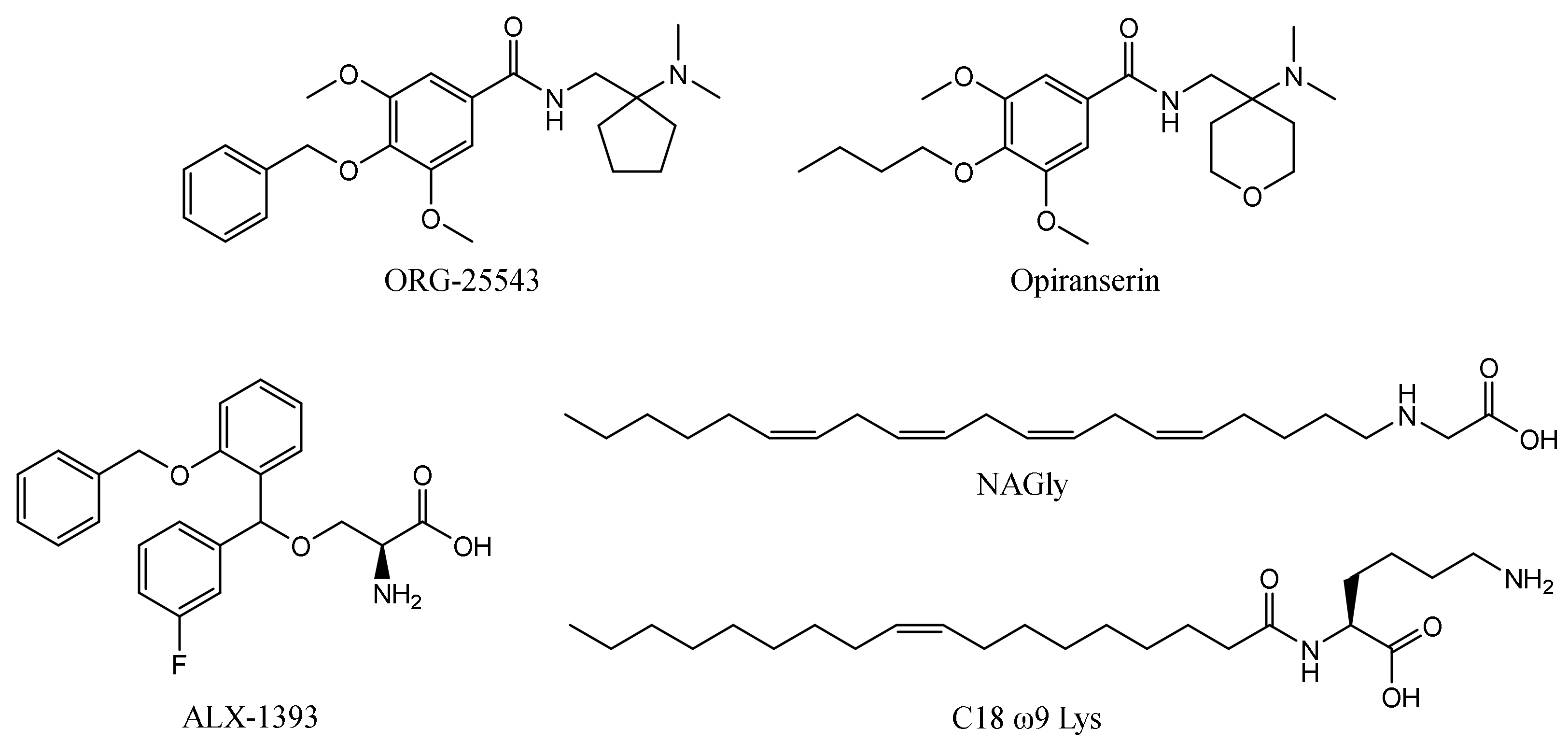
2.6. Binding Mode of GlyT-2 Inhibitors
3. Methods
3.1. Homology Modeling
3.2. Docking Studies
3.3. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Legendre, P. The glycinergic inhibitory synapse. Cell. Mol. Life Sci. 2001, 58, 760–793. [Google Scholar] [CrossRef] [PubMed]
- Zafra, F.; Giménez, C. Glycine transporters and synaptic function. IUBMB Life 2008, 60, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Köhr, G. NMDA receptor function: Subunit composition versus spatial distribution. Cell Tissue Res. 2006, 326, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Kristensen, A.S.; Andersen, J.; Jørgensen, T.N.; Sørensen, L.; Eriksen, J.; Loland, C.J.; Strømgaard, K.; Gether, U.; Jorgensen, T.N.; Sorensen, L.; et al. SLC6 neurotransmitter transporters: Structure, function, and regulation. Pharmacol. Rev. 2011, 63, 585–640. [Google Scholar] [CrossRef]
- Cioffi, C.L. Glycine transporter-1 inhibitors: A patent review (2011–2016). Expert Opin. Ther. Pat. 2018, 28, 197–210. [Google Scholar] [CrossRef]
- Al-Khrasani, M.; Mohammadzadeh, A.; Balogh, M.; Király, K.; Barsi, S.; Hajnal, B.; Köles, L.; Zádori, Z.S.; Harsing, L.G. Glycine transporter inhibitors: A new avenue for managing neuropathic pain. Brain Res. Bull. 2019, 152, 143–158. [Google Scholar] [CrossRef]
- Porter, R.A.; Dawson, L.A. GlyT-1 inhibitors: From hits to clinical candidates. Small Mol. Ther. Schizophr. 2015, 13, 51–100. [Google Scholar] [CrossRef]
- Marques, B.L.; Oliveira-lima, O.C.; Carvalho, G.A.; De Almeida, R.; Izidoro, R.; Cambraia, R.; Marra, E.; Ribeiro, R.; Klempin, F.; Ulrich, H.; et al. Neuroscience and biobehavioral reviews neurobiology of glycine transporters: From molecules to behavior. Neurosci. Biobehav. Rev. 2020, 118, 97–110. [Google Scholar] [CrossRef]
- Harvey, R.J.; Yee, B.K. Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat. Rev. Drug Discov. 2013, 12, 866–885. [Google Scholar] [CrossRef]
- Cioffi, C.L. Inhibition of glycine re-uptake: A potential approach for treating pain by augmenting glycine-mediated spinal neurotransmission and blunting central nociceptive signaling. Biomolecules 2021, 11, 864. [Google Scholar] [CrossRef] [PubMed]
- López-Corcuera, B.; Arribas-González, E.; Aragón, C. Hyperekplexia-associated mutations in the neuronal glycine transporter 2. Neurochem. Int. 2019, 123, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, S.; Kim, A.; Yoon, J.; Jang, K.; Lee, D.H.; Cho, S.; Lee, S.R.; Yu, K.; Chung, J. Characteristics of a novel nonopioid analgesic, VVZ-149 injections in healthy volunteers: A first-in-class, first-in-human. J. Clin. Pharmacol. 2017, 58, 61–73. [Google Scholar] [CrossRef]
- Song, I.; Cho, S.; Nedeljkovic, S.S.; Lee, S.R.; Lee, C.; Kim, J.; Bai, S.J. Role of VVZ-149, a novel analgesic molecule, in the affective component of pain: Results from an exploratory proof-of-concept study of postoperative pain following laparoscopic and robotic-laparoscopic gastrectomy. Pain Med. 2021, 22, 2037–2049. [Google Scholar] [CrossRef]
- Atkinson, B.N.; Bell, S.C.; De Vivo, M.; Kowalski, L.R.; Lechner, S.M.; Ognyanov, V.I.; Tham, C.-S.; Tsai, C.; Jia, J.; Ashton, D.; et al. ALX 5407: A potent, selective inhibitor of the hGlyT1 glycine transporter. Mol. Pharmacol. 2001, 60, 1414–1420. [Google Scholar] [CrossRef]
- Mezler, M.; Hornberger, W.; Mueller, R.; Schmidt, M.; Amberg, W.; Braje, W.; Ochse, M.; Schoemaker, H.; Behl, B. Inhibitors of GlyT1 affect glycine transport via discrete binding sites. Mol. Pharmacol. 2009, 75, 258. [Google Scholar] [CrossRef] [PubMed]
- Harsing, L., Jr.; Juranyi, Z.; Gacsalyi, I.; Tapolcsanyi, P.; Czompa, A.; Matyus, P. Glycine transporter type-1 and its inhibitors. Curr. Med. Chem. 2006, 13, 1017–1044. [Google Scholar] [CrossRef]
- Kopec, K.; Flood, D.G.; Gasior, M.; McKenna, B.A.W.; Zuvich, E.; Schreiber, J.; Salvino, J.M.; Durkin, J.T.; Ator, M.A.; Marino, M.J. Glycine transporter (GlyT1) inhibitors with reduced residence time increase prepulse inhibition without inducing hyperlocomotion in DBA/2 mice. Biochem. Pharmacol. 2010, 80, 1407–1417. [Google Scholar] [CrossRef]
- Alberati, D.; Moreau, J.-L.; Lengyel, J.; Hauser, N.; Mory, R.; Borroni, E.; Pinard, E.; Knoflach, F.; Schlotterbeck, G.; Hainzl, D.; et al. Glycine reuptake inhibitor RG1678: A pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology 2012, 62, 1152–1161. [Google Scholar] [CrossRef]
- Rosenbrock, H.; Desch, M.; Kleiner, O.; Dorner-Ciossek, C.; Schmid, B.; Keller, S.; Schlecker, C.; Moschetti, V.; Goetz, S.; Liesenfeld, K.-H.; et al. Evaluation of pharmacokinetics and pharmacodynamics of BI 425809, a novel GlyT1 inhibitor: Translational studies. Clin. Transl. Sci. 2018, 11, 616–623. [Google Scholar] [CrossRef]
- Fleischhacker, W.W.; Podhorna, J.; Gröschl, M.; Hake, S.; Zhao, Y.; Huang, S.; Keefe, R.S.E.; Desch, M.; Brenner, R.; Walling, D.P.; et al. Efficacy and safety of the novel glycine transporter inhibitor BI 425809 once daily in patients with schizophrenia: A double-blind, randomised, placebo-controlled phase 2 study. Lancet Psychiatry 2021, 8, 191–201. [Google Scholar] [CrossRef]
- Bugarski-Kirola, D.; Iwata, N.; Sameljak, S.; Reid, C.; Blaettler, T.; Millar, L.; Marques, T.R.; Garibaldi, G.; Kapur, S. Efficacy and safety of adjunctive bitopertin versus placebo in patients with suboptimally controlled symptoms of schizophrenia treated with antipsychotics: Results from three phase 3, randomised, double-blind, parallel-group, placebo-controlled, multicent. Lancet Psychiatry 2016, 3, 1115–1128. [Google Scholar] [CrossRef]
- Herdon, H.J.; Roberts, J.C.; Coulton, S.; Porter, R.A. Pharmacological characterisation of the GlyT-1 glycine transporter using two novel radioligands. Neuropharmacology 2010, 59, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, C.L.; Liu, S.; Wolf, M.A.; Guzzo, P.R.; Sadalapure, K.; Parthasarathy, V.; Loong, D.T.J.; Maeng, J.-H.; Carulli, E.; Fang, X.; et al. Synthesis and biological evaluation of N-((1-(4-(sulfonyl)piperazin-1-yl)cycloalkyl)methyl)benzamide inhibitors of glycine transporter-1. J. Med. Chem. 2016, 59, 8473–8494. [Google Scholar] [CrossRef] [PubMed]
- Sheffler, D.J.; Nedelcovych, M.T.; Williams, R.; Turner, S.C.; Duerk, B.B.; Robbins, M.R.; Jadhav, S.B.; Niswender, C.M.; Jones, C.K.; Conn, P.J.; et al. Novel GlyT1 inhibitor chemotypes by scaffold hopping. Part 2: Development of a [3.3.0]-based series and other piperidine bioisosteres. Bioorg. Med. Chem. Lett. 2014, 24, 1062–1066. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.K.; Sheffler, D.J.; Williams, R.; Jadhav, S.B.; Felts, A.S.; Morrison, R.D.; Niswender, C.M.; Daniels, J.S.; Conn, P.J.; Lindsley, C.W. Novel GlyT1 inhibitor chemotypes by scaffold hopping. Part 1: Development of a potent and CNS penetrant [3.1.0]-based lead. Bioorg. Med. Chem. Lett. 2014, 24, 1067–1070. [Google Scholar] [CrossRef] [Green Version]
- Amberg, W.; Lange, U.E.W.; Ochse, M.; Pohlki, F.; Behl, B.; Relo, A.L.; Hornberger, W.; Hoft, C.; Mezler, M.; Sydor, J.; et al. Discovery of novel aminotetralines and aminochromanes as selective and competitive glycine transporter 1 (GlyT1) inhibitors. J. Med. Chem. 2018, 61, 7503–7524. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Brewer, J.T.; Li, H.; Lao, Y.; Amberg, W.; Behl, B.; Akritopoulou-Zanze, I.; Dietrich, J.; Lange, U.E.W.; et al. De novo design, synthesis, and biological evaluation of 3,4-disubstituted pyrrolidine sulfonamides as potent and selective glycine transporter 1 competitive inhibitors. J. Med. Chem. 2018, 61, 7486–7502. [Google Scholar] [CrossRef]
- Benito-Muñoz, C.; Perona, A.; Felipe, R.; Pérez-Siles, G.; Núñez, E.; Aragón, C.; López-Corcuera, B. Structural Determinants of the neuronal glycine transporter 2 for the selective inhibitors ALX1393 and ORG25543. ACS Chem. Neurosci. 2021, 12, 1860–1872. [Google Scholar] [CrossRef]
- Frangos, Z.J.; Cantwell Chater, R.P.; Vandenberg, R.J. Glycine transporter 2: Mechanism and allosteric modulation. Front. Mol. Biosci. 2021, 8, 1–13. [Google Scholar] [CrossRef]
- Caulfield, W.L.; Collie, I.T.; Dickins, R.S.; Epemolu, O.; McGuire, R.; Hill, D.R.; McVey, G.; Morphy, J.R.; Rankovic, Z.; Sundaram, H. The first potent and selective inhibitors of the glycine transporter type 2. J. Med. Chem. 2001, 44, 2679–2682. [Google Scholar] [CrossRef] [PubMed]
- Mingorance-Le Meur, A.; Ghisdal, P.; Mullier, B.; De Ron, P.; Downey, P.; Van Der Perren, C.; Declercq, V.; Cornelis, S.; Famelart, M.; Van Asperen, J.; et al. Reversible inhibition of the glycine transporter GlyT2 circumvents acute toxicity while preserving efficacy in the treatment of pain. Br. J. Pharmacol. 2013, 170, 1053–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostyn, S.N.; Sarker, S.; Muthuraman, P.; Raja, A.; Shimmon, S.; Rawling, T.; Cioffi, C.L.; Vandenberg, R.J. Photoswitchable ORG25543 congener enables optical control of glycine transporter 2. ACS Chem. Neurosci. 2020, 11, 1250–1258. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.M.; Bisogno, T.; Petros, T.J.; Chang, S.Y.; Zavitsanos, P.A.; Zipkin, R.E.; Sivakumar, R.; Coop, A.; Maeda, D.Y.; de Petrocellis, L.; et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J. Biol. Chem. 2001, 276, 42639–42644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostyn, S.N.; Rawling, T.; Mohammadi, S.; Shimmon, S.; Frangos, Z.J.; Sarker, S.; Yousuf, A.; Vetter, I.; Ryan, R.M.; Christie, M.J.; et al. Development of an N-acyl amino acid that selectively inhibits the glycine transporter 2 to produce analgesia in a rat model of chronic pain. J. Med. Chem. 2019, 62, 2466–2484. [Google Scholar] [CrossRef] [PubMed]
- Mostyn, S.N.; Carland, J.E.; Shimmon, S.; Ryan, R.M.; Rawling, T.; Vandenberg, R.J. Synthesis and characterization of novel acyl-glycine inhibitors of GlyT2. ACS Chem. Neurosci. 2017, 8, 1949–1959. [Google Scholar] [CrossRef]
- Carland, J.E.; Mansfield, R.E.; Ryan, R.M.; Vandenberg, R.J. Oleoyl-l-carnitine inhibits glycine transport by GlyT2. Br. J. Pharmacol. 2013, 168, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Goehring, A.; Wang, K.H.; Penmatsa, A.; Ressler, R.; Gouaux, E. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 2013, 503, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.A.; Gouaux, E. Structural basis for recognition of diverse antidepressants by the human serotonin transporter. Nat. Struct. Mol. Biol. 2018, 25, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Benito-Muñoz, C.; Perona, A.; Abia, D.; dos Santos, H.G.; Núñez, E.; Aragón, C.; López-Corcuera, B. Modification of a putative third sodium site in the glycine transporter GlyT2 influences the chloride dependence of substrate transport. Front. Mol. Neurosci. 2018, 11, 1–18. [Google Scholar] [CrossRef]
- Gross, S.P.; Stavans, J.; Heisler, M.G.; Shapiro, B.E.; Meyerowitz, E.M.; Mjolsness, E.; Hogetsu, T.; Hara, N.; Calder, G.; Fox, S.; et al. A competitive inhibitor traps LeuT in an open-to-out conformation. Science 2008, 2454, 1655–1661. [Google Scholar]
- Krishnamurthy, H.; Gouaux, E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 2012, 481, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.T.; Zhang, Y.W.; Campbell, S.D.; Rudnick, G. Ibogaine, a noncompetitive inhibitor of serotonin transport, acts by stabilizing the cytoplasm-facing state of the transporter. J. Biol. Chem. 2007, 282, 29441–29447. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.A.; Yang, D.; Zhao, Z.; Wen, P.C.; Yoshioka, C.; Tajkhorshid, E.; Gouaux, E. Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 2019, 569, 141–145. [Google Scholar] [CrossRef]
- Shahsavar, A.; Stohler, P.; Bourenkov, G.; Zimmermann, I.; Siegrist, M.; Guba, W.; Pinard, E.; Sinning, S.; Seeger, M.A.; Schneider, T.R.; et al. Structural insights into the inhibition of glycine reuptake. Nature 2021, 591, 677–681. [Google Scholar] [CrossRef]
- Wilson, K.A.; Mostyn, S.N.; Frangos, Z.J.; Shimmon, S.; Rawling, T.; Vandenberg, R.J.; O’Mara, M.L. The allosteric inhibition of glycine transporter 2 by bioactive lipid analgesics is controlled by penetration into a deep lipid cavity. J. Biol. Chem. 2021, 296, 100282. [Google Scholar] [CrossRef]
- Mostyn, S.N.; Wilson, K.A.; Schumann-Gillett, A.; Frangos, Z.J.; Shimmon, S.; Rawling, T.; Ryan, R.M.; O’Mara, M.L.; Vandenberg, R.J. Identification of an allosteric binding site on the human glycine transporter, GlyT2, for bioactive lipid analgesics. eLife 2019, 8, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Carland, J.E.; Thomas, M.; Mostyn, S.N.; Subramanian, N.; O’Mara, M.L.; Ryan, R.M.; Vandenberg, R.J. Molecular determinants for substrate interactions with the glycine transporter GlyT2. ACS Chem. Neurosci. 2018, 9, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Łątka, K.; Jończyk, J.; Bajda, M. Structure modeling of γ-aminobutyric acid transporters—Molecular basics of ligand selectivity. Int. J. Biol. Macromol. 2020, 158, 1380–1389. [Google Scholar] [CrossRef]
- Łątka, K.; Jończyk, J.; Bajda, M. γ-aminobutyric acid transporters as relevant biological target: Their function, structure, inhibitors and role in the therapy of different diseases. Int. J. Biol. Macromol. 2020, 158, 750–772. [Google Scholar] [CrossRef]
- Vandenberg, R.J.; Shaddick, K.; Ju, P. Molecular basis for substrate discrimination by glycine transporters. J. Biol. Chem. 2007, 282, 14447–14453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Núñez, E.; Martínez-Maza, R.; Geerlings, A.; Aragón, C.; López-Corcuera, B. Transmembrane domains 1 and 3 of the glycine transporter GLYT1 contain structural determinants of N[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)-propyl]sarcosine specificity. Neuropharmacology 2005, 49, 922–934. [Google Scholar] [CrossRef]
- Aubrey, K.R.; Vandenberg, R.J. N [3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine (NFPS) is a selective persistent inhibitor of glycine transport. Br. J. Pharmacol. 2001, 134, 1429–1436. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-W.; Uchendu, S.; Leone, V.; Bradshaw, R.T.; Sangwa, N.; Forrest, L.R.; Rudnick, G. Chloride-dependent conformational changes in the GlyT1 glycine transporter. Proc. Natl. Acad. Sci. USA 2021, 118, e2017431118. [Google Scholar] [CrossRef]
- Beuming, T.; Shi, L.; Javitch, J.A.; Weinstein, H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 2006, 70, 1630–1642. [Google Scholar] [CrossRef] [Green Version]
- Skovstrup, S.; Taboureau, O.; Bräuner-Osborne, H.; Jørgensen, F.S. Homology modelling of the GABA transporter and analysis of tiagabine binding. ChemMedChem 2010, 5, 986–1000. [Google Scholar] [CrossRef]
- Zaręba, P.; Sałat, K.; Höfner, G.C.; Łątka, K.; Bajda, M.; Latacz, G.; Kotniewicz, K.; Rapacz, A.; Podkowa, A.; Maj, M.; et al. Development of tricyclic N-benzyl-4-hydroxybutanamide derivatives as inhibitors of GABA transporters mGAT1-4 with anticonvulsant, antinociceptive, and antidepressant activity. Eur. J. Med. Chem. 2021, 221, 113512. [Google Scholar] [CrossRef] [PubMed]
- Gryzło, B.; Zaręba, P.; Malawska, K.; Mazur, G.; Rapacz, A.; Łątka, K.; Höfner, G.C.; Latacz, G.; Bajda, M.; Sałat, K.; et al. Novel functionalized amino acids as inhibitors of GABA transporters with analgesic activity. ACS Chem. Neurosci. 2021, 12, 3073–3100. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MM-GBSA * dGbind (kcal/mol) | p Value ** | ||
|---|---|---|---|---|
| Outward-Open | Inward-Open | |||
| non-competitive inhibitors | compound 1 | −54.36 ± 0.42 | −73.69 ± 0.80 | p < 0.0001 |
| bitopertin | −55.88 ± 0.73 | −60.33 ± 1.07 | p = 0.0014 | |
| ORG-24598 | −45.14 ± 1.20 | −51.67 ± 0.79 | p < 0.0001 | |
| ALX-5407 | −65.78 ± 1.04 | −66.18 ± 0.71 | p = 0.7519 | |
| competitive inhibitors | SSR-504734 | −63.93 ± 0.88 | −50.80 ± 0.75 | p < 0.0001 |
| compound 2 | −74.40 ± 1.19 | −60.29 ± 0.84 | p < 0.0001 | |
| compound 3 | −68.56 ± 0.77 | −45.57 ± 0.82 | p < 0.0001 | |
| Compound | MM-GBSA * dGbind (kcal/mol) | p Value ** | |
|---|---|---|---|
| Outward-Open | Inward-Open | ||
| ORG-25543 | −65.90 ± 1.02 | −61.31 ± 0.84 | p = 0.0012 |
| ALX-1393 | −68.03 ± 0.94 | −39.54 ± 0.96 | p < 0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łątka, K.; Bajda, M. Analysis of Binding Determinants for Different Classes of Competitive and Noncompetitive Inhibitors of Glycine Transporters. Int. J. Mol. Sci. 2022, 23, 8050. https://doi.org/10.3390/ijms23148050
Łątka K, Bajda M. Analysis of Binding Determinants for Different Classes of Competitive and Noncompetitive Inhibitors of Glycine Transporters. International Journal of Molecular Sciences. 2022; 23(14):8050. https://doi.org/10.3390/ijms23148050
Chicago/Turabian StyleŁątka, Kamil, and Marek Bajda. 2022. "Analysis of Binding Determinants for Different Classes of Competitive and Noncompetitive Inhibitors of Glycine Transporters" International Journal of Molecular Sciences 23, no. 14: 8050. https://doi.org/10.3390/ijms23148050
APA StyleŁątka, K., & Bajda, M. (2022). Analysis of Binding Determinants for Different Classes of Competitive and Noncompetitive Inhibitors of Glycine Transporters. International Journal of Molecular Sciences, 23(14), 8050. https://doi.org/10.3390/ijms23148050