
Glycidyl Methacrylate-Based Copolymers as Healing Agents of Waterborne Polyurethanes

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
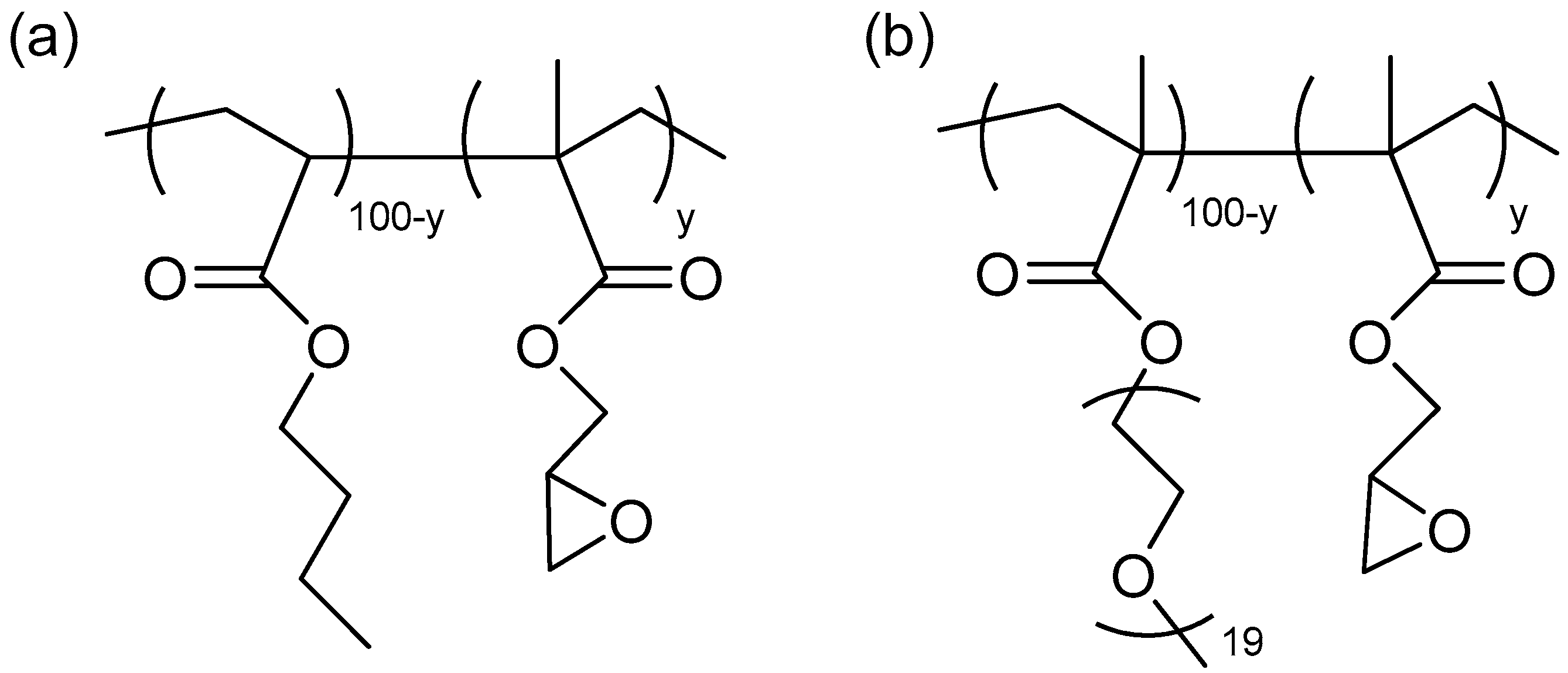
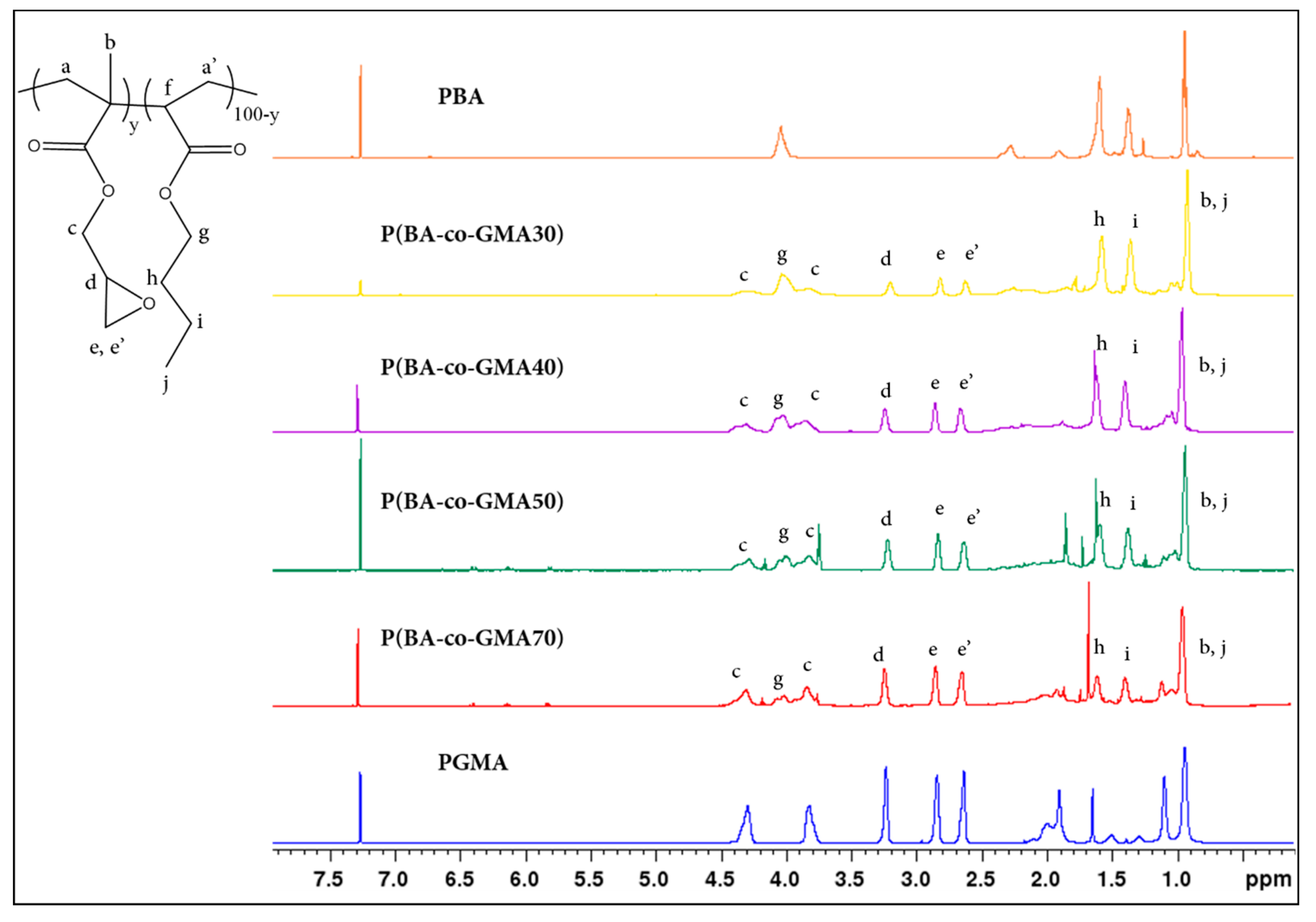
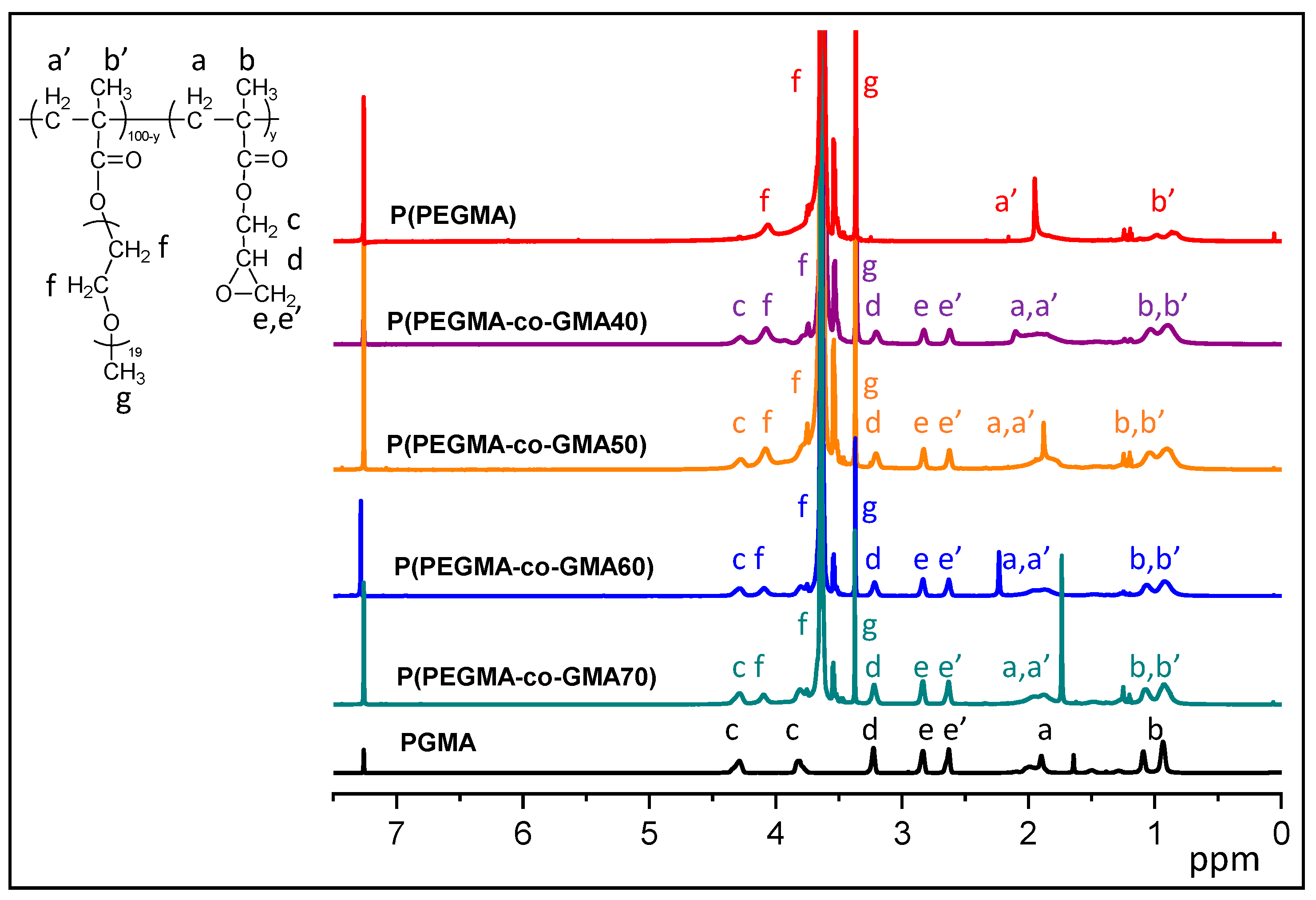
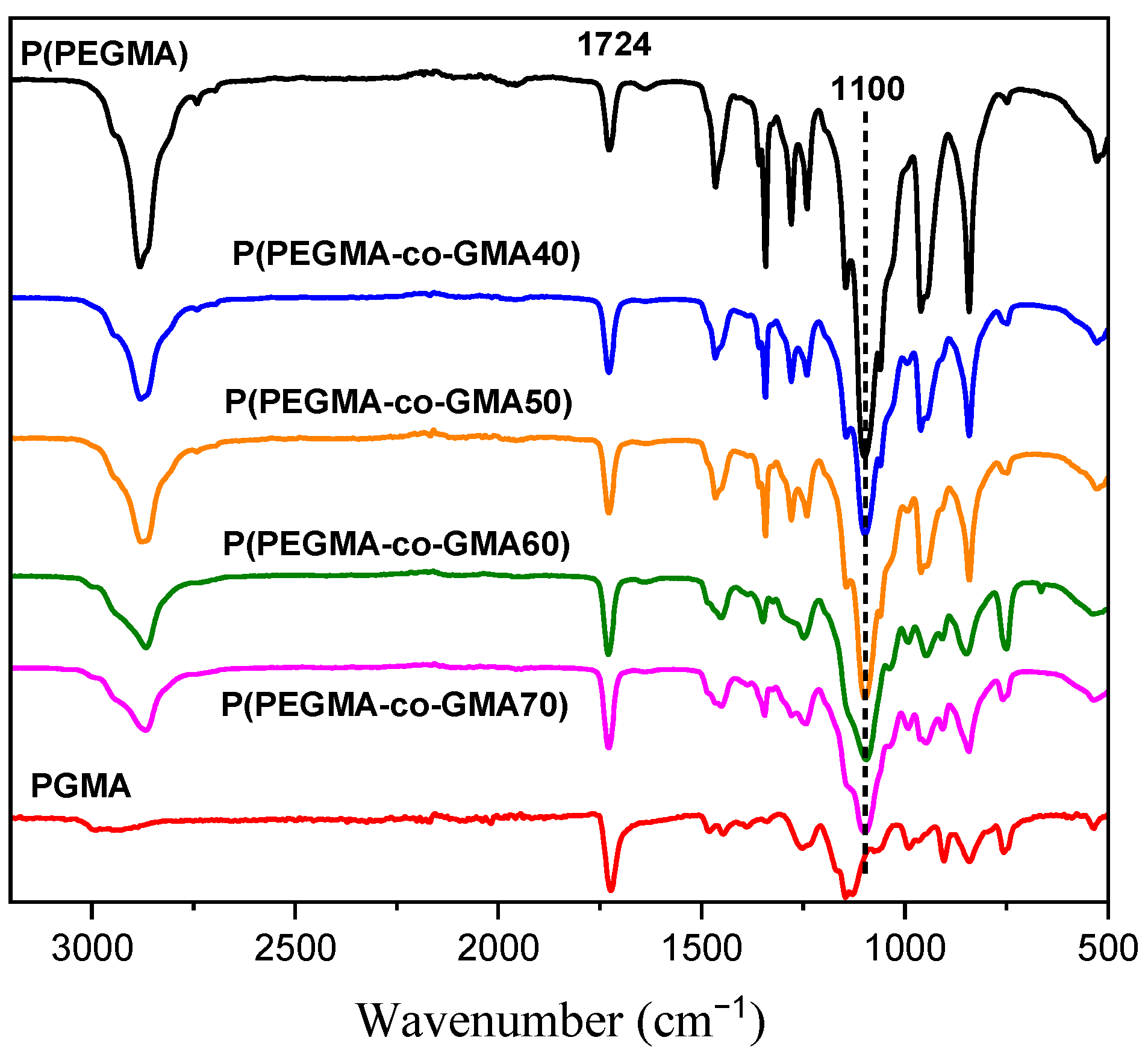
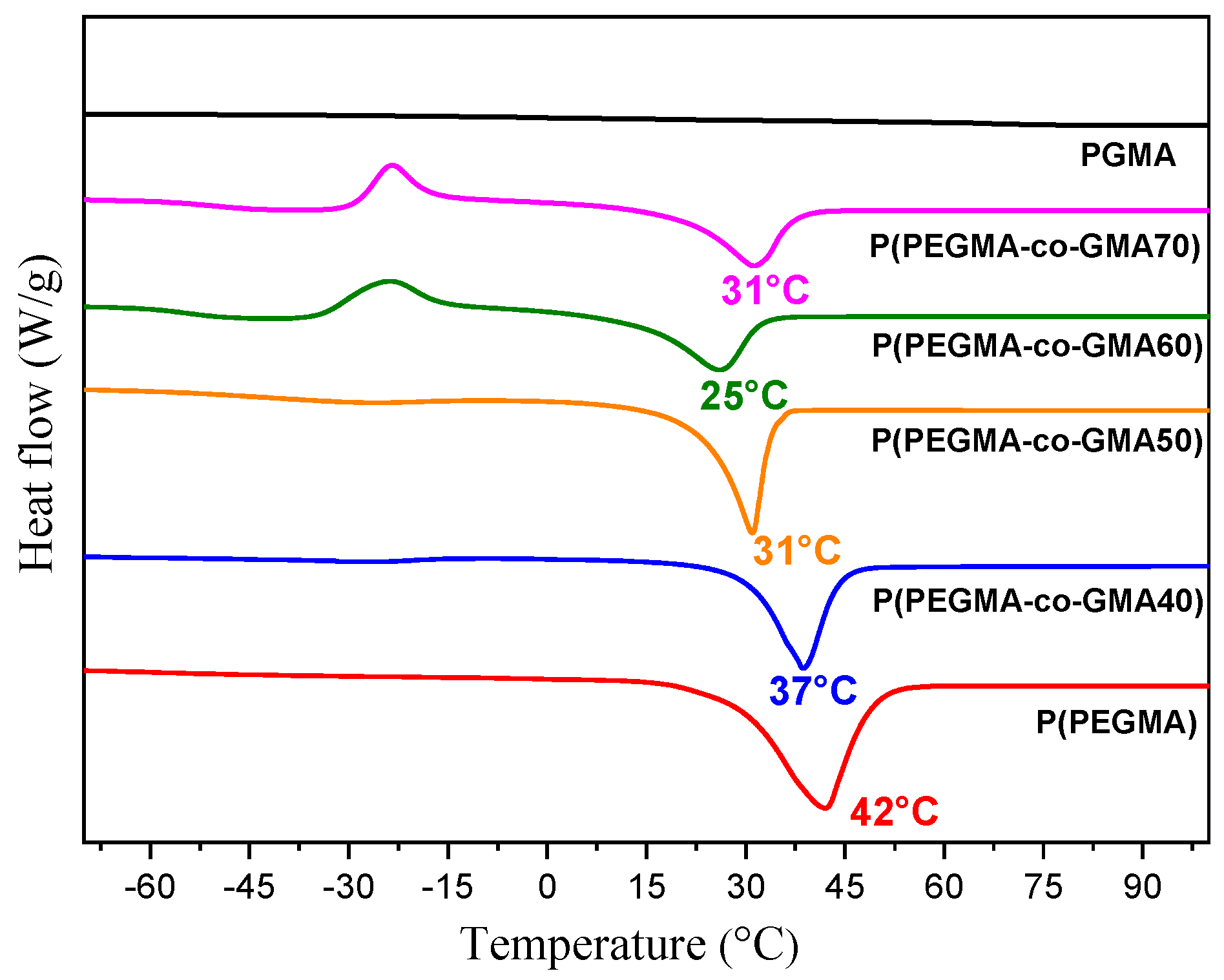
2.1. Copolymer Characterization
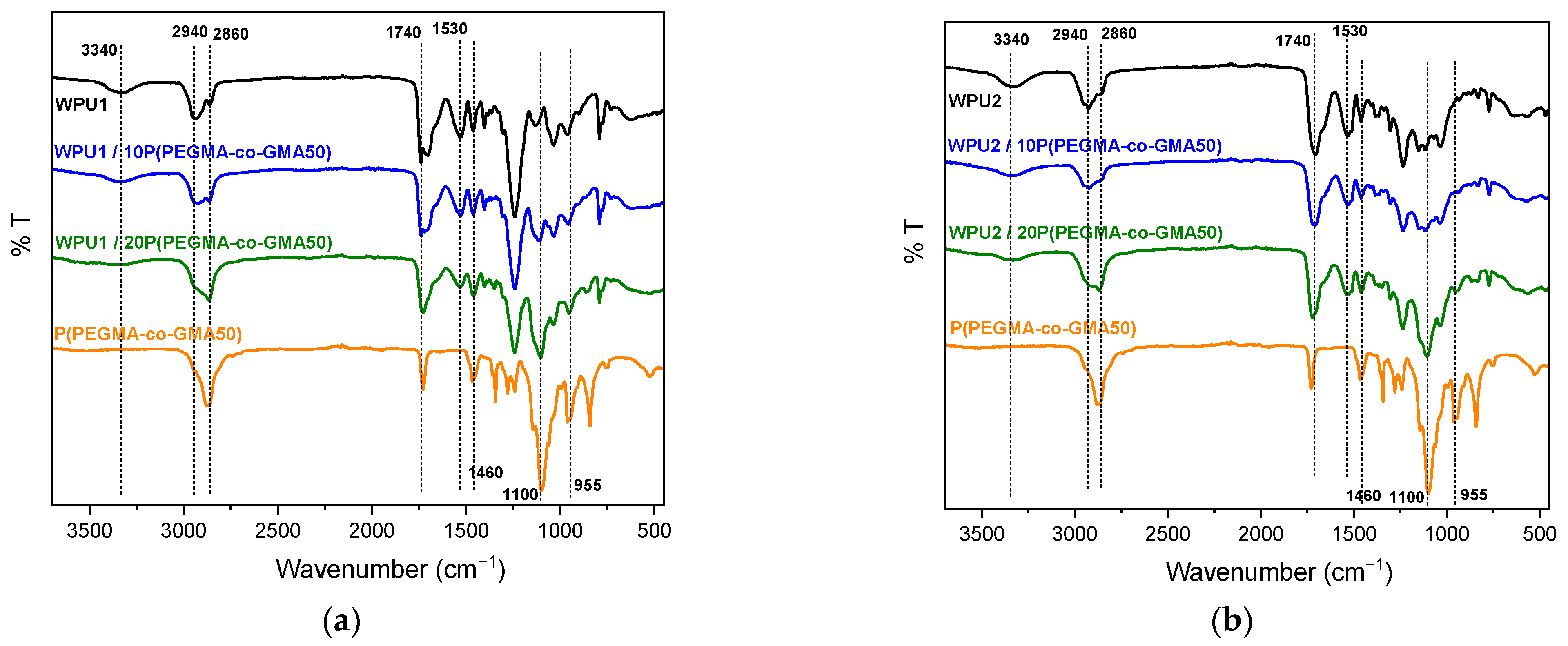
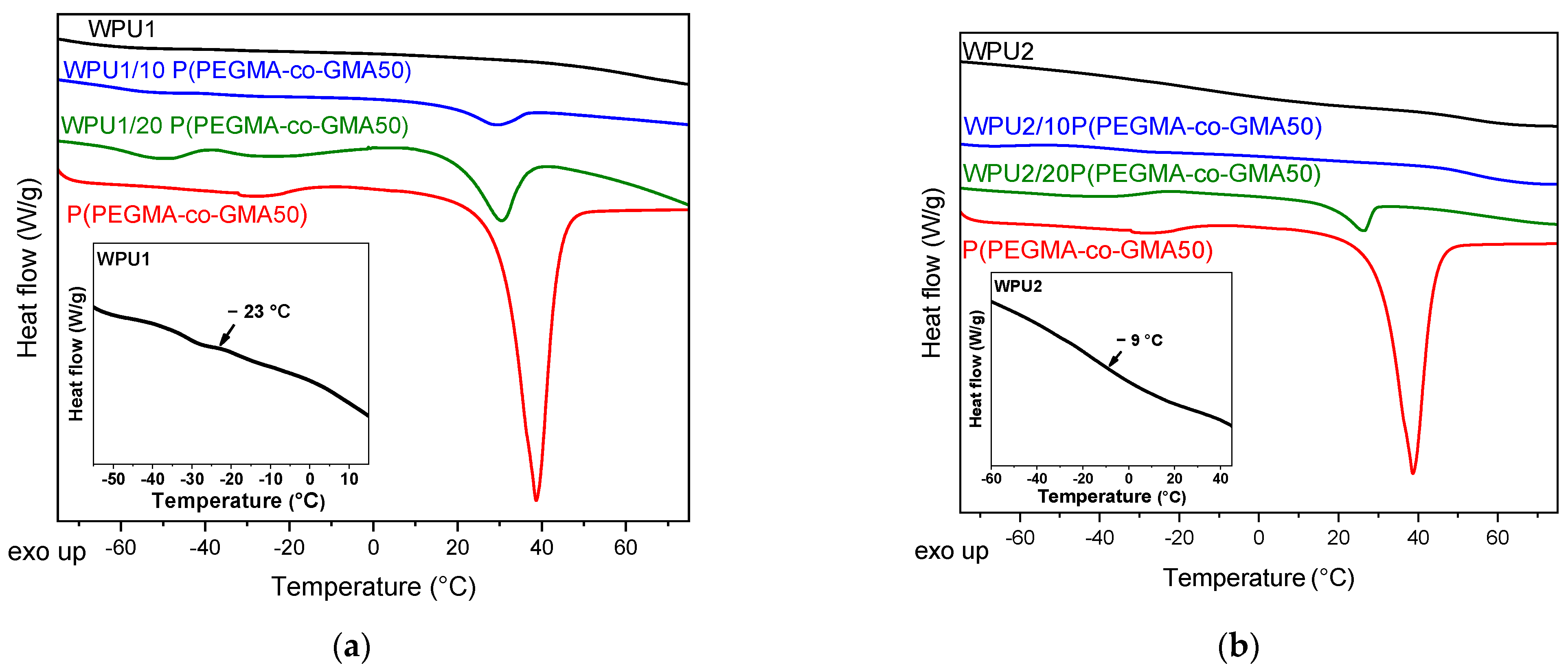
2.2. Investigation of WPU/Copolymer Films
2.3. Self-Healing Tests
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Copolymers
3.2.1. Synthesis of P(BA-co-GMAy) Copolymers
3.2.2. Synthesis of P(PEGMA-co-GMAy) Copolymers
3.3. Preparation of Polyurethane/Polymer Films
3.4. Characterization Techniques
3.4.1. Proton Nuclear Magnetic Resonance (1H-NMR)
3.4.2. Size Exclusion Chromatography (SEC)
3.4.3. Attenuated Total Reflection Fourier Transform Infrared Spectroscopy (ATR-FTIR)
3.4.4. Differential Scanning Calorimetry (DSC)
3.4.5. Self-Healing Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Urban, M.W. Self-healing polymeric materials. Chem. Soc. Rev. 2013, 42, 7446–7467. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.K.; Kessler, M.R. Self-healing polymer nanocomposite materials: A review. Polymer 2015, 69, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Bekas, D.G.; Tsirka, K.; Baltzis, D.; Paipetis, A.S. Self-healing materials: A review of advances in materials, evaluation, characterization and monitoring techniques. Compos. B Eng. 2016, 87, 92–119. [Google Scholar] [CrossRef]
- Wang, S.; Urban, M.W. Self-healing polymers. Nat. Rev. Mater. 2020, 5, 562–583. [Google Scholar] [CrossRef]
- Islam, S.; Bhat, G. Progress and challenges in self-healing composite materials. Mater. Adv. 2021, 2, 1896–1926. [Google Scholar] [CrossRef]
- Kahar, N.N.F.N.M.N.; Osman, A.F.; Alosime, E.; Arsat, N.; Azman, N.A.M.; Syamsir, A.; Itam, Z.; Hamid, Z.A.A. The Versatility of Polymeric Materials as Self-Healing Agents for Various Types of Applications: A Review. Polymers 2021, 13, 1194. [Google Scholar] [CrossRef]
- White, S.R.; Sottos, N.R.; Geubelle, P.H.; Moore, J.S.; Kessler, M.R.; Sriram, S.R.; Brown, E.N.; Viswanathan, S. Autonomic healing of polymer composites. Nature 2001, 409, 794–797. [Google Scholar] [CrossRef]
- Zhu, D.Y.; Rong, M.Z.; Zhang, M.Q. Self-healing polymeric materials based on microencapsulated healing agents: From design to preparation. Prog. Polym. Sci. 2015, 49–50, 175–220. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, X.; Urban, M.W. Chemical and physical aspects of self-healing materials. Prog. Polym. Sci. 2015, 49–50, 34–59. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Polymer engineering based on reversible covalent chemistry:A promising innovative pathway towards new materials and newfunctionalities. Prog. Polym. Sci. 2018, 80, 39–93. [Google Scholar] [CrossRef]
- Wang, Z.; Lu, X.; Sun, S.; Yu, C.; Xia, H. Preparation, characterization and properties of intrinsic self-healing elastomers. J. Mater. Chem. B 2019, 7, 4876–4926. [Google Scholar] [CrossRef]
- Willocq, B.; Odent, J.; Dubois, P.; Raquez, J.-M. Advances in intrinsic self-healing polyurethanes and related composites. RSC Adv. 2020, 10, 13766–13782. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-L.; Chuo, T.-W. Self-healing polymers based on thermally reversible Diels–Alder chemistry. Polym. Chem. 2013, 4, 2194–2205. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Y.; Chen, Q.; Fu, L.; Tao, L.; Wei, Y. Injectable and Self-Healing Chitosan Hydrogel Based on Imine Bonds: Design and Therapeutic Applications. Int. J. Mol. Sci. 2018, 19, 2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Chen, D.A. Novel Self-Healing Polyurethane Based on Disulfide Bonds. Macromol. Chem. Phys. 2016, 217, 1191–1196. [Google Scholar] [CrossRef]
- Jian, X.; Hu, Y.; Zhou, W.; Xiao, L. Self-healing polyurethane based on disulfide bond and hydrogen bond. Polym. Adv. Technol. 2018, 29, 463–469. [Google Scholar] [CrossRef]
- Changa, K.; Jia, H.; Gu, S.-Y. A transparent, highly stretchable, self-healing polyurethane based on disulfide bonds. Eur. Polym. J. 2019, 112, 822–831. [Google Scholar] [CrossRef]
- Cash, J.J.; Kubo, T.; Bapat, A.P.; Sumerlin, B.S. Room-Temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters. Macromolecules 2015, 48, 2098–2106. [Google Scholar] [CrossRef]
- Zhu, D.; Ye, Q.; Lu, X.; Lu, Q. Self-healing polymers with PEG oligomer side chains based on multiple H-bonding and adhesion properties. Polym. Chem. 2015, 6, 5086. [Google Scholar] [CrossRef]
- Wittmer, A.; Brinkmann, A.; Stenzel, V.; Hartwig, A.; Koschek, K. Moisture-Mediated Intrinsic Self-Healing of Modified Polyurethane Urea Polymers. J. Polym. Sci. A. Polym. Chem. 2018, 56, 537–548. [Google Scholar] [CrossRef]
- Li, Y.; Li, W.; Sun, A.; Jing, M.; Liu, X.; Wei, L.; Wu, K.; Fu, Q. A self-reinforcing and self-healing elastomer with high strength, unprecedented toughness and room-temperature reparability. Mater. Horiz. 2021, 8, 267–275. [Google Scholar] [CrossRef]
- Xiao, Y.; Huang, H.; Peng, X. Synthesis of self-healing waterborne polyurethanes containing sulphonate groups. RSC Adv. 2017, 7, 20093–20100. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Zhang, D.; Lu, F.; Yuan, W.; Xu, X.; Zhang, Q.; Liu, H.; Shao, Q.; Guo, Z.; Huang, Y. Multistimuli-Responsive Intrinsic Self-Healing Epoxy Resin Constructed by Host−Guest Interactions. Macromolecules 2018, 51, 5294–5303. [Google Scholar] [CrossRef]
- Zhang, F.; Ju, P.; Pan, M.; Zhang, D.; Huang, Y.; Li, G.; Li, X. Self-healing mechanisms in smart protective coatings: A review. Corros. Sci. 2018, 144, 74–88. [Google Scholar] [CrossRef]
- Díez-García, I.; Eceiza, A.; Tercjak, A. Self-Healable Nanocomposites with Enhanced Thermal Stability by Incorporation of TiO2 Nanoparticles to Waterborne Poly(urethane-urea) Matrices Based on Amphiphilic Triblock Copolymers. J. Phys. Chem. C 2019, 123, 21290–21298. [Google Scholar] [CrossRef]
- Díez-García, I.; Eceiza, A.; Tercjak, A. Improvement of Mechanical Properties and Self-Healing Effciency by Ex-Situ Incorporation of TiO2 Nanoparticles to a Waterborne Poly(Urethane-Urea). Polymers 2019, 11, 1209. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Jiang, Y.; Hu, J. Collagen incorporation into waterborne polyurethane improves breathability, mechanical property, and self-healing ability. Compos. Part A Appl. Sci. Manuf. 2020, 133, 105854. [Google Scholar] [CrossRef]
- Ha, Y.-M.; Kim, Y.N.; Jung, Y.C. Rapid and Local Self-Healing Ability of Polyurethane Nanocomposites Using Photothermal Polydopamine-Coated Graphene Oxide Triggered by Near-Infrared Laser. Polymers 2021, 13, 1274. [Google Scholar] [CrossRef]
- Song, S.; Yang, H.; Cui, Y.; Tang, Y.; Chen, Y.; Yang, B.; Yuan, J.; Huang, J. Mussel-inspired, self-healing polymer blends. Polymer 2020, 198, 122528. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Hailstone, R.K.; Lewis, C.L. Thermoplastic Blend Exhibiting Shape Memory Assisted Self-Healing Functionality. ACS Appl. Mater. Interfaces 2020, 12, 46733–46742. [Google Scholar] [CrossRef]
- Peng, H.; Du, X.; Cheng, X.; Wang, H.; Du, Z. Room-temperature self-healable and stretchable waterborne polyurethane film fabricated via multiple hydrogen bonds. Prog. Org. Coat. 2021, 151, 106081. [Google Scholar] [CrossRef]
- Muzammil, E.M.; Khan, A.; Stuparu, M.C. Post-polymerization modification reactions of poly(glycidyl methacrylate)s. RSC Adv. 2017, 7, 55874–55884. [Google Scholar] [CrossRef] [Green Version]
- Varley, R.J.; Craze, D.A.; Mouritz, A.P.; Wang, C.H. Thermoplastic Healing in Epoxy Networks: Exploring Performance and Mechanism of Alternative Healing Agents. Macromol. Mater. Eng. 2013, 298, 1232–1242. [Google Scholar] [CrossRef]
- Wang, H.P.; Yuan, Y.C.; Rong, M.Z.; Zhang, M.Q. Self-Healing of Thermoplastics via Living Polymerization. Macromolecules 2010, 43, 595–598. [Google Scholar] [CrossRef]
- Meng, L.M.; Yuan, Y.C.; Rong, M.Z.; Zhang, M.Q. A dual mechanism single-component self-healing strategy for polymers. J. Mater. Chem. 2010, 20, 6030–6038. [Google Scholar] [CrossRef]
- Urban, M.W.; Davydovich, D.; Yang, Y.; Demir, T.; Zhang, Y.; Casabianca, L. Key-and-lock commodity self-healing copolymers. Science 2018, 362, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Davydovich, D.; Urban, M.W. Water accelerated self-healing of hydrophobic copolymers. Nat. Commun. 2020, 11, 5743. [Google Scholar] [CrossRef]
- Valette, L.; Massardier, V.; Pascault, J.-P.; Magny, B. Synthesis and Photopolymerization of Acrylic Acrylate Copolymers. J. Appl. Polym. Sci. 2002, 86, 753–763. [Google Scholar] [CrossRef]
- Safa, K.D.; Nasirtabrizi, M.H. Ring opening reactions of glycidyl methacrylate copolymers to introduce bulky organosilicon side chain substituents. Polym. Bull. 2006, 57, 293–304. [Google Scholar] [CrossRef]
- de la Fuente, J.L.; Canamero, P.F.; Fernandez-Garcia, M. Synthesis and Characterization of Glycidyl Methacrylate/Butyl Acrylate Copolymers Obtained at a Low Temperature by Atom Transfer Radical Polymerization. J. Polym. Sci. A. Polym. Chem. 2006, 44, 1807–1816. [Google Scholar] [CrossRef]
- Cañamero, P.F.; de la Fuente, J.L.; Fernández-García, M. Curing kinetic study using a well-controlled multifunctional copolymer based on glycidyl methacrylate. Eur. Polym. J. 2009, 45, 2665–2673. [Google Scholar] [CrossRef]
- Venault, A.; Liou, C.-S.; Yeh, L.-C.; Jhong, J.-F.; Huang, J.; Chang, Y. Turning Expanded Poly(tetrafluoroethylene) Membranes into Potential Skin Wound Dressings by Grafting a Bioinert Epoxylated PEGMA Copolymer. ACS Biomater. Sci. Eng. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Laskar, P.; Dey, J.; Ghosh, S.K. Evaluation of zwitterionic polymersomes spontaneously formed by pH-sensitive and biocompatible PEG based random copolymers asdrug delivery systems. Colloids Surf. B Biointerfaces 2016, 139, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, J.; Yu, J.; Zhao, J.; Tang, J. Silver nanoparticles well-dispersed in amine-functionalized, one-pot made vesicles as an effective antibacterial agent. Mater. Sci. Eng. C 2016, 60, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Zholobko, O.; Hammed, A.; Zakharchenko, A.; Borodinov, N.; Luzinov, I.; Urbanowicz, B.; Patsahan, T.; Ilnytskyi, J.; Minko, S.; Pryor, S.W.; et al. Biomimetic Cellulosomes Assembled on Molecular Brush Scaffolds: Random Complexes vs Enzyme Mixtures. ACS Appl. Polym. Mater. 2021, 3, 1840–1853. [Google Scholar] [CrossRef]
- Daigle, J.-C.; Asakawa, Y.; Vijh, A.; Hovington, P.; Armand, M.; Zaghib, K. Exceptionally stable polymer electrolyte for a lithium battery based on cross-linking by a residue-free process. J. Power Source 2016, 332, 213–221. [Google Scholar] [CrossRef]
- Yu, T.-Y.; Yeh, S.-C.; Lee, J.-Y.; Wu, N.-L.; Jeng, R.-J. Epoxy-Based Interlocking Membranes for All Solid-State Lithium Ion Batteries: The Effects of Amine Curing Agents on Electrochemical Properties. Polymers 2021, 13, 3244. [Google Scholar] [CrossRef]
- Luo, D.; Li, Y.; Yang, M. Preparation and Characterization of Novel Crosslinked Poly[glycidyl methacrylate–poly(ethylene glycol) methyl ether methacrylate] as Gel Polymer Electrolytes. J. Appl. Polym. Sci. 2011, 120, 2979–2984. [Google Scholar] [CrossRef]
- Xiu, K.M.; Cai, Q.; Li, J.S.; Yang, X.P.; Yang, W.T.; Xu, F.J. Anti-fouling surfaces by combined molecular self-assembly and surface-initiated ATRP for micropatterning active proteins. Colloids Surf. B Biointerfaces 2012, 90, 177–183. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, G.; Jia, X.; Liu, D.; Wyman, I. Preparation of Multipurpose Polyvinylidene Fluoride Membranes via a Spray-Coating Strategy Using Waterborne Polymers. ACS Appl. Mater. Interfaces 2021, 13, 4485–4498. [Google Scholar] [CrossRef]
- Druvari, D.; Koromilas, N.D.; Lainioti, G.C.; Bokias, G.; Vasilopoulos, G.; Vantarakis, A.; Baras, I.; Dourala, N.; Kallitsis, J.K. Polymeric Quaternary Ammonium-Containing Coatings with Potential Dual Contact-Based and Release-Based Antimicrobial Activity. ACS Appl. Mater. Interfaces 2016, 8, 35593–35605. [Google Scholar] [CrossRef]
- Tsagdi, A.; Druvari, D.; Panagiotaras, D.; Avramidis, P.; Bekiari, V.; Kallitsis, J.K. Polymeric Coatings Based on Water-Soluble Trimethylammonium Copolymers for Antifouling Applications. Molecules 2020, 25, 1678. [Google Scholar] [CrossRef] [Green Version]
- Lainioti, G.C.; Savva, P.; Druvari, D.; Avramidis, P.; Panagiotaras, D.; Karellou, E.I.E.; Kallitsis, J.K. Cross-linking of antimicrobial polymers with hexamethylene diamine to prevent biofouling in marine applications. Prog. Org. Coat. 2021, 157, 106336. [Google Scholar] [CrossRef]
- Tzoumani, I.; Lainioti, G.C.; Aletras, A.J.; Zainescu, G.; Stefan, S.; Meghea, A.; Kallitsis, J.K. Modification of Collagen Derivatives with Water-Soluble Polymers for the Development of Cross-Linked Hydrogels for Controlled Release. Materials 2019, 12, 4067. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Qiu, W.Z.; Wu, Z.L.; Ren, P.F.; Zheng, Q.; Xu, Z.K. Water-Triggered Self-Healing Coatings of Hydrogen-Bonded Complexes for High Binding Affinity and Antioxidative Property. Adv. Mater. Interfaces 2016, 3, 1600167. [Google Scholar] [CrossRef]
- Kim, K.-H.; Mai, H.-N.; Hyun, D.-C.; Lee, D.-H. New Autonomous Water-Enabled Self-Healing Coating Material with Antibacterial-Agent-Releasing Properties. Pharmaceutics 2022, 14, 1005. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Cha, S.-H. Enhancement of Self-Healing Property by Introducing Ethylene Glycol Group into Thermally Reversible Diels-Alder Reaction Based Self-Healable Materials. Macromol. Res. 2017, 25, 640–647. [Google Scholar] [CrossRef]
- Pulikkalparambil, H.; Varghese, S.A.; Siengchin, S.; Parameswaranpillai, J. Thermally mendable and improved hydrophilic bioepoxy/PEG-PPG-PEG blends for coating application. Mater. Res. Express 2019, 6, 025307. [Google Scholar] [CrossRef]
- Mattia, J.; Painter, P. A Comparison of Hydrogen Bonding and Order in a Polyurethane and Poly(urethane-urea) and Their Blends with Poly(ethylene glycol). Macromolecules 2007, 40, 1546–1554. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Q. Synthesis of block copolymer poly (n-butyl acrylate)-b-polystyrene by DPE seeded emulsion polymerization with monodisperse latex particles and morphology of self-assembly film surface. J. Colloid Interf. Sci. 2012, 374, 54–60. [Google Scholar] [CrossRef]
- Athawale, V.D.; Raut, S.S. New interpenetrating polymer networks based on uralkyd/poly(glycidyl methacrylate). Eur. Polym. J. 2002, 38, 2033–2040. [Google Scholar] [CrossRef]
- Gunaydin, O.; Yilmaz, F. Copolymers of Glycidyl Methacrylate with 3-Methylthienyl Methacrylate: Synthesis, Characterization and Reactivity Ratios. Polym. J. 2007, 6, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Borodinov, N.; Gil, D.; Savchak, M.; Gross, C.E.; Yadavalli, N.S.; Ma, R.; Tsukruk, V.V.; Minko, S.; Vertegel, A.; Luzinov, I. En route to practicality of the polymer grafting technology: One-step interfacial modification with amphiphilic molecular brushes. ACS Appl. Mater. Interfaces 2018, 10, 13941–13952. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.B.; Fan, L.Y.; Sheng, Y.; Mao, J.; Chao, G.T.; Li, J.; Tu, M.J.; Qian, Z.Y. Synthesis of Biodegradable Poly(butylene terephthalate)/poly(ethylene glycol) (PBT/PEG) Multiblock Copolymers and Preparation of Indirubin Loaded Microspheres. Polym. Bull. 2005, 53, 147–154. [Google Scholar] [CrossRef]
- Vassiliadou, O.; Chrysostomou, V.; Pispas, S.; Klonos, P.A.; Kyritsis, A. Molecular dynamics and crystallization in polymers based on ethylene glycol methacrylates (EGMAs) with melt memory characteristics: From linear oligomers to comb-like polymers. Soft Matter 2021, 17, 1284–1298. [Google Scholar] [CrossRef]
- Cao, S.; Li, S.; Li, M.; Xu, L.; Ding, H.; Xia, J.; Zhang, M.; Huang, K. A thermal self-healing polyurethane thermoset based on phenolic urethane. Polym. J. 2017, 49, 775–781. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Z.; Fan, W.; Yang, K.; Li, Z. Preparation of renewable gallic acid-based self-healing waterborne polyurethane with dynamic phenol–carbamate network: Toward superior mechanical properties and shape memory function. J. Mater. Sci. 2022, 57, 5679–5696. [Google Scholar] [CrossRef]
- Handique, J.; Saikia, B.J.; Dolui, S.K. Designing Microencapsulation Based Self-Healing Methylmethacrylate-Glycidyl Methacrylate Copolymer. Pol. Sci. Ser. A 2019, 61, 577–588. [Google Scholar] [CrossRef]
- Guo, Y.K.; Chen, L.; Xu, D.G.; Zhong, J.R.; Yue, G.Z.; Astruc, D.; Shuai, M.B.; Zhao, P.X. The Dual Functional Epoxy Material with Autonomic Damage Indication and Self-healing. RSC Adv. 2016, 6, 65067–65071. [Google Scholar] [CrossRef]
- Li, X.; Ni, C.; Ma, F.; Yao, B.; Zhu, C. Preparation of poly(N-Butyl Acrylate-Co-Glycidyl Methacrylate) and its Application in Enhancement of Epoxy Resin. Polym. Plast. Technol. Eng. 2014, 53, 262–267. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymers | Feed Composition (% w GMA) | Feed Composition (% mol GMA) | 1H-NMR Composition (% mol GMA) | Mn (g/mol) | Mw (g/mol) | PDI |
|---|---|---|---|---|---|---|
| P(BA-co-GMA30) | 32 | 30 | 30 | 14,000 | 28,000 | 2.0 |
| P(BA-co-GMA40) | 42 | 40 | 46 | 23,000 | 55,000 | 2.4 |
| P(BA-co-GMA50) | 53 | 50 | 55 | 24,000 | 52,000 | 2.2 |
| P(BA-co-GMA70) | 72 | 70 | 67 | 21,000 | 45,000 | 2.1 |
| Polymers | Feed Composition (% w GMA) | Feed Composition (% mol GMA) | 1H-NMR Composition (% mol GMA) | Mn (g/mol) | Mw (g/mol) | PDI (Mw/Mn) |
|---|---|---|---|---|---|---|
| P(PEGMA-co-GMA40) | 9 | 40 | 45 | 60,000 | 105,000 | 1.7 |
| P(PEGMA-co-GMA50) | 13 | 50 | 54 | 235,000 | 470,000 | 2.0 |
| P(PEGMA-co-GMA60) | 18 | 60 | 67 | 80,000 | 220,000 | 2.7 |
| P(PEGMA-co-GMA70) | 26 | 70 | 74 | 110,000 | 340,000 | 3.1 |
| Polymers | Tg (°C) | Tc (°C) | ΔHc (J/g) | Tm (°C) | ΔHm (J/g) | %Xc | %X’c |
|---|---|---|---|---|---|---|---|
| P(PEGMA) | −70 | - | - | 42 | 120.0 | 61.0 | 61.0 |
| PGMA | 70 | - | - | - | - | - | - |
| P(PEGMA-co-GMA40) | −43 | - | - | 37 | 84.0 | 42.6 | 47.3 |
| P(PEGMA-co-GMA50) | −45 | - | - | 31 | 99.2 | 50.3 | 59.2 |
| P(PEGMA-co-GMA60) | −55 | −24 | 39.6 | 25 | 77.7 | 19.3 | 25.1 |
| P(PEGMA-co-GMA70) | −50 | −23 | 33.8 | 31 | 73.2 | 20.0 | 28.6 |
| Sample | Tg (°C) | Tc (°C) | ΔHc (J/g) | Tm (°C) | ΔHm (J/g) | %X″c |
|---|---|---|---|---|---|---|
| Pure WPU films | ||||||
| WPU1 | −23 | - | - | - | - | - |
| WPU2 | −9 | - | - | - | - | - |
| Copolymers | ||||||
| P(PEGMA-co-GMA50) | −45 | - | - | 31 | 99.2 | 59.2 |
| P(PEGMA-co-GMA70) | −50 | −23 | 33.80 | 31 | 73.20 | 28.6 |
| WPU/copolymer films | ||||||
| WPU1/ 10 P(PEGMA-co-GMA50) | −37 | - | - | 28 | 6.04 | 36.0 |
| WPU1/ 20 P(PEGMA-co-GMA50) | −34 | - | - | 30.5 | 10.16 | 30.4 |
| WPU1/ 10 P(PEGMA-co-GMA70) * | −32 | - | - | - | - | - |
| WPU2/ 10 P(PEGMA-co-GMA50) | −28 | - | - | - | - | - |
| WPU2/ 20 P(PEGMA-co-GMA50) | −32 | - | - | 26.5 | 6.50 | 19.4 |
| WPU2/ 10 P(PEGMA-co-GMA70) * | −3 | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzoumani, I.; Soto Beobide, A.; Iatridi, Z.; Voyiatzis, G.A.; Bokias, G.; Kallitsis, J.K. Glycidyl Methacrylate-Based Copolymers as Healing Agents of Waterborne Polyurethanes. Int. J. Mol. Sci. 2022, 23, 8118. https://doi.org/10.3390/ijms23158118
Tzoumani I, Soto Beobide A, Iatridi Z, Voyiatzis GA, Bokias G, Kallitsis JK. Glycidyl Methacrylate-Based Copolymers as Healing Agents of Waterborne Polyurethanes. International Journal of Molecular Sciences. 2022; 23(15):8118. https://doi.org/10.3390/ijms23158118
Chicago/Turabian StyleTzoumani, Ioanna, Amaia Soto Beobide, Zacharoula Iatridi, George A. Voyiatzis, Georgios Bokias, and Joannis K. Kallitsis. 2022. "Glycidyl Methacrylate-Based Copolymers as Healing Agents of Waterborne Polyurethanes" International Journal of Molecular Sciences 23, no. 15: 8118. https://doi.org/10.3390/ijms23158118