
The Role of HDACs in the Response of Cancer Cells to Cellular Stress and the Potential for Therapeutic Intervention


Abstract
:1. Introduction
2. Histone Deacetylases (HDACs)
2.1. HDACs Classification
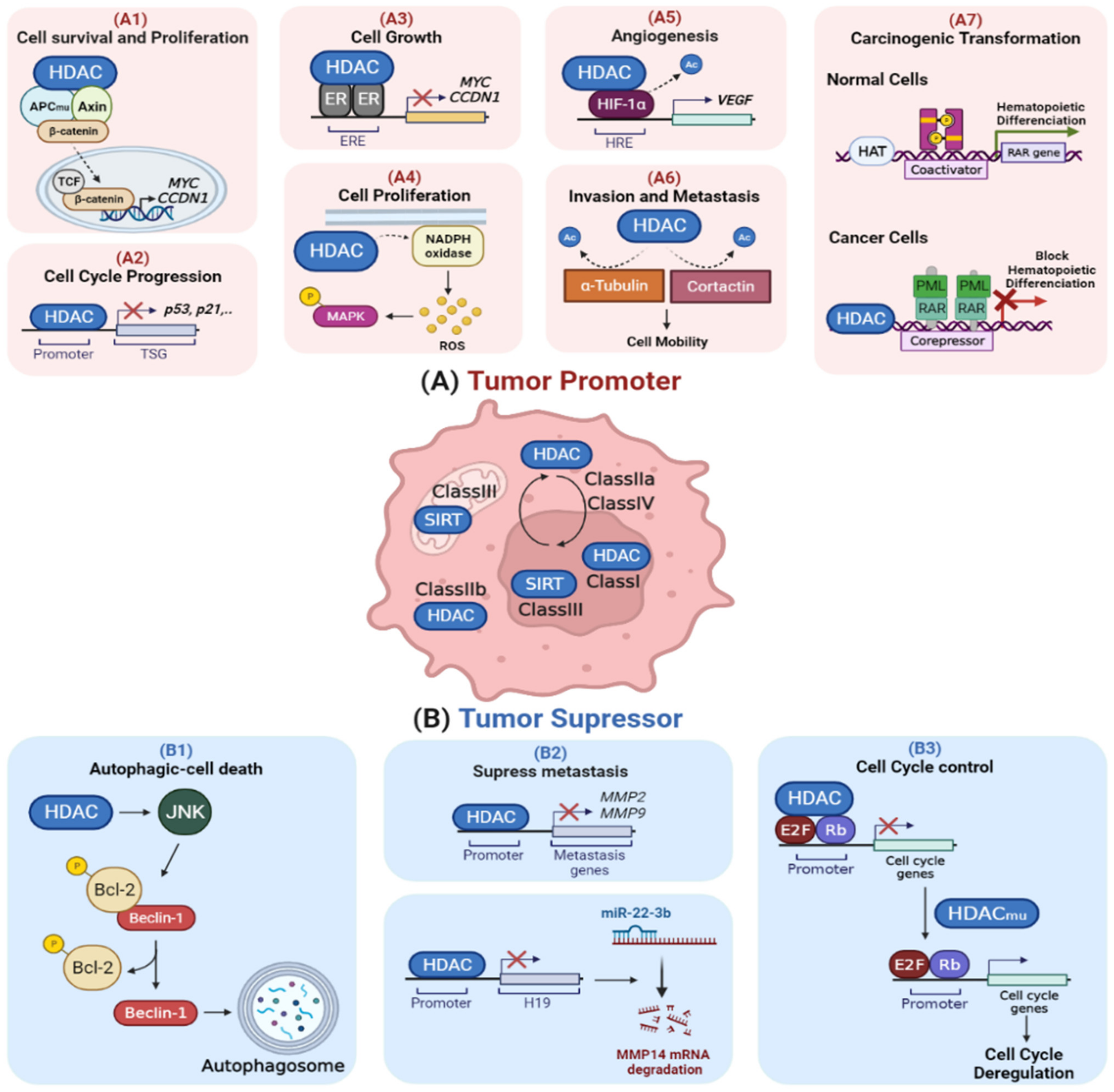
2.2. Dysregulation of HDACs in Cancer
3. Role of HDACs in Cellular Stress Response
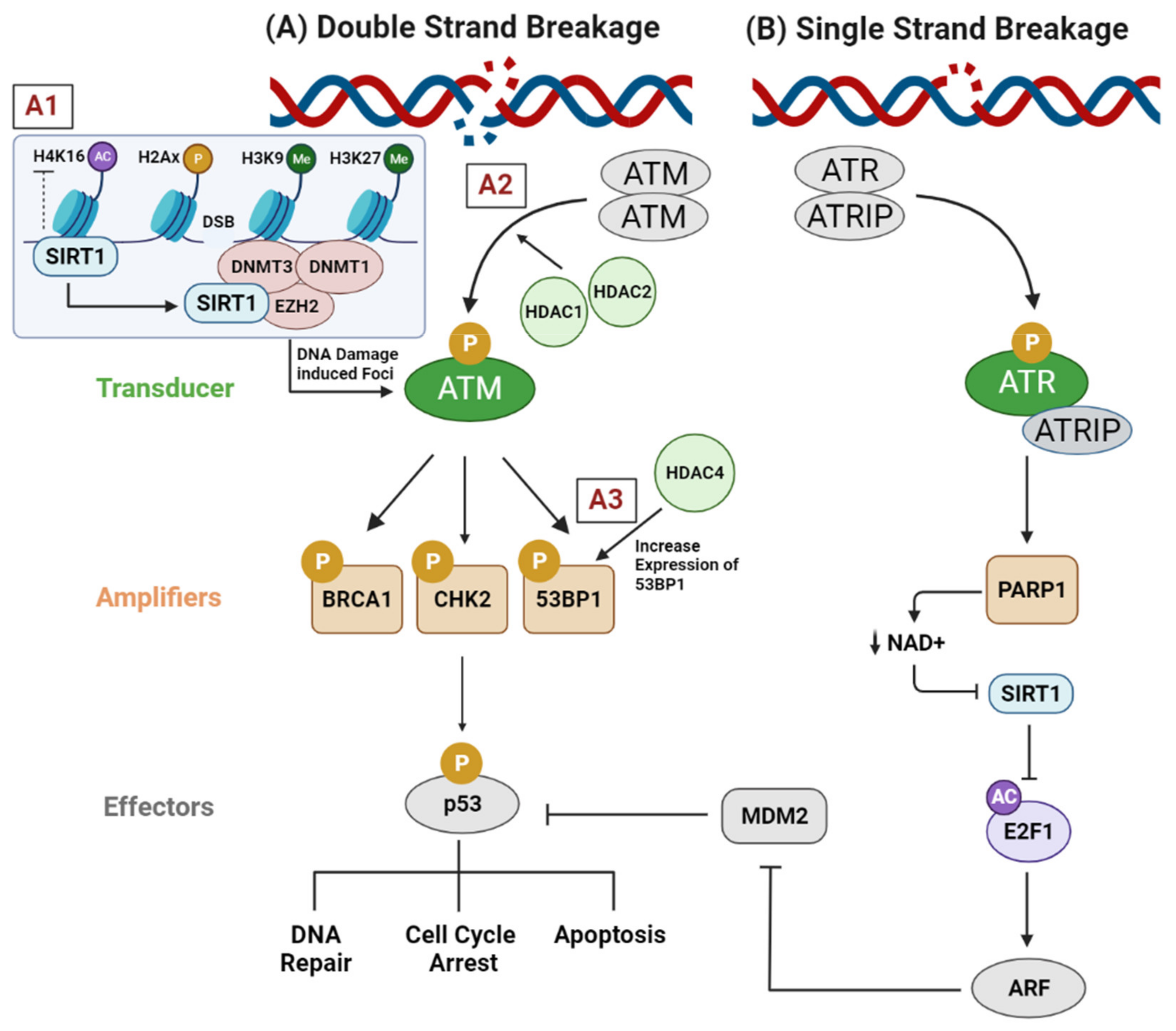
3.1. Genotoxic Stress (DNA Replication Stress, DNA Damage Response, and DNA Repair Pathways)
3.2. Proteotoxic Stress (Heat Shock Response and Endoplasmic Reticulum Stress)
3.3. Oxidative Stress
3.4. Metabolic Stress (Hypoglycemia and Hypoxia)
4. HDACs and Anti-Tumor Immune Response
Immunomodulatory Role of HDACs under Genotoxic Stress
5. Therapeutic Implications of HDAC Inhibitors
5.1. Clinical Trials of HDACs Inhibitors
5.2. HDAC Inhibitors in Combination Therapies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDACIs | Combination Drugs | Clinical Trial Phase | Clinical Trial ID | Cancer Type | Trial Description | Status | References |
|---|---|---|---|---|---|---|---|
| Vorinostat (SAHA) | Paclitaxel, Trastuzumab, Doxorubicin, Cyclo-phosphamide | I and II | NCT00574587 | Breast cancer, gastric cancer | To determine the optimal dose of vorinostat when combined with standard chemotherapy alone (or with trastuzumab when treating HER2-positive cancer). | Completed | [160] |
| Doxorubicin hydrochloride | I | NCT00331955 | Unspecified adult solid tumor | Vorinostat may help doxorubicin work better by making tumor cells more sensitive to the drug. | Completed | [161] | |
| Capecitabine and cisplatin | I and II | NCT01045538 | Non-small cell lung | Phase 1—maximum tolerated dose, Phase 2—response rate. | Completed | [162] | |
| Bortezomib | II | NCT00798720 | Cancer | The combination showed a weak anti-tumor activity as third-line therapy in NSCLC. | Completed | [163] | |
| Panobinostat (LBH589) | Trastuzumab/ Paclitaxel | I | NCT00788931 | HER-2-positive breast cancer, metastatic breast cancer | Combination is well tolerated. | Completed | [164] |
| Capecitabine Lapatinib | I | NCT00632489 | Lung vancer, head and neck cancer | Combination of Panobinostat and capecitabine is well tolerated at the recommended doses. | Completed | [165] | |
| Erlotinib | I | NCT00738751 | Lung adenocarcinoma | Panobinostat increases the sensitivity of lung adenocarcinoma cells to the antiproliferative effects of erlotinib. (synergism) | Completed | [166] | |
| Belinostat (PXD101) | Doxorubicin | I/II | NCT00878800 | Solid tumors and soft tissue sarcomas | No evidence of synergy between belinostat and doxorubicin in terms of objective tumour shrinkage. | Completed | [167] |
| Valproic Acid | Epirubicin 5-Fluorouracil Cyclo-phosphamide | I | NCT00246103 | Advanced neoplasms | Maximum tolerated dose and recommended phase II dose was VPA 140 mg/kg/d for 48 h followed by epirubicin 100 mg/m2. | Completed | [156] |
6. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poljšak, B.; Milisav, I. Clinical implications of cellular stress responses. Bosn. J. Basic Med. Sci. 2012, 12, 122–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xie, S. Therapeutic targeting of cellular stress responses in cancer. Thorac. Cancer 2018, 9, 1575–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.P. Epigenetics: Principles and Practice. Dig. Dis. 2011, 29, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.S.; Kumar, R. Chromatin remodeling in Cancer: A Gateway to regulate gene Transcription. Mol. Oncol. 2012, 6, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Esteller, M. The Role of Histone Deacetylases (HDACs) in Human Cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Cao, J.; Sun, L.; Aramsangtienchai, P.; Spiegelman, N.A.; Zhang, X.; Huang, W.; Seto, E.; Lin, H. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc. Natl. Acad. Sci. USA 2019, 116, 5487–5492. [Google Scholar] [CrossRef] [Green Version]
- Sanaei, M.; Kavoosi, F. Histone Deacetylases and Histone Deacetylase Inhibitors: Molecular Mechanisms of Action in Various Cancers. Adv. Biomed. Res. 2019, 8, 63. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of Site-Specific Histone Acetylation and Deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Pant, K.; Peixoto, E.; Richard, S.; Gradilone, S.A. Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangi-ocarcinoma. Cells 2020, 9, 780. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, F.; Chen, X.; Wang, J.; Zhao, Y.; Li, Y.; He, B. Zinc-dependent Deacetylase (HDAC) Inhibitors with Different Zinc Binding Groups. Curr. Top. Med. Chem. 2019, 19, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Nebbioso, A.; Altucci, L. Sirtuins and Disease: The Road Ahead. Front. Pharmacol. 2012, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaradonna, F.; Cirulli, C.; Palorini, R.; Votta, G.; Alberghina, L. New Insights into the Connection between Histone Deacetylases, Cell Metabolism, and Cancer. Antioxid. Redox Signal. 2015, 23, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.-G.; Munster, P. Histone deacetylase inhibitors for the treatment of breast cancer: Recent trial data. Clin. Investig. 2013, 3, 557–569. [Google Scholar] [CrossRef]
- De Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone Deacetylases (HDACs): Char-acterization of the Classical HDAC Family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone Deacetylases and Cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC Family: What Are the Cancer Relevant Targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of Histone Deacetylase 6 Acetylates and Disrupts the Chaperone Function of Heat Shock Protein 90: A Novel Basis for Antileukemia Activity of Histone Deacetylase Inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-S.; Lim, K.-H.; Guo, X.; Kawaguchi, Y.; Gao, Y.; Barrientos, T.; Ordentlich, P.; Wang, X.-F.; Counter, C.M.; Yao, T.-P. The Cytoplasmic Deacetylase HDAC6 Is Required for Efficient Oncogenic Tumorigenesis. Cancer Res. 2008, 68, 7561–7569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deakin, N.O.; Turner, C.E. Paxillin inhibits HDAC6 to regulate microtubule acetylation, Golgi structure, and polarized migration. J. Cell Biol. 2014, 206, 395–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [Green Version]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and Nuclear Recruitment of HDAC1 in Hormone Refractory Prostate Cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef]
- Zeng, L.-S.; Yang, X.-Z.; Wen, Y.-F.; Mai, S.-J.; Wang, M.-H.; Zhang, M.-Y.; Zheng, X.S.; Wang, H.-Y. Overexpressed HDAC4 is associated with poor survival and promotes tumor progression in esophageal carcinoma. Ageing 2016, 8, 1236–1248. [Google Scholar] [CrossRef] [Green Version]
- Kang, Z.-H.; Wang, C.-Y.; Zhang, W.-L.; Zhang, J.-T.; Yuan, C.-H.; Zhao, P.-W.; Lin, Y.-Y.; Hong, S.; Li, C.-Y.; Wang, L. Histone Deacetylase HDAC4 Promotes Gastric Cancer SGC-7901 Cells Progression via p21 Repression. PLoS ONE 2014, 9, e98894. [Google Scholar] [CrossRef] [Green Version]
- Hagelkruys, A.; Sawicka, A.; Rennmayr, M.; Seiser, C. The Biology of HDAC in Cancer: The Nuclear and Epigenetic Components. Handb. Exp. Pharmacol. 2011, 206, 13–37. [Google Scholar] [CrossRef]
- Minucci, S.; Nervi, C.; lo Coco, F.; Pelicci, P.G. Histone Deacetylases: A Common Molecular Target for Differentiation Treat-ment of Acute Myeloid Leukemias? Oncogene 2001, 20, 3110–3115. [Google Scholar] [CrossRef]
- Kawai, H.; Li, H.; Avraham, S.; Jiang, S.; Avraham, H.K. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor α. Int. J. Cancer 2003, 107, 353–358. [Google Scholar] [CrossRef]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.-P.; Göttlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.-I.; Iwase, H. HDAC6 Expression Is Correlated with Better Survival in Breast Cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osada, H.; Tatematsu, Y.; Saito, H.; Yatabe, Y.; Mitsudomi, T.; Takahashi, T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer 2004, 112, 26–32. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular Stress Responses: Cell Survival and Cell Death. Int. J. Cell Biol. 2010, 2010, 214074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhmoud, J.F.; Woolley, J.F.; al Moustafa, A.E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef] [PubMed]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone Deacetylase Regulation of ATM-Mediated DNA Damage Signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F.; Shiloh, Y. The Genetic Defect in Ataxia-Telangiectasia. Annu. Rev. Immunol. 1997, 15, 177–202. [Google Scholar] [CrossRef]
- Kao, G.D.; McKenna, W.G.; Guenther, M.G.; Muschel, R.J.; Lazar, M.A.; Yen, T. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J. Cell Biol. 2003, 160, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, Y.; Kato, J.; Ikeda, H. Silencing Factors Participate in DNA Repair and Recombination in Saccharomyces Cerevisiae [See Comments]. Nature 1997, 388, 900–903. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [Green Version]
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double Strand Breaks Can Initiate Gene Silencing and SIRT1-Dependent Onset of DNA Methylation in an Exogenous Promoter CpG Island. PLoS Genet. 2008, 4, e1000155. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Ji, L.; Chen, H.; Li, X.; Zhang, J.; Wang, X.; Wu, W.; Xu, Y.; Huang, F.; Cai, W.; et al. Acetylation of hMOF Modulates H4K16ac to Regulate DNA Repair Genes in Response to Oxidative Stress. Int. J. Biol. Sci. 2017, 13, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006, 8, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Orlando, G.; Khoronenkova, S.V.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. ARF induction in response to DNA strand breaks is regulated by PARP1. Nucleic Acids Res. 2013, 42, 2320–2329. [Google Scholar] [CrossRef]
- Stengel, K.R.; Hiebert, S.W. Class I HDACs Affect DNA Replication, Repair, and Chromatin Structure: Implications for Cancer Therapy. Antioxid. Redox Signal. 2015, 23, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabert, C.; Groth, A. Chromatin Replication and Epigenome Maintenance. Nat. Rev. Mol. Cell Biol. 2012, 13, 153–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, C.E.; Bhaskara, S.R.; Stengel, K.; Zhao, Y.; Sirbu, B.; Chagot, B.; Cortez, D.; Khabele, D.; Chazin, W.J.; Cooper, A.; et al. Inhibition of Histone Deacetylase 3 Causes Replication Stress in Cutaneous T Cell Lymphoma. PLoS ONE 2013, 8, e68915. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.-W.; Hiebert, S.W. Deletion of Histone Deacetylase 3 Reveals Critical Roles in S Phase Progression and DNA Damage Control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.-P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Gokhale, S.; Nyayanit, D.; Gadgil, C. A systems view of the protein expression process. Syst. Synth. Biol. 2011, 5, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tsai, Y.-H.; Tseng, S.-H. HDAC Inhibitors and RECK Modulate Endoplasmic Reticulum Stress in Tumor Cells. Int. J. Mol. Sci. 2017, 18, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.-T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.-P. HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activation of Glucocorticoid Receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Aldana-Masangkay, G.I. The Role of HDAC6 in Cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar]
- Krämer, O.H.; Mahboobi, S.; Sellmer, A. Drugging the HDAC6–HSP90 interplay in malignant cells. Trends Pharmacol. Sci. 2014, 35, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Fenger, J.; Murahari, S.; Bear, M.D.; Kulp, S.K.; Wang, D.; Chen, C.-S.; Kisseberth, W.C.; London, C.A. AR-42, a novel HDAC inhibitor, exhibits biologic activity against malignant mast cell lines via down-regulation of constitutively activated Kit. Blood 2010, 115, 4217–4225. [Google Scholar] [CrossRef] [Green Version]
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.-P.; Vourc’H, C.; Matthias, P.; et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21, 2172–2181. [Google Scholar] [CrossRef] [Green Version]
- Ahn, M.Y.; Kang, D.O.; Na, Y.J.; Yoon, S.; Choi, W.S.; Kang, K.W.; Chung, H.Y.; Jung, J.H.; Min, D.S.; Kim, H.S. Histone deacetylase inhibitor, apicidin, inhibits human ovarian cancer cell migration via class II histone deacetylase 4 silencing. Cancer Lett. 2012, 325, 189–199. [Google Scholar] [CrossRef]
- Jeon, H.W.; Lee, Y.M. Inhibition of Histone Deacetylase Attenuates Hypoxia-Induced Migration and Invasion of Cancer Cells via the Restoration of RECK Expression. Mol. Cancer Ther. 2010, 9, 1361–1370. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, C.; Sheng, Z.; Horan, T.P.; Kitayama, H.; Maki, M.; Hitomi, K.; Kitaura, Y.; Takai, S.; Sasahara, R.M.; Horimoto, A.; et al. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc. Natl. Acad. Sci. USA 1998, 95, 13221–13226. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Tseng, S.-H. The potential of RECK inducers as antitumor agents for glioma. Anticancer Res. 2012, 32, 2991–2998. [Google Scholar] [PubMed]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.-E.; Lee, S.-W.; Moon, E.-J.; Kim, H.-S.; Lee, S.-K.; Chung, H.Y.; et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001, 7, 437–443. [Google Scholar] [CrossRef]
- Lawless, M.W.; O’Byrne, K.J.; Gray, S.G. Oxidative stress induced lung cancer and COPD: Opportunities for epigenetic therapy. J. Cell. Mol. Med. 2009, 13, 2800–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawless, M.W.; O’Byrne, K.J.; Gray, S.G. Targeting Oxidative Stress in Cancer. Expert Opin. Ther. Targets 2010, 14, 1225–1245. [Google Scholar] [CrossRef] [PubMed]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Kawai, Y.; Garduño, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-Deacetylation of the Transcription Factor Nrf2 (Nuclear Factor Erythroid 2-related Factor 2) Regulates Its Transcriptional Activity and Nucleocytoplasmic Localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.-H.; Qu, J.; Shen, X. NF-κB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Dohi, Y.; Ikura, T.; Hoshikawa, Y.; Katoh, Y.; Ota, K.; Nakanome, A.; Muto, A.; Omura, S.; Ohta, T.; Ito, A.; et al. Bach1 inhibits oxidative stress–induced cellular senescence by impeding p53 function on chromatin. Nat. Struct. Mol. Biol. 2008, 15, 1246–1254. [Google Scholar] [CrossRef]
- Kim, S.-H.; Jeong, J.-W.; Park, J.A.; Lee, J.-W.; Seo, J.H.; Jung, B.-K.; Bae, M.-K.; Kim, K.-W. Regulation of the HIF-1α stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.-G.; Kong, G.; Lee, M.-O. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1α protein by recruiting histone deacetylase 1. EMBO J. 2006, 25, 1231–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Tamamizu-Kato, S.; Shibasaki, F. Histone Deacetylase 7 Associates with Hypoxia-inducible Factor 1α and Increases Transcriptional Activity. J. Biol. Chem. 2004, 279, 41966–41974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraoka, N.; Allen, E.; Apel, I.J.; Gyetko, M.R.; Weiss, S.J. Matrix Metalloproteinases Regulate Neovascularization by Acting as Pericellular Fibrinolysins. Cell 1998, 95, 365–377. [Google Scholar] [CrossRef] [Green Version]
- Potente, M.; Ghaeni, L.; Baldessari, D.; Mostoslavsky, R.; Rossig, L.; Dequiedt, F.; Haendeler, J.; Mione, M.; Dejana, E.; Alt, F.W.; et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007, 21, 2644–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.; Cardoso, S.M.; Clish, C.; et al. SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1α Destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [Green Version]
- White, E. Role of the Metabolic Stress Responses of Apoptosis and Autophagy in Tumor Suppression. In Oncogenes Meet Metabolism; Springer: Berlin/Heidelberg, Germany, 2007; pp. 23–24. [Google Scholar]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- De Berardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Mak, T.W. The current state of cancer metabolism. Nat. Cancer 2016, 16, 613–614. [Google Scholar] [CrossRef]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tu, B.; Wang, H.; Cao, Z.; Tang, M.; Zhang, C.; Gu, B.; Li, Z.; Wang, L.; Yang, Y.; et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl. Acad. Sci. USA 2014, 111, 10684–10689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, A.; Das, S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl. Acad. Sci. USA 2016, 113, E538–E547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Yung, W.K.A.; Lu, Z. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, K.S.; Lukey, M.J.; Wang, X.; Blank, B.; Druso, J.E.; Lin, M.-C.J.; Stalnecker, C.A.; Zhang, C.; Abril, Y.N.; Erickson, J.W.; et al. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 26625–26632. [Google Scholar] [CrossRef]
- Li, M.; Chiang, Y.-L.; Lyssiotis, C.A.; Teater, M.R.; Hong, J.Y.; Shen, H.; Wang, L.; Hu, J.; Jing, H.; Chen, Z.; et al. Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 2019, 35, 916–931.e9. [Google Scholar] [CrossRef]
- Wang, B.; Ye, Y.; Yang, X.; Liu, B.; Wang, Z.; Chen, S.; Jiang, K.; Zhang, W.; Jiang, H.; Mustonen, H.K.; et al. SIRT 2-dependent IDH 1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep. 2020, 21, e48183. [Google Scholar] [CrossRef]
- Kutil, Z.; Novakova, Z.; Meleshin, M.; Mikesova, J.; Schutkowski, M.; Barinka, C. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem. Biol. 2018, 13, 685–693. [Google Scholar] [CrossRef]
- Wang, W.; Zeng, Y.; Wu, B.; Deiters, A.; Liu, W.R. A Chemical Biology Approach to Reveal Sirt6-targeted Histone H3 Sites in Nucleosomes. ACS Chem. Biol. 2016, 11, 1973–1981. [Google Scholar] [CrossRef] [Green Version]
- Kutil, Z.; Mikešová, J.; Zessin, M.; Meleshin, M.; Nováková, Z.; Alquicer, G.; Kozikowski, A.; Sippl, W.; Bařinka, C.; Schutkowski, M. Continuous Activity Assay for HDAC11 Enabling Reevaluation of HDAC Inhibitors. ACS Omega 2019, 4, 19895–19904. [Google Scholar] [CrossRef] [Green Version]
- Radogna, F.; Diederich, M. Stress-Induced Cellular Responses in Immunogenic Cell Death: Implications for Cancer Immuno-therapy. Biochem. Pharmacol. 2018, 153, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woan, K.V.; Sahakian, E.; Sotomayor, E.M.; Seto, E.; Villagra, A. Modulation of antigen-presenting cells by HDAC inhibitors: Implications in autoimmunity and cancer. Immunol. Cell Biol. 2011, 90, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, F.; Zaliani, A.; Bertolini, G.; Porro, G.; Pagani, P.; Pozzi, P.; Donà, G.; Fossati, G.; Sozzani, S.; Azam, T.; et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. USA 2002, 99, 2995–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balliu, M.; Guandalini, L.; Romanelli, M.N.; D’Amico, M.; Paoletti, F. HDAC -inhibitor (S)-8 disrupts HDAC 6- PP 1 complex prompting A375 melanoma cell growth arrest and apoptosis. J. Cell. Mol. Med. 2014, 19, 143–154. [Google Scholar] [CrossRef]
- Cheng, F.; Lienlaf, M.; Wang, H.-W.; Perez-Villarroel, P.; Lee, C.; Woan, K.; Rock-Klotz, J.; Sahakian, E.; Woods, D.; Pinilla-Ibarz, J.; et al. A Novel Role for Histone Deacetylase 6 in the Regulation of the Tolerogenic STAT3/IL-10 Pathway in APCs. J. Immunol. 2014, 193, 2850–2862. [Google Scholar] [CrossRef]
- Woan, K.; Lienlaf, M.; Perez-Villaroel, P.; Lee, C.; Cheng, F.; Knox, T.; Woods, D.M.; Barrios, K.; Powers, J.; Sahakian, E.; et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation. Mol. Oncol. 2015, 9, 1447–1457. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Feng, Y.; Zhou, J.; Cheung, O.K.-W.; Cao, J.; Wang, J.; Tang, W.; Tu, Y.; Xu, L.; Wu, F.; et al. A selective HDAC8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma. Sci. Transl. Med. 2021, 13, eaaz6804. [Google Scholar] [CrossRef]
- Li, X.; Su, X.; Liu, R.; Pan, Y.; Fang, J.; Cao, L.; Feng, C.; Shang, Q.; Chen, Y.; Shao, C.; et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene 2021, 40, 1836–1850. [Google Scholar] [CrossRef]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic Enhancement of Antigen Processing and Presentation Promotes Immune Recognition of Tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [Green Version]
- Manning, J.; Indrova, M.; Lubyova, B.; Pribylova, H.; Bieblova, J.; Hejnar, J.; Simova, J.; Jandlova, T.; Bubenik, J.; Reinis, M. Induction of MHC class I molecule cell surface expression and epigenetic activation of antigen-processing machinery components in a murine model for human papilloma virus 16-associated tumours. Immunology 2008, 123, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B. Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunol. Immunother. 2008, 57, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.H.; Gregorie, C.J.; Tomasi, T.B. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol. Immunother. 2008, 57, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroesen, M.; Gielen, P.R.; Brok, I.C.; Armandari, I.; Hoogerbrugge, P.M.; Adema, G.J. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 2014, 5, 6558–6572. [Google Scholar] [CrossRef] [Green Version]
- Frikeche, J.; Peric, Z.; Brissot, E.; Grégoire, M.; Gaugler, B.; Mohty, M. Impact of HDAC inhibitors on dendritic cell functions. Exp. Hematol. 2012, 40, 783–791. [Google Scholar] [CrossRef]
- Jung, I.; Lee, J.; Jeong, Y.-I.; Lee, C.-M.; Chang, J.; Jeong, S.; Chun, S.; Park, W.; Han, J.; Shin, Y.; et al. Apicidin, the Histone Deacetylase Inhibitor, Suppresses TH1 Polarization of Murine Bone Marrow-Derived Dendritic Cells. Int. J. Immunopathol. Pharmacol. 2009, 22, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Bode, K.A.; Schroder, K.; Hume, D.A.; Ravasi, T.; Heeg, K.; Sweet, M.J.; Dalpke, A.H. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology 2007, 122, 596–606. [Google Scholar] [CrossRef]
- Zhou, Y.; Jin, X.; Yu, H.; Qin, G.; Pan, P.; Zhao, J.; Chen, T.; Liang, X.; Sun, Y.; Wang, B.; et al. HDAC5 modulates PD-L1 expression and cancer immunity via p65 deacetylation in pancreatic cancer. Theranostics 2022, 12, 2080–2094. [Google Scholar] [CrossRef]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [Green Version]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR–dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [Green Version]
- Fine, J.H.; Chen, P.; Mesci, A.; Allan, D.S.; Gasser, S.; Raulet, D.H.; Carlyle, J.R. Chemotherapy-Induced Genotoxic Stress Promotes Sensitivity to Natural Killer Cell Cytotoxicity by Enabling Missing-Self Recognition. Cancer Res. 2010, 70, 7102–7113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, C.-H.; Keum, J.-H.; Yang, K.; Nam, J.; Kim, M.-J.; Kim, S.-H.; Kang, C.-D.; Oh, S.-O.; Kim, C.-D.; Park, Y.-S.; et al. Synergistic enhancement of NK cell-mediated cytotoxicity by combination of histone deacetylase inhibitor and ionizing radiation. Radiat. Oncol. 2014, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, N.I.; Niimi, A.; Isono, M.; Oike, T.; Sato, H.; Nakano, T.; Shibata, A. Inhibition of the HDAC/Suv39/G9a pathway restores the expression of DNA damage-dependent major histocompatibility complex class I-related chain A and B in cancer cells. Oncol. Rep. 2017, 38, 693–702. [Google Scholar] [CrossRef] [Green Version]
- A Marks, P. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Investig. Drugs 2010, 19, 1049–1066. [Google Scholar] [CrossRef] [PubMed]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2001, 98, 10833–10838. [Google Scholar] [CrossRef] [Green Version]
- Xue, K.; Gu, J.J.; Zhang, Q.; Mavis, C.; Hernandez-Ilizaliturri, F.J.; Czuczman, M.S.; Guo, Y. Vorinostat, a histone deacetylase (HDAC) inhibitor, promotes cell cycle arrest and re-sensitizes rituximab- and chemo-resistant lymphoma cells to chemotherapy agents. J. Cancer Res. Clin. Oncol. 2015, 142, 379–387. [Google Scholar] [CrossRef]
- E Gryder, B.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Futur. Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [Green Version]
- Scuto, A.; Kirschbaum, M.; Kowolik, C.; Kretzner, L.; Juhasz, A.; Atadja, P.; Pullarkat, V.; Bhatia, R.; Forman, S.; Yen, Y.; et al. The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph− acute lymphoblastic leukemia cells. Blood 2008, 111, 5093–5100. [Google Scholar] [CrossRef] [Green Version]
- Maiso, P.; Carvajal-Vergara, X.; Ocio, E.M.; López-Pérez, R.; Mateo, G.; Gutiérrez, N.; Atadja, P.; Pandiella, A.; Miguel, J.F.S. The Histone Deacetylase Inhibitor LBH589 Is a Potent Antimyeloma Agent that Overcomes Drug Resistance. Cancer Res. 2006, 66, 5781–5789. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.; Li, J.; Atadja, P.; Bhalla, K.; Haura, E.B. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor–dependent human lung cancer cells. Mol. Cancer Ther. 2007, 6, 2515–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, D.C.; Kutko, M.C.; Glick, R.D.; Butler, L.M.; Heller, G.; A Rifkind, R.; A Marks, P.; Richon, V.M.; La Quaglia, M.P. The histone deacetylase inhibitor, CBHA, inhibits growth of human neuroblastoma xenografts in vivo, alone and synergistically with all-trans retinoic acid. Cancer Res. 2001, 61, 3591–3594. [Google Scholar] [PubMed]
- Coffey, D.C.; Kutko, M.C.; Glick, R.D.; Swendeman, S.L.; Butler, L.; Rifkind, R.; Marks, P.A.; Richon, V.M.; LaQuaglia, M.P. Histone deacetylase inhibitors and retinoic acids inhibit growth of human neuroblastoma in vitro. Med. Pediatr. Oncol. 2000, 35, 577–581. [Google Scholar] [CrossRef]
- Varghese, S.; Senanayake, T.; Stewart, T.M.; Doering, K.; Fraser, A.; Casero, R.A., Jr.; Woster, P.M. Polyaminohydroxamic Acids and Polyaminobenzamides as Isoform Selective Histone Deacetylase Inhibitors. J. Med. Chem. 2008, 51, 2447–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buyandelger, B.; E Bar, E.; Hung, K.-S.; Chen, R.-M.; Chiang, Y.-H.; Liou, J.-P.; Huang, H.-M.; Wang, J.-Y. Histone deacetylase inhibitor MPT0B291 suppresses Glioma Growth in vitro and in vivo partially through acetylation of p53. Int. J. Biol. Sci. 2020, 16, 3184–3199. [Google Scholar] [CrossRef]
- Bolden, J.E.; Shi, W.; Jankowski, K.; Kan, C.-Y.; Cluse, L.; Martin, B.P.; MacKenzie, K.L.; Smyth, G.K.; Johnstone, R.W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4, e519. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef] [Green Version]
- Mahboobi, S.; Dove, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Design of Chimeric Histone Deacetylase- and Tyrosine Kinase-Inhibitors: A Series of Imatinib Hybrides as Potent Inhibitors of Wild-Type and Mutant BCR-ABL, PDGF-Rβ, and Histone Deacetylases. J. Med. Chem. 2009, 52, 2265–2279. [Google Scholar] [CrossRef]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent Progress in Histone Deacetylase Inhibitors as Anticancer Agents. Curr. Med. Chem. 2020, 27, 2449–2493. [Google Scholar] [CrossRef]
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Gandara, D.R.; et al. A Phase II Trial of Vorinostat (Suberoylanilide Hydroxamic Acid) in Metastatic Breast Cancer: A California Cancer Consortium Study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, R.P.; Rankin, C.; Hoff, P.M.G.; Gold, P.J.; Billingsley, K.G.; Chapman, R.A.; Wong, L.; Ward, J.H.; Abbruzzese, J.L.; Blanke, C.D. Phase II trial of romidepsin (NSC-630176) in previously treated colorectal cancer patients with advanced disease: A Southwest Oncology Group study (S0336). Investig. New Drugs 2008, 27, 469–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.-A.; Tsao, M.S.; et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Chun, P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch. Pharmacal Res. 2015, 38, 933–949. [Google Scholar] [CrossRef]
- Bradley, D.; Rathkopf, D.; Dunn, R.; Stadler, W.M.; Liu, G.; Smith, D.; Pili, R.; Zwiebel, J.; I Scher, H.; Hussain, M. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862). Cancer 2009, 115, 5541–5549. [Google Scholar] [CrossRef] [Green Version]
- Undevia, S.D.; Janisch, L.; Schilsky, R.L.; Loury, D.; Balasubramanian, S.; Mani, C.; Sirisawad, M.; Buggy, J.J.; Miller, R.A.; Ratain, M.J. Phase I study of the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of the histone deacetylase inhibitor (HDACi) PCI-24781. J. Clin. Oncol. 2008, 26, 14514. [Google Scholar] [CrossRef]
- Nemunaitis, J.J.; Orr, D.; Eager, R.; Cunningham, C.C.; Williams, A.; Mennel, R.; Grove, W.; Olson, S. Phase I Study of Oral CI-994 in Combination with Gemcitabine in Treatment of Patients with Advanced Cancer. Cancer J. 2003, 9, 58–66. [Google Scholar] [CrossRef]
- Prakash, S.; Foster, B.J.; Meyer, M.; Wozniak, A.; Heilbrun, L.K.; Flaherty, L.; Zalupski, M.; Radulovic, L.; Valdivieso, M.; Lorusso, P.M. Chronic Oral Administration of CI-994: A Phase I Study. Investig. New Drugs 2001, 19, 1–11. [Google Scholar] [CrossRef]
- Leoni, F.; Fossati, G.; Lewis, E.C.; Lee, J.-K.; Porro, G.; Pagani, P.; Modena, D.; Moras, M.L.; Pozzi, P.; Reznikov, L.L.; et al. The Histone Deacetylase Inhibitor ITF2357 Reduces Production of Pro-Inflammatory Cytokines In Vitro and Systemic Inflammation In Vivo. Mol. Med. 2005, 11, 1–15. [Google Scholar] [CrossRef]
- Kummar, S.; Gutierrez, M.; Gardner, E.R.; Donovan, E.; Hwang, K.; Chung, E.J.; Lee, M.-J.; Maynard, K.; Kalnitskiy, M.; Chen, A.; et al. Phase I Trial of MS-275, a Histone Deacetylase Inhibitor, Administered Weekly in Refractory Solid Tumors and Lymphoid Malignancies. Clin. Cancer Res. 2007, 13, 5411–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouladi, M.; Furman, W.L.; Chin, T.; Iii, B.B.F.; Dudkin, L.; Stewart, C.F.; Krailo, M.D.; Speights, R.; Ingle, A.M.; Houghton, P.J.; et al. Phase I Study of Depsipeptide in Pediatric Patients With Refractory Solid Tumors: A Children’s Oncology Group Report. J. Clin. Oncol. 2006, 24, 3678–3685. [Google Scholar] [CrossRef] [PubMed]
- Stadler, W.M.; Margolin, K.; Ferber, S.; McCulloch, W.; Thompson, J.A. A Phase II Study of Depsipeptide in Refractory Metastatic Renal Cell Cancer. Clin. Genitourin. Cancer 2006, 5, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvic, M.; Dummer, R.; Becker, J.C.; Poulalhon, N.; Romero, P.O.; Bernengo, M.G.; Lebbé, C.; Assaf, C.; Squier, M.; Williams, D.; et al. Panobinostat activity in both bexarotene-exposed and -naïve patients with refractory cutaneous T-cell lymphoma: Results of a phase II trial. Eur. J. Cancer 2012, 49, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, A.; Hatake, K.; Matsui, K.; Sakajiri, S.; Hirashima, T.; Tanii, H.; Kobayashi, K.; Yamamoto, N. A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Investig. New Drugs 2011, 30, 1096–1106. [Google Scholar] [CrossRef]
- Takebe, N.; Beumer, J.H.; Kummar, S.; Kiesel, B.F.; Dowlati, A.; O’Sullivan Coyne, G.; Piekarz, R.; Rubinstein, L.; Fogli, L.K.; Vaishampayan, U.; et al. A phase I pharmacokinetic study of belinostat in patients with advanced cancers and varying degrees of liver dysfunction. Br. J. Clin. Pharmacol. 2019, 85, 2499–2511. [Google Scholar] [CrossRef]
- Yeo, W.; Chung, H.C.; Chan, S.L. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: A multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J. Clin. Oncol. 2012, 30, 3361–3367. [Google Scholar]
- Crump, M.; Coiffier, B.; Jacobsen, E.D.; Sun, L.; Ricker, J.L.; Xie, H.; Frankel, S.R.; Randolph, S.S.; Cheson, B.D. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann. Oncol. 2008, 19, 964–969. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Com-bating Cancer. Oncotarget 2017, 8, 38022. [Google Scholar] [CrossRef] [Green Version]
- Stiborova, M.; Eckschlager, T.; Poljakova, J.; Hrabeta, J.; Adam, V.; Kizek, R.; Frei, E. The synergistic effects of DNA-targeted chemotherapeutics and histone deacetylase inhibitors as therapeutic strategies for cancer treatment. Curr. Med. Chem. 2012, 19, 4218–4238. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-S.; Wang, Y.-C.; Yang, H.-C.; Huang, P.-H.; Kulp, S.K.; Yang, C.-C.; Lu, Y.-S.; Matsuyama, S.; Chen, C.-Y. Histone Deacetylase Inhibitors Sensitize Prostate Cancer Cells to Agents that Produce DNA Double-Strand Breaks by Targeting Ku70 Acetylation. Cancer Res. 2007, 67, 5318–5327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantin, V.R.; Richon, V.M. Mechanisms of Resistance to Histone Deacetylase Inhibitors and Their Therapeutic Implications. Clin. Cancer Res. 2007, 13, 7237–7242. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munster, P.; Marchion, D.; Bicaku, E.; Schmitt, M.; Lee, J.H.; DeConti, R.; Simon, G.; Fishman, M.; Minton, S.; Garrett, C.; et al. Phase I Trial of Histone Deacetylase Inhibition by Valproic Acid Followed by the Topoisomerase II Inhibitor Epirubicin in Advanced Solid Tumors: A Clinical and Translational Study. J. Clin. Oncol. 2007, 25, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, B.; Xu, H.; Sun, Y.; Shi, Y.; Luo, Y.; Jia, H.; Wang, F. Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene 2017, 36, 6863–6872. [Google Scholar] [CrossRef]
- Robey, R.W.; Chakraborty, A.R.; Basseville, A.; Luchenko, V.; Bahr, J.; Zhan, Z.; Bates, S.E. Histone Deacetylase Inhibitors: Emerging Mechanisms of Resistance. Mol. Pharm. 2011, 8, 2021–2031. [Google Scholar] [CrossRef]
- Nolan, L.; Johnson, P.W.M.; Ganesan, A.; Packham, G.; Crabb, S.J. Will Histone Deacetylase Inhibitors Require Combination with Other Agents to Fulfil Their Therapeutic Potential? Br. J. Cancer 2008, 99, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Hershman, D.L.; Pellegrino, C.M.; Andreopoulou, E.; Makower, D.; Fineberg, S.; Bhalla, K.N.; Kalinsky, K.; Fehn, K.; Huang, M.; et al. Phase I-II study of the histone deacetytase inhibitor vorinostat plus sequential weekly paclitaxel and doxorubicin-cyclophosphamide in locally advanced breast cancer. J. Clin. Oncol. 2014, 32, 598. [Google Scholar] [CrossRef]
- Schneider, B.J.; Kalemkerian, G.P.; Bradley, D.; Smith, D.C.; Egorin, M.J.; Daignault, S.; Dunn, R.; Hussain, M. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Investig. New Drugs 2010, 30, 249–257. [Google Scholar] [CrossRef]
- Yoo, C.; Ryu, M.-H.; Na, Y.-S.; Ryoo, B.-Y.; Lee, C.-W.; Kang, Y.-K. Vorinostat in combination with capecitabine plus cisplatin as a first-line chemotherapy for patients with metastatic or unresectable gastric cancer: Phase II study and biomarker analysis. Br. J. Cancer 2016, 114, 1185–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, T.; Campbell, T.C.; Zhang, C.; Kim, K.M.; Kolesar, J.M.; Oettel, K.R.; Blank, J.H.; Robinson, E.G.; Ahuja, H.G.; Kirschling, R.J.; et al. Vorinostat and bortezomib as third-line therapy in patients with advanced non-small cell lung cancer: A Wisconsin Oncology Network Phase II study. Investig. New Drugs 2014, 32, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Conte, P.; Campone, M.; Pronzato, P.; Amadori, D.; Frank, R.; Schuetz, F.; Rea, D.; Wardley, A.; Britten, C.; Elias, A. Phase I trial of panobinostat (LBH589) in combination with trastuzumab in pretreated HER2-positive metastatic breast cancer (mBC): Preliminary safety and tolerability results. J. Clin. Oncol. 2009, 27, 1081. [Google Scholar] [CrossRef]
- Peacock, N.W.; Jones, S.F.; Yardley, D.A.; Bendell, J.C.; Infante, J.R.; Murphy, P.B.; Burris, H.A. A phase I study of panobinostat (LBH589) with capecitabine with or without lapatinib. J. Clin. Oncol. 2010, 28, 1115. [Google Scholar] [CrossRef]
- Gray, J.E.; Haura, E.B.; Chiappori, A.; Tanvetyanon, T.; Williams, C.C.; Pinder, M.C.; Neuger, A.; Giglia, J.L.; Bepler, G.; Altiok, S. Phase I study of LBH589 in combination with erlotinib for advanced aerodigestive tract cancers. J. Clin. Oncol. 2010, 28, e13016. [Google Scholar] [CrossRef]
- Vitfell-Rasmussen, J.; Judson, I.; Safwat, A.; Jones, R.L.; Rossen, P.B.; Lind-Hansen, M.; Knoblauch, P.; Krarup-Hansen, A. A Phase I/II Clinical Trial of Belinostat (PXD101) in Combination with Doxorubicin in Patients with Soft Tissue Sarcomas. Sarcoma 2016, 2016, 2090271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Romano, E.; Romero, P. The therapeutic promise of disrupting the PD-1/PD-L1 immune checkpoint in cancer: Unleashing the CD8 T cell mediated anti-tumor activity results in significant, unprecedented clinical efficacy in various solid tumors. J. Immunother. Cancer 2015, 3, 15. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Orillion, A.; Pili, R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics 2016, 8, 415–428. [Google Scholar] [CrossRef]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Epigenetics 2017, 9, 59. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, Y.; Lee, M.-J.; Lee, S.; Tomita, S.; Chumsri, S.; Cruickshank, S.; Ordentlich, P.; Trepel, J.B. The interplay of epigenetic therapy and immunity in locally recurrent or metastatic estrogen receptor-positive breast cancer: Correlative analysis of ENCORE 301, a randomized, placebo-controlled phase II trial of exemestane with or without entinostat. OncoImmunology 2016, 5, e1219008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Waschke, B.C.; Woolaver, R.A.; Chen, Z.; Zhang, G.; Piscopio, A.D.; Liu, X.; Wang, J.H. Histone Deacetylase Inhibition Sensitizes PD1 Blockade–Resistant B-cell Lymphomas. Cancer Immunol. Res. 2019, 7, 1318–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 68, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef] [Green Version]
- Rihawi, k.; Gelsomino, F.; Sperandi, F.; Melotti, B.; Fiorentino, M.; Casolari, L.; Ardizzoni, A. Pembrolizumab in the treatment of metastatic non-small cell lung cancer: A review of current evidence. Ther. Adv. Respir. Dis. 2017, 11, 353–373. [Google Scholar] [CrossRef] [Green Version]
- Pili, R.; Quinn, D.; Hahn, N.M.; Albany, C.; Logan, T.F.; Drake, C.G. A phase I/Ib, open-label, dose-finding study to evaluate safety, pharmacodynamics, and efficacy of pembrolizumab (MK-3475) in combination with vorinostat in patients with advanced renal or urothelial cell carcinoma. J. Clin. Oncol. 2016, 34, TPS4581. [Google Scholar] [CrossRef]
- Forero, A.; Stroyakovskiy, D.; Cha, E.; Cruickshank, S.; Hasapidis, J.; Meyers, M.L.; Slamon, D.J. Abstract OT2-01-12: ENCORE 602: A randomized, placebo-controlled, double-blind, multicenter phase 2 study (with a phase 1b lead-in) of atezolizumab with or without entinostat in patients with advanced triple negative breast cancer (aTNBC). Cancer Res. 2017, 77, OT2-01. [Google Scholar] [CrossRef]
- Van Tilburg, C.M.; Witt, R.; Pajtler, K.W.; Christoph, P.; Poschke, I.; Platten, M.; Harting, I.; Sedlaczek, O.; Freitag, A.; Meyrath, D.; et al. INFORM2 exploratory multinational phase I/II combination study of nivolumab and entinostat in children and adolescents with refractory high-risk malignancies: INFORM2 NivEnt. J. Clin. Oncol. 2019, 37, TPS10065. [Google Scholar] [CrossRef]



| Class | Member | Location | Substrates | Inhibitors | References |
|---|---|---|---|---|---|
| Class I | HDAC 1 HDAC 2 HDAC 3 HDAC 8 | Nuclear | Histones |
| [14] |
| Class IIa | HDAC4 HDAC5 HDAC7 HDAC9 | Nuclear/cytoplasmic | Histones |
| [15] |
| Class IIb | HDAC 6 HDAC 10 | Nuclear/cytoplasmic | Histones; α-tubulin; Hsp90 |
| [16] |
| Class III | Sirtuins Sir2 | Nuclear/cytoplasmic | Mitochondrial Histones; Tubulin; p53; TAF |
| [17] |
| Class IV | HDAC 11 | Nuclear | Rpd3 protein |
| [18] |
| HDACIs | Clinical Trial Phase | Clinical Trial ID | Cancer Types | Trial Description | Status | References |
|---|---|---|---|---|---|---|
| MS-275 (Entinostat) | I | NCT00020579 | Refractory solid tumors and lymphoid | Well tolerated at a dose of 6 mg/m2, administered weekly with food for 4 weeks every 6 weeks | Completed | [142] |
| Romidepsin (Depsipeptid) | I | NCT00053963 | Solid tumors | Well tolerated in children with refractory solid tumors | Completed | [143] |
| II | NCT00106613 | Renal cell carcinoma | Did not have sufficient activity. | Completed | [144] | |
| I | NCT00077337 | Colorectal cancer | Romidepsin at dose of 13 mg/m2 as a 4 h iv infusion on days 1, 8, and 15 of a 28-day cycle was ineffective in treatment of metastatic colon cancer | Completed | [133] | |
| Panobinostat (LBH589) | II | NCT00667862 | Hormone refractory prostate cancer | Panobinostat did not show a sufficient level of clinical activity to undergo further investigation in CRPC | Completed | [145] |
| II | NCT00425555 | Cutaneous T-cell lymphoma | Panobinostat was generally well tolerated with no major safety concerns. | Completed | [146] | |
| I | NCT00412997 | Solid tumors | Doses well tolerated up to 20 mg in Japanese patients | Completed | [147] | |
| Belinostat (PXD 101) | I | NCT01273155 | Adult primary hepatocellular carcinoma and advanced adult primary liver cancer | Increased belinostat exposure accompanied hepatic dysfunction | Completed | [148] |
| I and II | NCT00321594 | Localized unresectable adult primary liver cancer and recurrent adult primary liver cancer | Phase I—belinostat tolerated at maximum dose of 1200 mg/m2/day Phase II—will start with 1200 mg/m2/day | Completed | [149] | |
| Vorinostat | I | NCT00097929 | Relapsed diffuse large B-cell lymphoma | Limited activity against relapsed DLBCL | Completed | [150] |
| II | NCT00132067 | Primary peritoneal cavity recurrent ovarian epithelial cancer | Vorinostat well tolerated with minimal activity as a single agent | Completed | [135] |
| HDACIs | Immune Checkpoint Inhibitor | Clinical Trial Phase | Clinical Trial ID | Cancer Types | Trial Description | Status | References |
|---|---|---|---|---|---|---|---|
| Vorinostat (SAHA) | Pembrolizumab | I/II | NCT02638090 | Lung cancer/stage IV NSCLC | - | Recruiting | [178] |
| I/Ib | NCT02619253 | Renal cell carcinoma and urinary bladder neoplasms | - | Recruiting | [179] | ||
| Entinostat | Atezolizumab | I/II | NCT02708680 | Breast cancer | Combination therapy resulted in more toxicity | Completed | [180] |
| Nivolumab | I/II | NCT03838042 | Central nervous system tumors, solid tumors | - | Recruiting | [181] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alseksek, R.K.; Ramadan, W.S.; Saleh, E.; El-Awady, R. The Role of HDACs in the Response of Cancer Cells to Cellular Stress and the Potential for Therapeutic Intervention. Int. J. Mol. Sci. 2022, 23, 8141. https://doi.org/10.3390/ijms23158141
Alseksek RK, Ramadan WS, Saleh E, El-Awady R. The Role of HDACs in the Response of Cancer Cells to Cellular Stress and the Potential for Therapeutic Intervention. International Journal of Molecular Sciences. 2022; 23(15):8141. https://doi.org/10.3390/ijms23158141
Chicago/Turabian StyleAlseksek, Rahma K., Wafaa S. Ramadan, Ekram Saleh, and Raafat El-Awady. 2022. "The Role of HDACs in the Response of Cancer Cells to Cellular Stress and the Potential for Therapeutic Intervention" International Journal of Molecular Sciences 23, no. 15: 8141. https://doi.org/10.3390/ijms23158141