
Therapeutic Potential of Fingolimod and Dimethyl Fumarate in Non-Small Cell Lung Cancer Preclinical Models



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
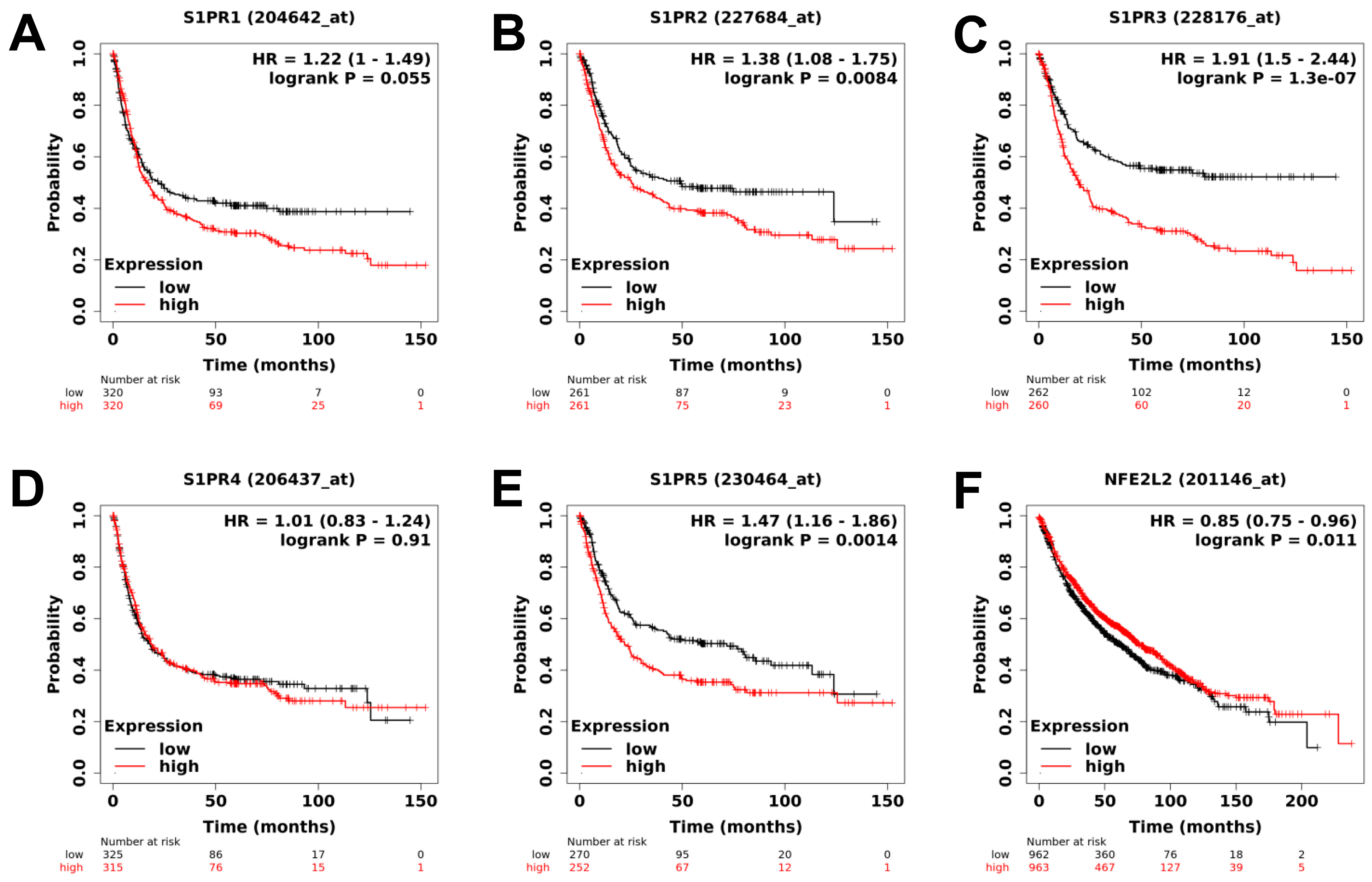
2.1. Analysis of Public Clinical Data: Expression of Downstream Target of Fingolimod and DMF Are Correlated with Better Prognostic Value in Lung Cancer Cohorts
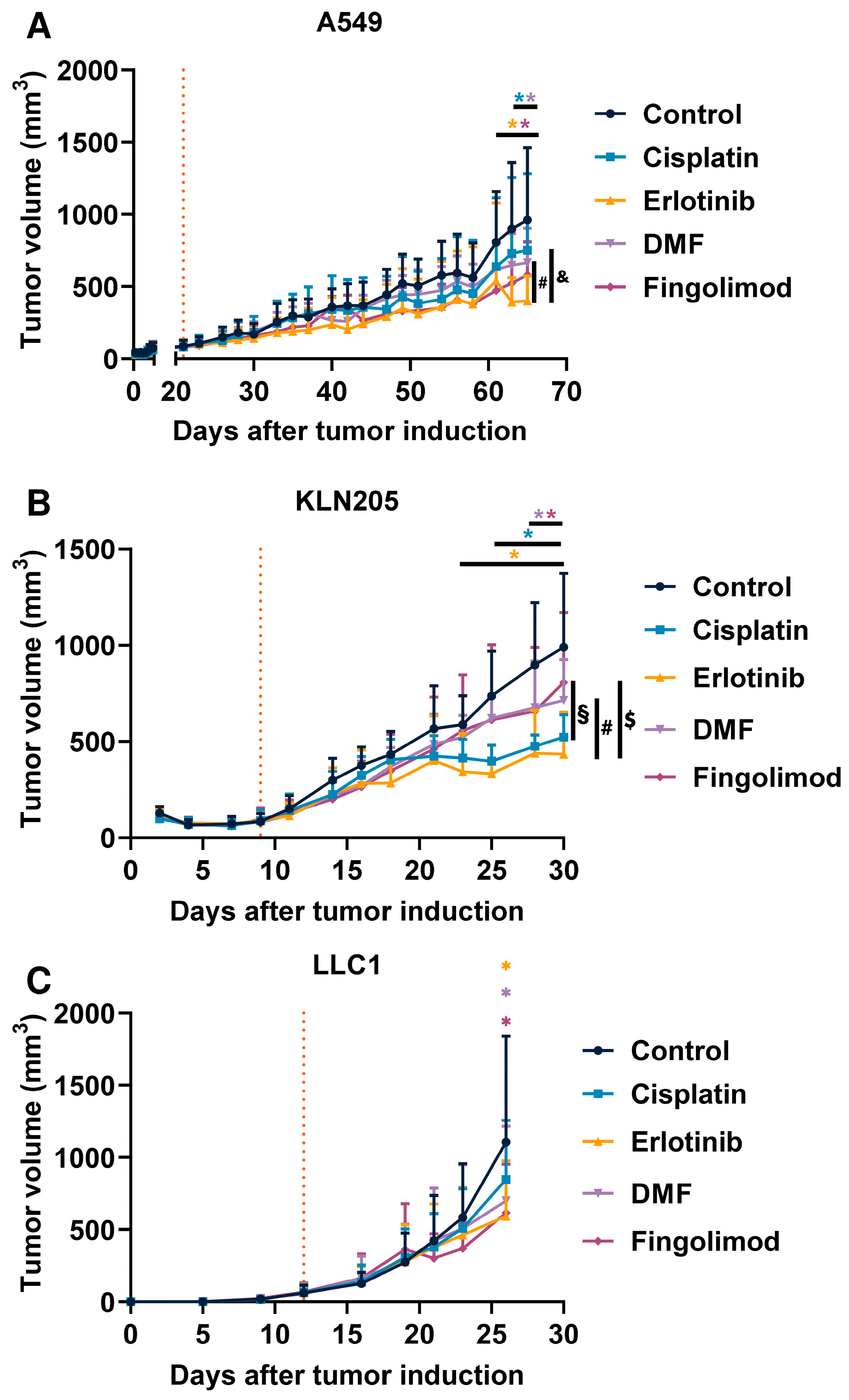
2.2. Fingolimod and DMF Decreased NSCLC Tumor Growth In Vivo in Immunocompetent and Immunodeficient Mouse Models
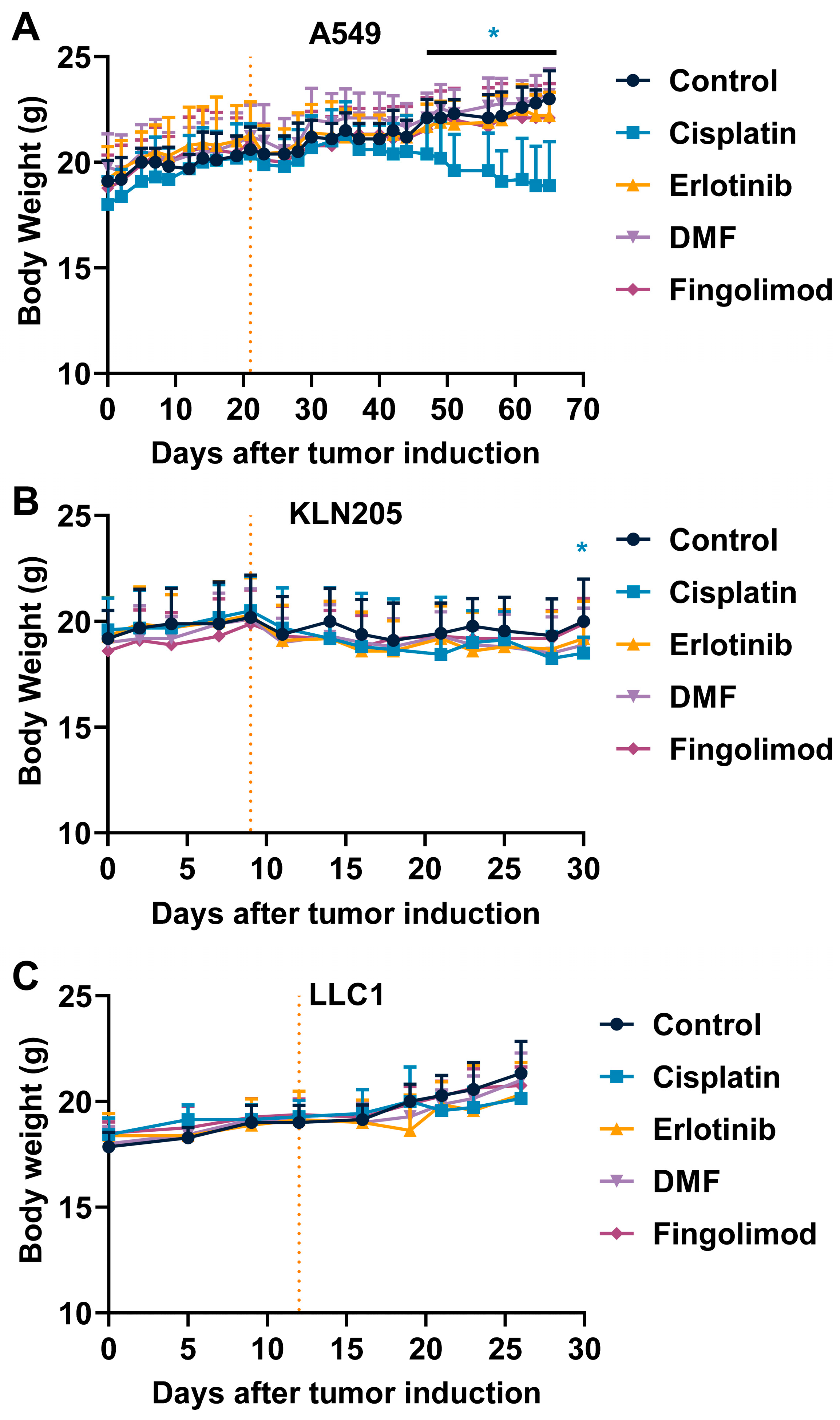
2.3. Fingolimod and DMF Are Well Tolerated In Vivo
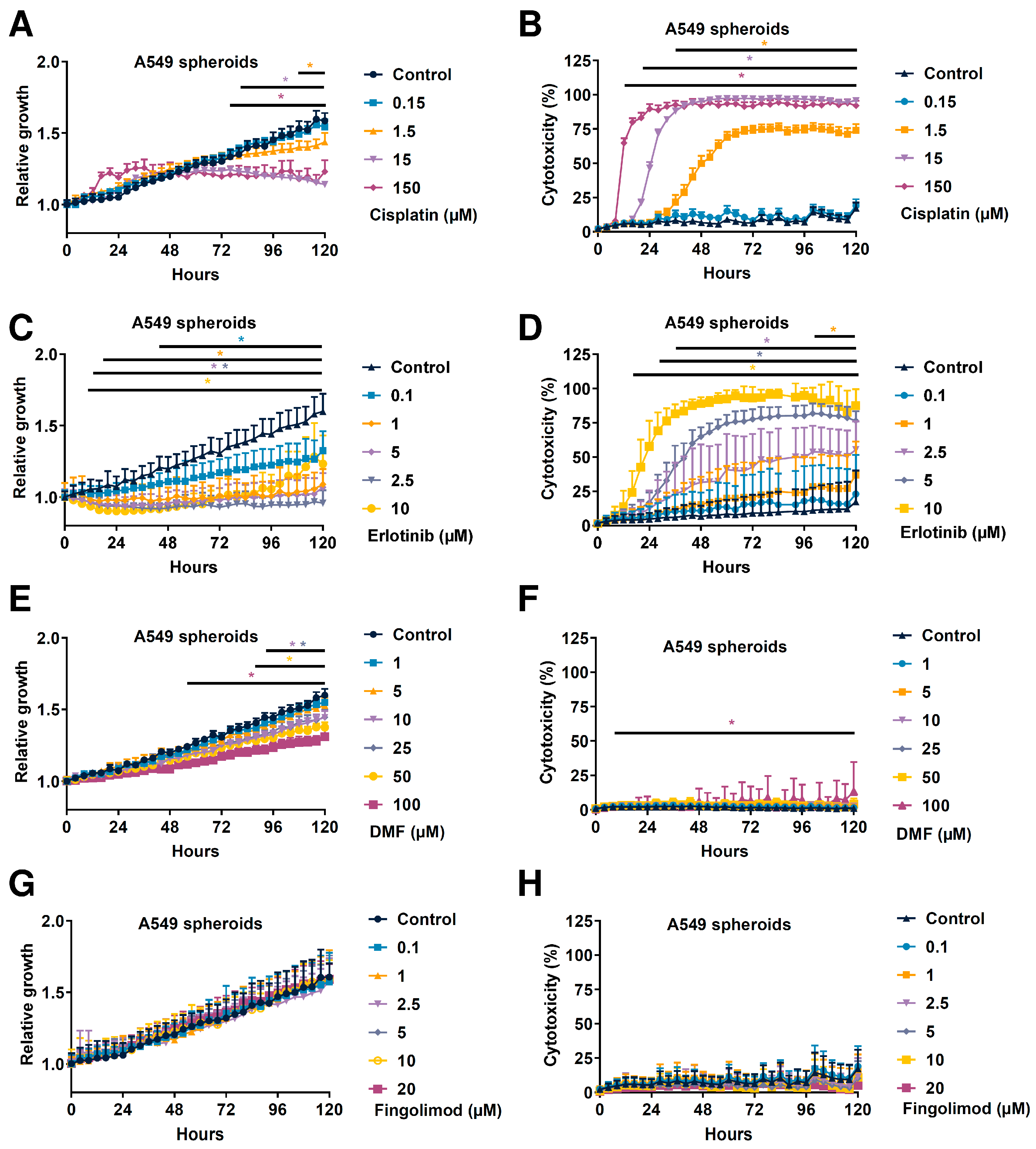
2.4. Fingolimod and DMF Differentially Affected In Vitro A549 Spheroid 3D Viability
2.5. Fingolimod and DMF Reduced In Vitro A549 Cell Growth When Cultured as 2D Monolayer
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Cells and Cell Culture
4.3. 2D Cell Growth Assay
4.4. 3D Tumor Spheroid Assay
4.5. Subcutaneous Graft Animal Model
4.6. Animal Ethical Consideration and Limit Points
4.7. Treatments
4.8. Patient Survival Analysis
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DMF | Dimethyl Fumarate |
| DMSO | DiMethyl SulfOxide |
| EGFR | Epidermal Growth Factor |
| EMT | Epithelial-to-Mesenchymal Transition |
| NRF2 | Nuclear factor E2-Related Factor 2 |
| OS | Overall Survival |
| NSCLC | Non-Small Cell Lung Cancer |
| PEG | PolyEthylene Glycol |
| S1P | Sphingosine-1-Phosphate |
| S1PR | Sphingosine-1-Phosphate Receptor |
| SD | Standard Deviation |
| TME | Tumor MicroEnvironment |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non–Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Sato, M.; Shames, D.S.; Gazdar, A.F.; Minna, J.D. A Translational View of the Molecular Pathogenesis of Lung Cancer. J. Thorac. Oncol. 2007, 2, 327–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodard, G.A.; Jones, K.D.; Jablons, D.M. Lung Cancer Staging and Prognosis. Cancer Treat. Res. 2016, 170, 47–75. [Google Scholar] [CrossRef]
- Hubbard, M.O.; Fu, P.; Margevicius, S.; Dowlati, A.; Linden, P.A. Five-Year Survival Does Not Equal Cure in Non–Small Cell Lung Cancer: A Surveillance, Epidemiology, and End Results–Based Analysis of Variables Affecting 10- to 18-Year Survival. J. Thorac. Cardiovasc. Surg. 2012, 143, 1307–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung Cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef]
- Parashar, B.; Arora, S.; Wernicke, A.G. Radiation Therapy for Early Stage Lung Cancer. Semin. Intervent. Radiol. 2013, 30, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.H.; Johnson, J.R.; Chen, Y.-F.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Erlotinib (Tarceva) Tablets. Oncologist 2005, 10, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H. Comparison of Four Chemotherapy Regimens for Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of Cytotoxic Action and Molecular Basis of Resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Amable, L. Cisplatin Resistance and Opportunities for Precision Medicine. Pharmacol. Res 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Ji, M.; Liu, Y.; Li, Q.; Li, X.; Ning, Z.; Zhao, W.; Shi, H.; Jiang, J.; Wu, C. PD-1/PD-L1 Expression in Non-Small-Cell Lung Cancer and Its Correlation with EGFR/KRAS Mutations. Cancer Biol. Ther. 2016, 17, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Salama, R.; Gadgeel, S.M.; Sarkar, F.H.; Ahmad, A. Erlotinib Resistance in Lung Cancer: Current Progress and Future Perspectives. Front. Pharmacol. 2013, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharm. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharm. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Roh, M.S. Molecular Pathology of Lung Cancer: Current Status and Future Directions. Tuberc. Respir. Dis. 2014, 77, 49. [Google Scholar] [CrossRef] [Green Version]
- Landi, L.; Cappuzzo, F. Experience with Erlotinib in the Treatment of Non-Small Cell Lung Cancer. Ther. Adv. Respir Dis. 2015, 9, 146–163. [Google Scholar] [CrossRef]
- Wang, W.; Bai, L.; Li, W.; Cui, J. The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front. Oncol. 2020, 10, 605154. [Google Scholar] [CrossRef]
- Nagahashi, M.; Abe, M.; Sakimura, K.; Takabe, K.; Wakai, T. The Role of Sphingosine-1-phosphate in Inflammation and Cancer Progression. Cancer Sci. 2018, 109, 3671–3678. [Google Scholar] [CrossRef] [Green Version]
- Pyne, N.J.; Pyne, S. Sphingosine 1-Phosphate and Cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Geffken, K.; Spiegel, S. SPHINGOSINE KINASE 1 IN BREAST CANCER. Adv. Biol. Regul. 2018, 67, 59–65. [Google Scholar] [CrossRef]
- Patmanathan, S.N.; Yap, L.F.; Murray, P.G.; Paterson, I.C. The Antineoplastic Properties of FTY720: Evidence for the Repurposing of Fingolimod. J. Cell Mol. Med. 2015, 19, 2329–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tintore, M.; Vidal-Jordana, A.; Sastre-Garriga, J. Treatment of Multiple Sclerosis—Success from Bench to Bedside. Nat. Rev. Neurol. 2019, 15, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.; Valesky, E.M.; Kippenberger, S.; Kaufmann, R. Dimethyl Fumarate—Only an Anti-Psoriatic Medication? JDDG J. Dtsch. Dermatol. Ges. 2012, 10, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Bomprezzi, R. Dimethyl Fumarate in the Treatment of Relapsing–Remitting Multiple Sclerosis: An Overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Saidu, N.E.B.; Noé, G.; Cerles, O.; Cabel, L.; Kavian-Tessler, N.; Chouzenoux, S.; Bahuaud, M.; Chéreau, C.; Nicco, C.; Leroy, K.; et al. Dimethyl Fumarate Controls the NRF2/DJ-1 Axis in Cancer Cells: Therapeutic Applications. Mol. Cancer 2017, 16, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl Fumarate, a Two-Edged Drug: Current Status and Future Directions. Med. Res. Rev. 2019, 39, 1923–1952. [Google Scholar] [CrossRef]
- White, C.; Alshaker, H.; Cooper, C.; Winkler, M.; Pchejetski, D. The Emerging Role of FTY720 (Fingolimod) in Cancer Treatment. Oncotarget 2016, 7, 23106–23127. [Google Scholar] [CrossRef] [Green Version]
- Rupp, T.; Pelouin, O.; Genest, L.; Legrand, C.; Froget, G.; Castagné, V. Therapeutic Potential of Fingolimod in Triple Negative Breast Cancer Preclinical Models. Transl. Oncol. 2021, 14, 100926. [Google Scholar] [CrossRef]
- Rosa, R.; Marciano, R.; Malapelle, U.; Formisano, L.; Nappi, L.; D’Amato, C.; D’Amato, V.; Damiano, V.; Marfè, G.; Vecchio, S.D.; et al. Sphingosine Kinase 1 Overexpression Contributes to Cetuximab Resistance in Human Colorectal Cancer Models. Clin. Cancer Res. 2013, 19, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Pchejetski, D.; Bohler, T.; Brizuela, L.; Sauer, L.; Doumerc, N.; Golzio, M.; Salunkhe, V.; Teissié, J.; Malavaud, B.; Waxman, J.; et al. FTY720 (Fingolimod) Sensitizes Prostate Cancer Cells to Radiotherapy by Inhibition of Sphingosine Kinase-1. Cancer Res. 2010, 70, 8651–8661. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.W.Y.; Man, K.; Sun, C.K.; Lee, T.K.; Poon, R.T.P.; Fan, S.T. Effects of a Novel Immunomodulating Agent, FTY720, on Tumor Growth and Angiogenesis in Hepatocellular Carcinoma. Mol. Cancer 2005, 4, 1430–1438. [Google Scholar] [CrossRef] [Green Version]
- Kreitzburg, K.M.; Fehling, S.C.; Landen, C.N.; Gamblin, T.L.; Vance, R.B.; Arend, R.C.; Katre, A.A.; Oliver, P.G.; van Waardenburg, R.C.A.M.; Alvarez, R.D.; et al. FTY720 Enhances the Anti-Tumor Activity of Carboplatin and Tamoxifen in a Patient-Derived Xenograft Model of Ovarian Cancer. Cancer Lett. 2018, 436, 75–86. [Google Scholar] [CrossRef]
- Ota, K.; Okuma, T.; Lorenzo, A.D.; Yokota, A.; Hino, H.; Kazama, H.; Moriya, S.; Takano, N.; Hiramoto, M.; Miyazawa, K. Fingolimod Sensitizes EGFR Wild-type Non-small Cell Lung Cancer Cells to Lapatinib or Sorafenib and Induces Cell Cycle Arrest. Oncol. Rep. 2019, 42, 231–242. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Spiegel, S.; Poklepovic, A.; Dent, P. Fingolimod Augments Pemetrexed Killing of Non-Small Cell Lung Cancer and Overcomes Resistance to ERBB Inhibition. Cancer Biol. Ther. 2018, 20, 597–607. [Google Scholar] [CrossRef]
- Liu, H.; Gu, Y.; Wang, H.; Yin, J.; Zheng, G.; Zhang, Z.; Lu, M.; Wang, C.; He, Z. Overexpression of PP2A Inhibitor SET Oncoprotein Is Associated with Tumor Progression and Poor Prognosis in Human Non-Small Cell Lung Cancer. Oncotarget 2015, 6, 14913–14925. [Google Scholar] [CrossRef] [Green Version]
- Saddoughi, S.A.; Gencer, S.; Peterson, Y.K.; Ward, K.E.; Mukhopadhyay, A.; Oaks, J.; Bielawski, J.; Szulc, Z.M.; Thomas, R.J.; Selvam, S.P.; et al. Sphingosine Analogue Drug FTY720 Targets I2PP2A/SET and Mediates Lung Tumour Suppression via Activation of PP2A-RIPK1-Dependent Necroptosis. EMBO Mol. Med. 2013, 5, 105. [Google Scholar] [CrossRef]
- Li, Y.; Hu, T.; Chen, T.; Yang, T.; Ren, H.; Chen, M. Combination Treatment of FTY720 and Cisplatin Exhibits Enhanced Antitumour Effects on Cisplatin-Resistant Non-Small Lung Cancer Cells. Oncol. Rep. 2018, 39, 565–572. [Google Scholar] [CrossRef]
- Xie, X.; Zhao, Y.; Ma, C.-Y.; Xu, X.-M.; Zhang, Y.-Q.; Wang, C.-G.; Jin, J.; Shen, X.; Gao, J.-L.; Li, N.; et al. Dimethyl Fumarate Induces Necroptosis in Colon Cancer Cells through GSH Depletion/ROS Increase/MAPKs Activation Pathway. Br. J. Pharmacol. 2015, 172, 3929–3943. [Google Scholar] [CrossRef] [Green Version]
- Kastrati, I.; Siklos, M.I.; Calderon-Gierszal, E.L.; El-Shennawy, L.; Georgieva, G.; Thayer, E.N.; Thatcher, G.R.J.; Frasor, J. Dimethyl Fumarate Inhibits the Nuclear Factor ΚB Pathway in Breast Cancer Cells by Covalent Modification of P65 Protein. J. Biol. Chem. 2016, 291, 3639–3647. [Google Scholar] [CrossRef] [Green Version]
- Loewe, R.; Valero, T.; Kremling, S.; Pratscher, B.; Kunstfeld, R.; Pehamberger, H.; Petzelbauer, P. Dimethylfumarate Impairs Melanoma Growth and Metastasis. Cancer Res. 2006, 66, 11888–11896. [Google Scholar] [CrossRef] [Green Version]
- Valero, T.; Steele, S.; Neumüller, K.; Bracher, A.; Niederleithner, H.; Pehamberger, H.; Petzelbauer, P.; Loewe, R. Combination of Dacarbazine and Dimethylfumarate Efficiently Reduces Melanoma Lymph Node Metastasis. J. Investig. Dermatol. 2010, 130, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Stepanovska, B.; Huwiler, A. Targeting the S1P Receptor Signaling Pathways as a Promising Approach for Treatment of Autoimmune and Inflammatory Diseases. Pharmacol. Res. 2020, 154, 104170. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in Cancers: A Double-Edged Sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Hill, D.P.; Harper, A.; Malcolm, J.; McAndrews, M.S.; Mockus, S.M.; Patterson, S.E.; Reynolds, T.; Baker, E.J.; Bult, C.J.; Chesler, E.J.; et al. Cisplatin-Resistant Triple-Negative Breast Cancer Subtypes: Multiple Mechanisms of Resistance. BMC Cancer 2019, 19, 1039. [Google Scholar] [CrossRef] [Green Version]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D Tumor Spheroid Models for in Vitro Therapeutic Screening: A Systematic Approach to Enhance the Biological Relevance of Data Obtained. Sci. Rep. 2016, 6, 19103. [Google Scholar] [CrossRef]
- Phung, Y.T.; Barbone, D.; Broaddus, V.C.; Ho, M. Rapid Generation of In Vitro Multicellular Spheroids for the Study of Monoclonal Antibody Therapy. J. Cancer 2011, 2, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Sant, S.; Johnston, P.A. The Production of 3D Tumor Spheroids for Cancer Drug Discovery. Drug Discov. Today: Technol. 2017, 23, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Schmid, G.; Guba, M.; Ischenko, I.; Papyan, A.; Joka, M.; Schrepfer, S.; Bruns, C.J.; Jauch, K.-W.; Heeschen, C.; Graeb, C. The Immunosuppressant FTY720 Inhibits Tumor Angiogenesis via the Sphingosine 1-Phosphate Receptor 1. J. Cell. Biochem. 2007, 101, 259–270. [Google Scholar] [CrossRef]
- Jacobi, N.; Seeboeck, R.; Hofmann, E.; Schweiger, H.; Smolinska, V.; Mohr, T.; Boyer, A.; Sommergruber, W.; Lechner, P.; Pichler-Huebschmann, C.; et al. Organotypic Three-Dimensional Cancer Cell Cultures Mirror Drug Responses in Vivo: Lessons Learned from the Inhibition of EGFR Signaling. Oncotarget 2017, 8, 107423. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, V.; Yakisich, J.S.; Kulkarni, Y.; Azad, N.; Iyer, A.K.V. Chemoresistance of Lung Cancer Cells: 2D and 3D In Vitro Models for Anticancer Drug Screening; IntechOpen: London, UK, 2018; ISBN 978-1-78984-350-7. [Google Scholar]
- Melissaridou, S.; Wiechec, E.; Magan, M.; Jain, M.V.; Chung, M.K.; Farnebo, L.; Roberg, K. The Effect of 2D and 3D Cell Cultures on Treatment Response, EMT Profile and Stem Cell Features in Head and Neck Cancer. Cancer Cell Int. 2019, 19, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, F.; Moriya, Y.; Ohnuki, T.; Ryo, M.; Takahashi, W. Neurological Safety of Fingolimod: An Updated Review. Clin. Exp. Neuroimmunol. 2017, 8, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, J.P.; Müller-Decker, K.; Schroeder, A.; Brechmann, M.; Möbs, M.; Géraud, C.; Assaf, C.; Goerdt, S.; Krammer, P.H.; Gülow, K. Dimethyl Fumarate Restores Apoptosis Sensitivity and Inhibits Tumor Growth and Metastasis in CTCL by Targeting NF-ΚB. Blood 2016, 128, 805–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stafman, L.L.; Williams, A.P.; Marayati, R.; Aye, J.M.; Stewart, J.E.; Mroczek-Musulman, E.; Beierle, E.A. PP2A Activation Alone and in Combination with Cisplatin Decreases Cell Growth and Tumor Formation in Human HuH6 Hepatoblastoma Cells. PLoS ONE 2019, 14, e0214469. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.T.; Man, K.; Ho, J.W.; Sun, C.K.; Lee, T.K.; Zhao, Y.; Lo, C.M.; Poon, R.T.; Fan, S.T. Marked Suppression of Tumor Growth by FTY720 in a Rat Liver Tumor Model: The Significance of down-Regulation of Cell Survival Akt Pathway. Int. J. Oncol. 2007, 30, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Langlois, B.; Saupe, F.; Rupp, T.; Arnold, C.; van der Heyden, M.; Orend, G.; Hussenet, T. AngioMatrix, a Signature of the Tumor Angiogenic Switch-Specific Matrisome, Correlates with Poor Prognosis for Glioma and Colorectal Cancer Patients. Oncotarget 2014, 5, 10529–10545. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Ortega, M.; Carrera, A.C.; Garrido, A. Role of NRF2 in Lung Cancer. Cells 2021, 10, 1879. [Google Scholar] [CrossRef]
- Zhao, J.; Lin, X.; Meng, D.; Zeng, L.; Zhuang, R.; Huang, S.; Lv, W.; Hu, J. Nrf2 Mediates Metabolic Reprogramming in Non-Small Cell Lung Cancer. Front. Oncol. 2020, 10, 578315. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Gstalder, C.; Ader, I.; Cuvillier, O. FTY720 (Fingolimod) Inhibits HIF1 and HIF2 Signaling, Promotes Vascular Remodeling, and Chemosensitizes in Renal Cell Carcinoma Animal Model. Mol. Cancer Ther. 2016, 15, 2465–2474. [Google Scholar] [CrossRef] [Green Version]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- LaMontagne, K.; Littlewood-Evans, A.; Schnell, C.; O’Reilly, T.; Wyder, L.; Sanchez, T.; Probst, B.; Butler, J.; Wood, A.; Liau, G.; et al. Antagonism of Sphingosine-1-Phosphate Receptors by FTY720 Inhibits Angiogenesis and Tumor Vascularization. Cancer Res. 2006, 66, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, T.; Langlois, B.; Koczorowska, M.M.; Radwanska, A.; Sun, Z.; Hussenet, T.; Lefebvre, O.; Murdamoothoo, D.; Arnold, C.; Klein, A.; et al. Tenascin-C Orchestrates Glioblastoma Angiogenesis by Modulation of Pro- and Anti-Angiogenic Signaling. Cell Rep. 2016, 17, 2607–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, N.; Liu, Y.-R.; Li, Y.-M.; Yang, Y.-N.; Pan, L.; Wei, Y.-B.; Wang, P.-Y.; Li, Y.-J.; Xie, S.-Y. Cisplatin Decreases Cyclin D2 Expression via Upregulating MiR-93 to Inhibit Lung Adenocarcinoma Cell Growth. Mol. Med. Rep. 2019, 20, 3355–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wang, Y.; Yao, A.; Xu, Z.; Dou, H.; Shen, S.; Hou, Y.; Wang, T. Low Frequency Magnetic Fields Induce Autophagy-Associated Cell Death in Lung Cancer through MiR-486-Mediated Inhibition of Akt/MTOR Signaling Pathway. Sci. Rep. 2017, 7, 11776. [Google Scholar] [CrossRef] [Green Version]
- Bennett Saidu, N.E.; Bretagne, M.; Mansuet, A.L.; Just, P.-A.; Leroy, K.; Cerles, O.; Chouzenoux, S.; Nicco, C.; Damotte, D.; Alifano, M.; et al. Dimethyl Fumarate Is Highly Cytotoxic in KRAS Mutated Cancer Cells but Spares Non-Tumorigenic Cells. Oncotarget 2018, 9, 9088–9099. [Google Scholar] [CrossRef] [Green Version]
- García-Caballero, M.; Marí-Beffa, M.; Medina, M.Á.; Quesada, A.R. Dimethylfumarate Inhibits Angiogenesis in Vitro and in Vivo: A Possible Role for Its Antipsoriatic Effect? J. Invest. Dermatol. 2011, 131, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Feng, X.-D.; Yang, B.; Tong, R.-L.; Lu, Y.-J.; Chen, D.-Y.; Zhou, L.; Xie, H.-Y.; Zheng, S.-S.; Wu, J. Dimethyl Fumarate Suppresses Hepatocellular Carcinoma Progression via Activating SOCS3/JAK1/STAT3 Signaling Pathway. Am. J. Transl. Res. 2019, 11, 4713–4725. [Google Scholar]
- To, C.; Ringelberg, C.S.; Royce, D.B.; Williams, C.R.; Risingsong, R.; Sporn, M.B.; Liby, K.T. Dimethyl Fumarate and the Oleanane Triterpenoids, CDDO-Imidazolide and CDDO-Methyl Ester, Both Activate the Nrf2 Pathway but Have Opposite Effects in the A/J Model of Lung Carcinogenesis. Carcinogenesis 2015, 36, 769. [Google Scholar] [CrossRef] [Green Version]
- Ebenezer, D.L.; Fu, P.; Natarajan, V. Targeting Sphingosine-1-Phosphate Signaling in Lung Diseases. Pharmacol. Ther. 2016, 168, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.; Harikumar, K.B. Sphingosine 1-Phosphate: A Novel Target for Lung Disorders. Front. Immunol. 2017, 8, 296. [Google Scholar] [CrossRef] [Green Version]
- Motono, N.; Ueda, Y.; Shimasaki, M.; Iwai, S.; Iijima, Y.; Usuda, K.; Uramoto, H. Prognostic Impact of Sphingosine Kinase 1 in Nonsmall Cell Lung Cancer. Clin. Pathol. 2021, 14, 2632010X20988531. [Google Scholar] [CrossRef] [PubMed]
- Gachechiladze, M.; Tichý, T.; Kolek, V.; Grygárková, I.; Klein, J.; Mgebrishvili, G.; Kharaishvili, G.; Janíková, M.; Smičková, P.; Cierna, L.; et al. Sphingosine Kinase-1 Predicts Overall Survival Outcomes in Non-Small Cell Lung Cancer Patients Treated with Carboplatin and Navelbine. Oncol. Lett. 2019, 18, 1259–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourakis, S.; Timpani, C.A.; de Haan, J.B.; Gueven, N.; Fischer, D.; Rybalka, E. Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility? Pharmaceuticals 2020, 13, 306. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Chang, J.-Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Du, Y.; Wang, Z.; Wang, X.; Duan, J.; Wan, R.; Xu, J.; Zhang, P.; Wang, D.; Tian, Y.; et al. Upfront Dose-Reduced Chemotherapy Synergizes with Immunotherapy to Optimize Chemoimmunotherapy in Squamous Cell Lung Carcinoma. J. Immunother. Cancer 2020, 8, e000807. [Google Scholar] [CrossRef] [PubMed]
- Regmi, S.C.; Park, S.Y.; Kim, S.J.; Banskota, S.; Shah, S.; Kim, D.-H.; Kim, J.-A. The Anti-Tumor Activity of Succinyl Macrolactin A Is Mediated through the β-Catenin Destruction Complex via the Suppression of Tankyrase and PI3K/Akt. PLoS ONE 2015, 10, e0141753. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Yee, J.; Cho, Y.S.; Jang, H.W.; Han, J.M.; Gwak, H.S. Risk Factors for Erlotinib-Induced Hepatotoxicity: A Retrospective Follow-up Study. BMC Cancer 2018, 18, 988. [Google Scholar] [CrossRef]
- Liang, S.; Peng, X.; Li, X.; Yang, P.; Xie, L.; Li, Y.; Du, C.; Zhang, G. Silencing of CXCR4 Sensitizes Triple-Negative Breast Cancer Cells to Cisplatin. Oncotarget 2014, 6, 1020–1030. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.R.; Rocha, C.R.R.; Martins, D.J.; Fiore, A.P.Z.P.; Kinker, G.S.; Bruni-Cardoso, A.; Menck, C.F.M. ATR Mediates Cisplatin Resistance in 3D-Cultured Breast Cancer Cells via Translesion DNA Synthesis Modulation. Cell Death Dis. 2019, 10, 459. [Google Scholar] [CrossRef]
- Marvaso, G.; Barone, A.; Amodio, N.; Raimondi, L.; Agosti, V.; Altomare, E.; Scotti, V.; Lombardi, A.; Bianco, R.; Bianco, C.; et al. Sphingosine Analog Fingolimod (FTY720) Increases Radiation Sensitivity of Human Breast Cancer Cells in Vitro. Cancer Biol. Ther. 2014, 15, 797–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, T.; Genest, L.; Babin, D.; Legrand, C.; Hunault, M.; Froget, G.; Castagné, V. Anti-CTLA-4 and Anti-PD-1 Immunotherapies Repress Tumor Progression in Preclinical Breast and Colon Model with Independent Regulatory T Cells Response. Transl. Oncol. 2022, 20, 101405. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Han, F.; Cao, L.; Li, C.; Wang, J.; Li, Q.; Zheng, W.; Guo, Z.; Li, A.; Zhou, J. Dose-Response Relationship in Cisplatin-Treated Breast Cancer Xenografts Monitored with Dynamic Contrast-Enhanced Ultrasound. BMC Cancer 2015, 15, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Yoon, Y.; Votaw, J.; Goodman, M.M.; Williams, L.; Shim, H. Silencing of CXCR4 Blocks Breast Cancer Metastasis. Cancer Res. 2005, 65, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Cavazzoni, A.; Alfieri, R.R.; Cretella, D.; Saccani, F.; Ampollini, L.; Galetti, M.; Quaini, F.; Graiani, G.; Madeddu, D.; Mozzoni, P.; et al. Combined Use of Anti-ErbB Monoclonal Antibodies and Erlotinib Enhances Antibody-Dependent Cellular Cytotoxicity of Wild-Type Erlotinib-Sensitive NSCLC Cell Lines. Mol. Cancer 2012, 11, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friess, T.; Scheuer, W.; Hasmann, M. Combination Treatment with Erlotinib and Pertuzumab against Human Tumor Xenografts Is Superior to Monotherapy. Clin. Cancer Res. 2005, 11, 5300–5309. [Google Scholar] [CrossRef] [Green Version]
- Azuma, H.; Takahara, S.; Ichimaru, N.; Wang, J.D.; Itoh, Y.; Otsuki, Y.; Morimoto, J.; Fukui, R.; Hoshiga, M.; Ishihara, T.; et al. Marked Prevention of Tumor Growth and Metastasis by a Novel Immunosuppressive Agent, FTY720, in Mouse Breast Cancer Models. Cancer Res. 2002, 62, 1410–1419. [Google Scholar]
- Woo, S.M.; Seo, B.R.; Min, K.; Kwon, T.K. FTY720 Enhances TRAIL-Mediated Apoptosis by up-Regulating DR5 and down-Regulating Mcl-1 in Cancer Cells. Oncotarget 2015, 6, 11614–11626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An Online Survival Analysis Tool to Rapidly Assess the Effect of 22,277 Genes on Breast Cancer Prognosis Using Microarray Data of 1,809 Patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rupp, T.; Debasly, S.; Genest, L.; Froget, G.; Castagné, V. Therapeutic Potential of Fingolimod and Dimethyl Fumarate in Non-Small Cell Lung Cancer Preclinical Models. Int. J. Mol. Sci. 2022, 23, 8192. https://doi.org/10.3390/ijms23158192
Rupp T, Debasly S, Genest L, Froget G, Castagné V. Therapeutic Potential of Fingolimod and Dimethyl Fumarate in Non-Small Cell Lung Cancer Preclinical Models. International Journal of Molecular Sciences. 2022; 23(15):8192. https://doi.org/10.3390/ijms23158192
Chicago/Turabian StyleRupp, Tristan, Solène Debasly, Laurie Genest, Guillaume Froget, and Vincent Castagné. 2022. "Therapeutic Potential of Fingolimod and Dimethyl Fumarate in Non-Small Cell Lung Cancer Preclinical Models" International Journal of Molecular Sciences 23, no. 15: 8192. https://doi.org/10.3390/ijms23158192
APA StyleRupp, T., Debasly, S., Genest, L., Froget, G., & Castagné, V. (2022). Therapeutic Potential of Fingolimod and Dimethyl Fumarate in Non-Small Cell Lung Cancer Preclinical Models. International Journal of Molecular Sciences, 23(15), 8192. https://doi.org/10.3390/ijms23158192