
Analysis of Givinostat/ITF2357 Treatment in a Rat Model of Neonatal Hypoxic-Ischemic Brain Damage


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Effect of Givinostat on IL-1β Expression in Primary Glial Cells Exposed to LPS
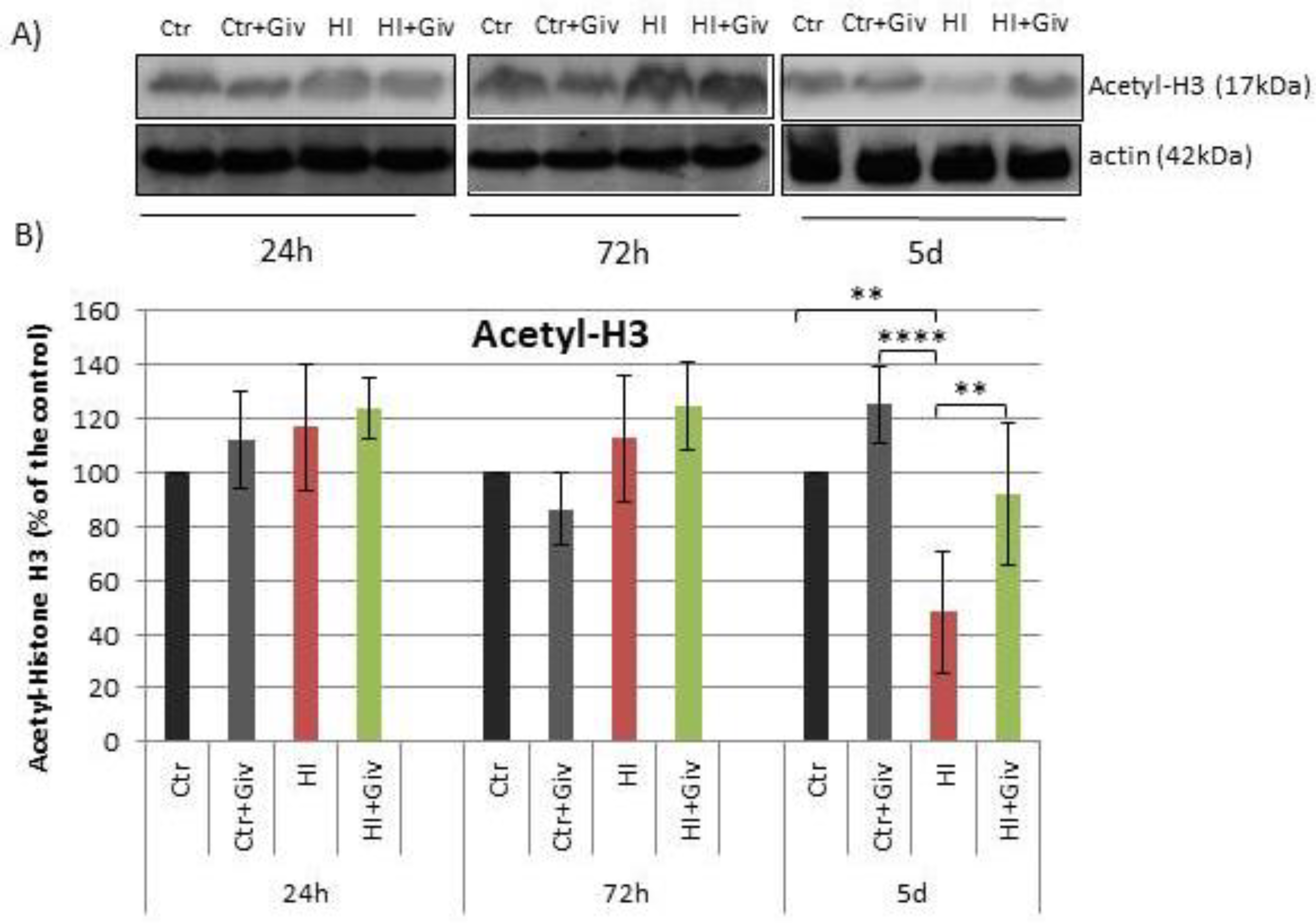
2.2. The Effect of Givinostat on the Acetylation of Histone H3
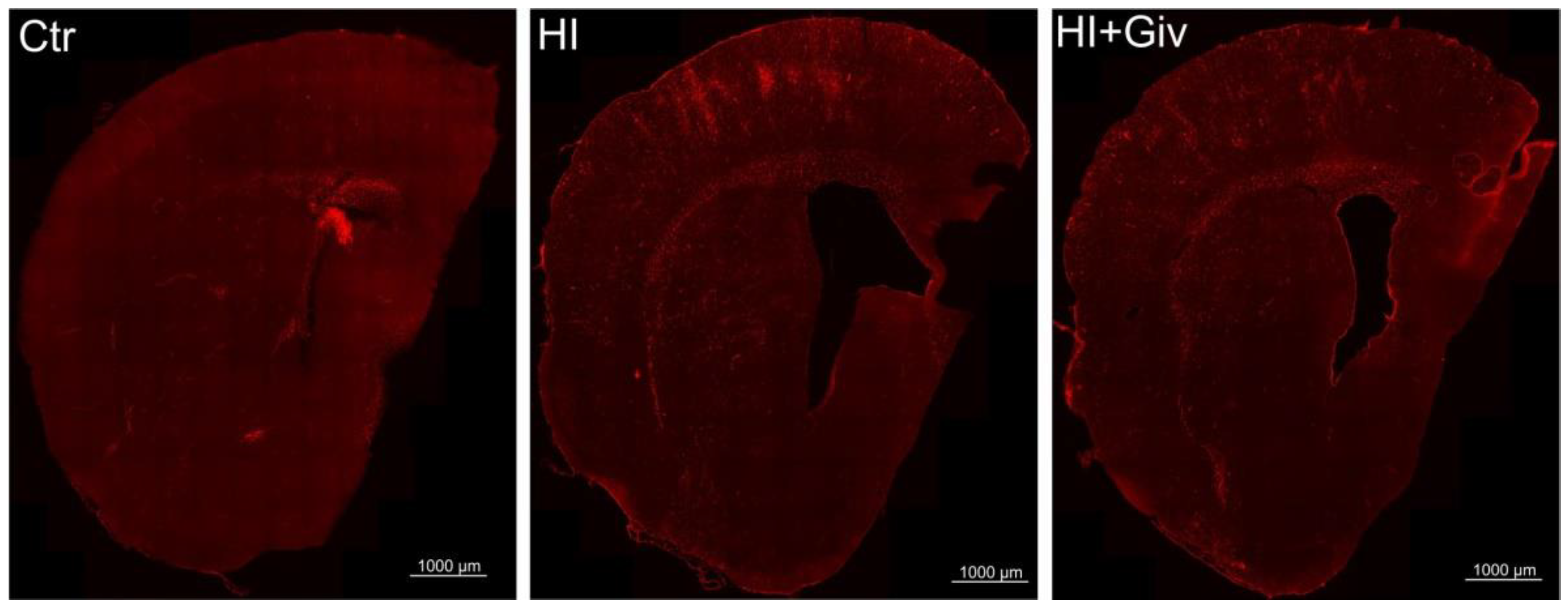
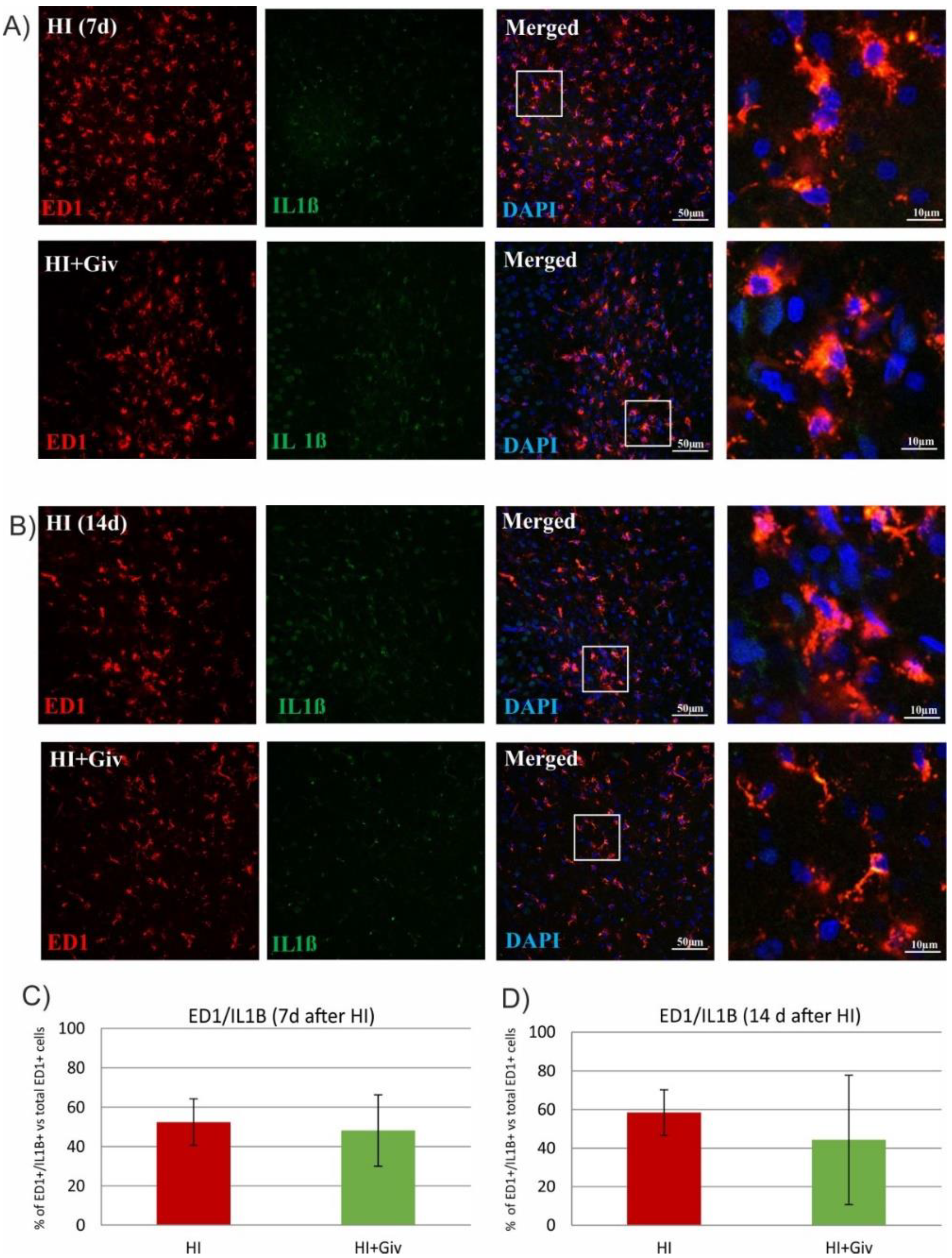
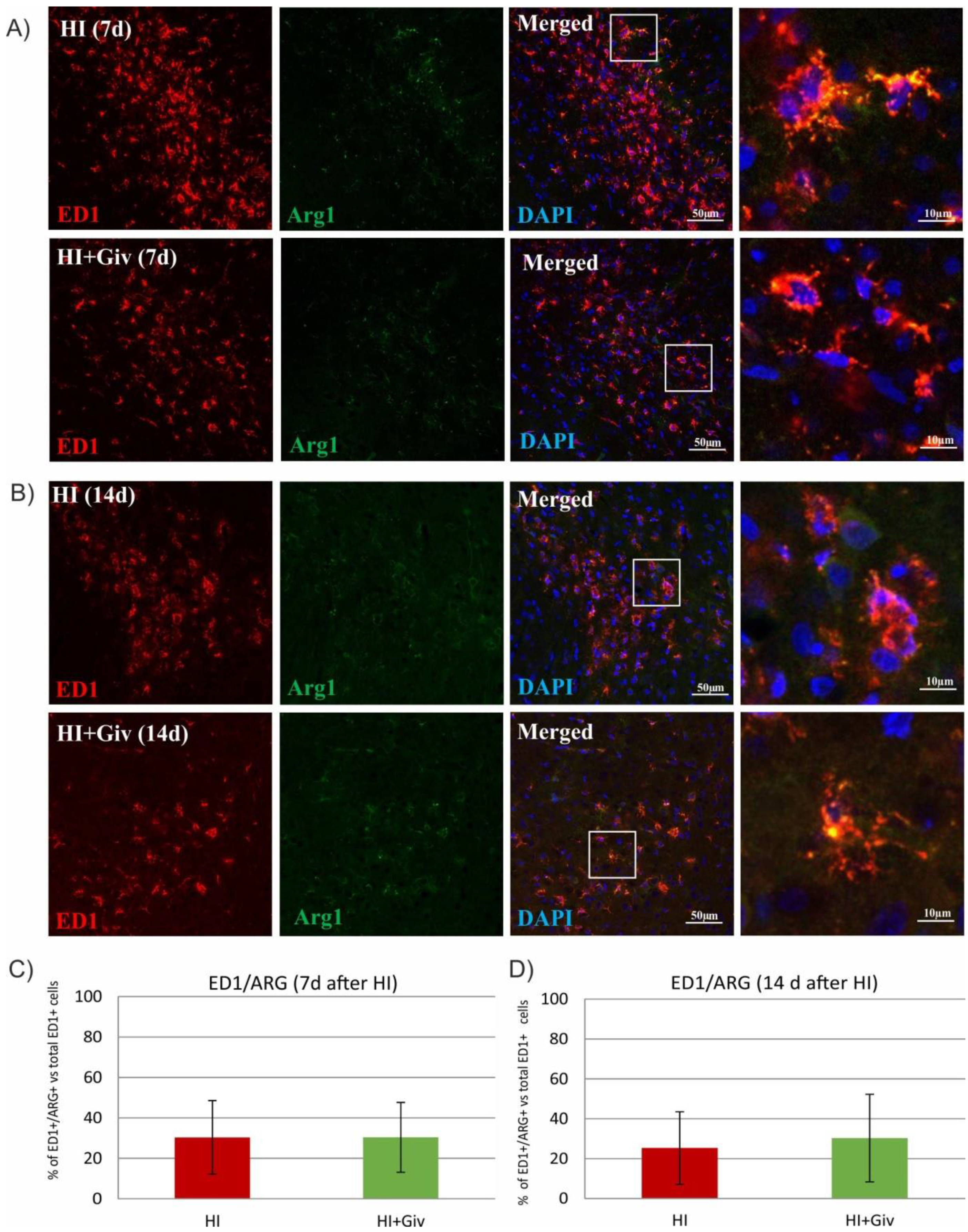
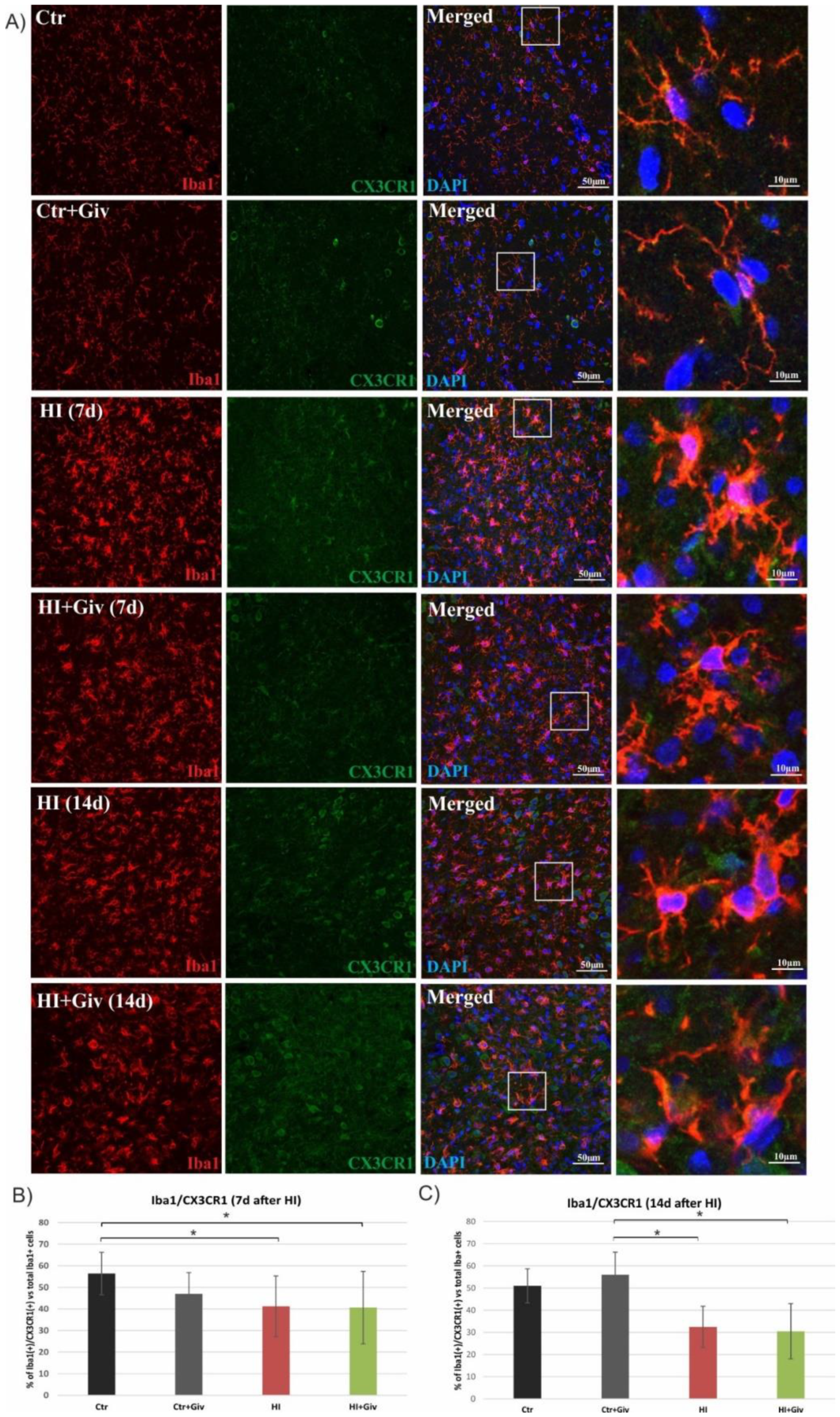
2.3. The Effect of Hypoxia-Ischemia and Givinostat Treatment on the Activation and Polarization of Microglia
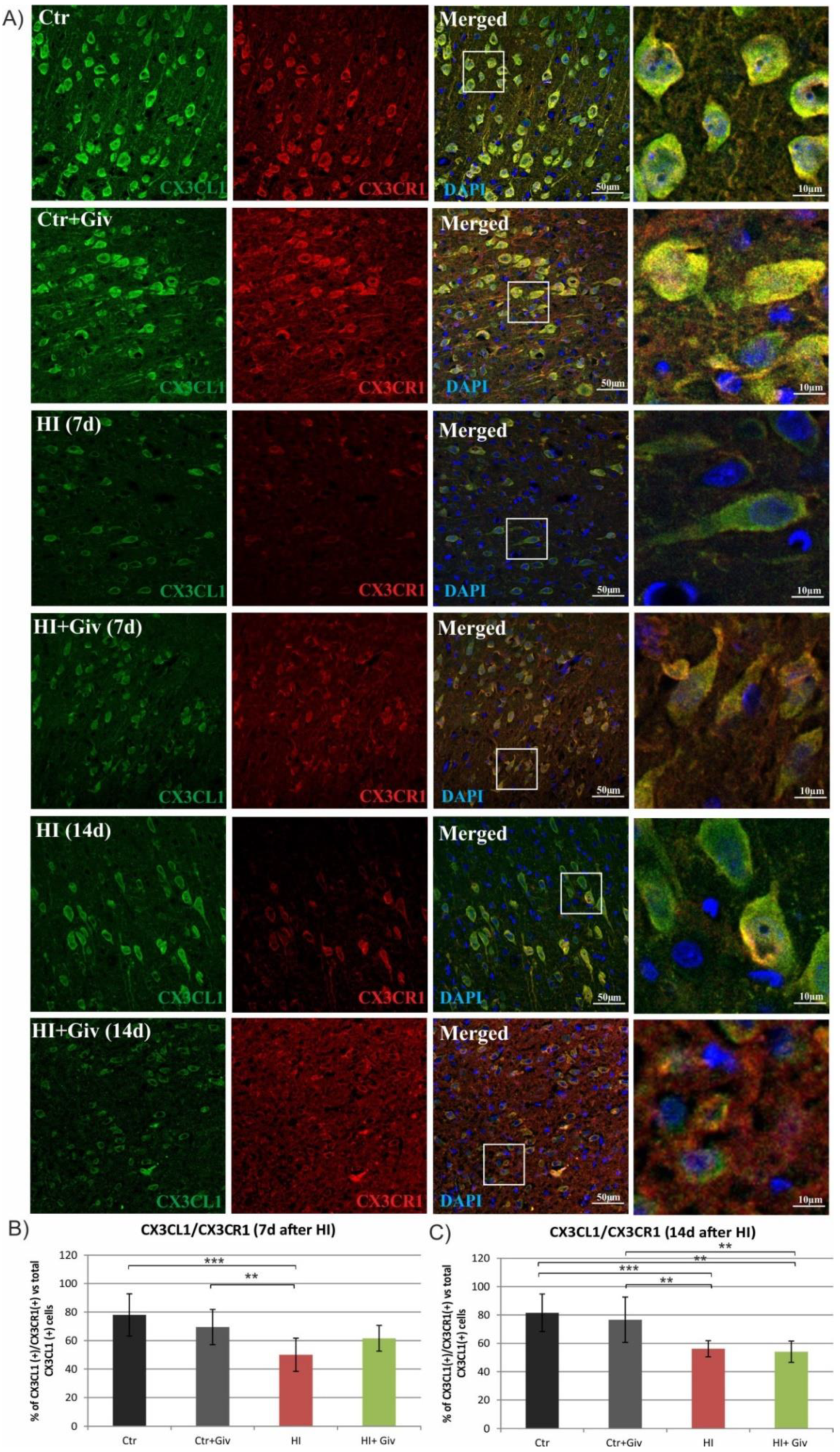
2.4. The Effect of Hypoxia-Ischemia and Givinostat Treatment on Microglia–Neuron Interactions
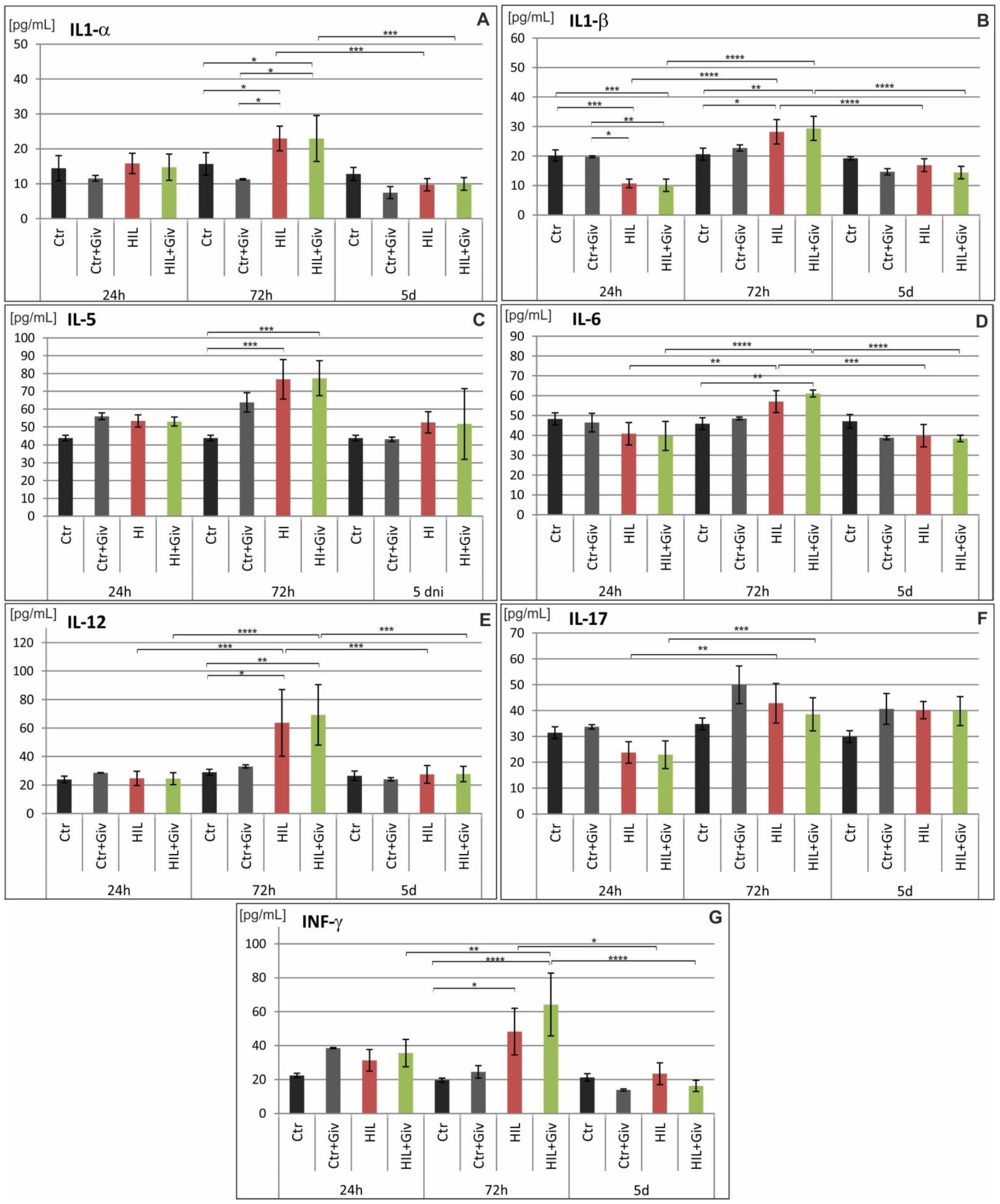
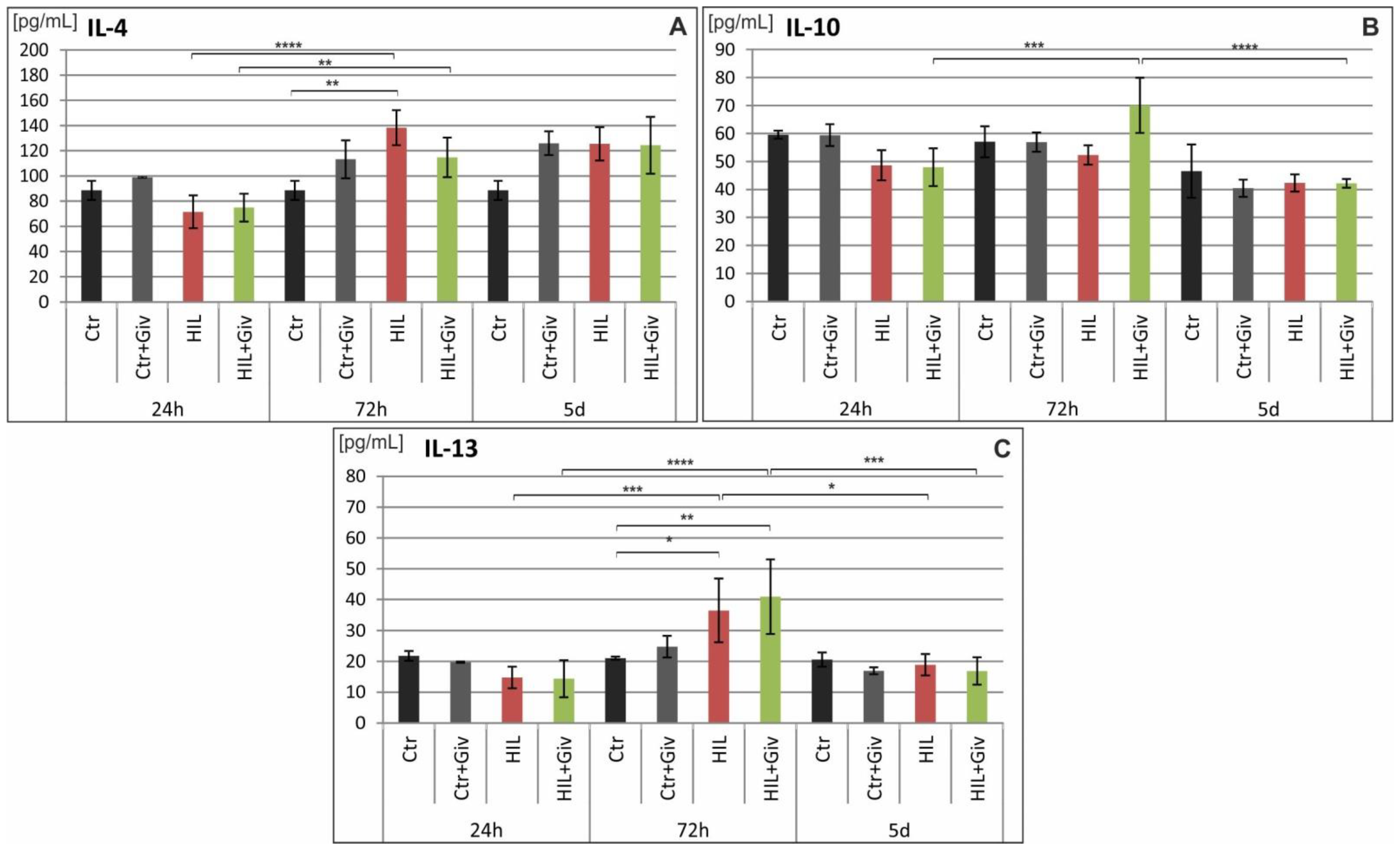
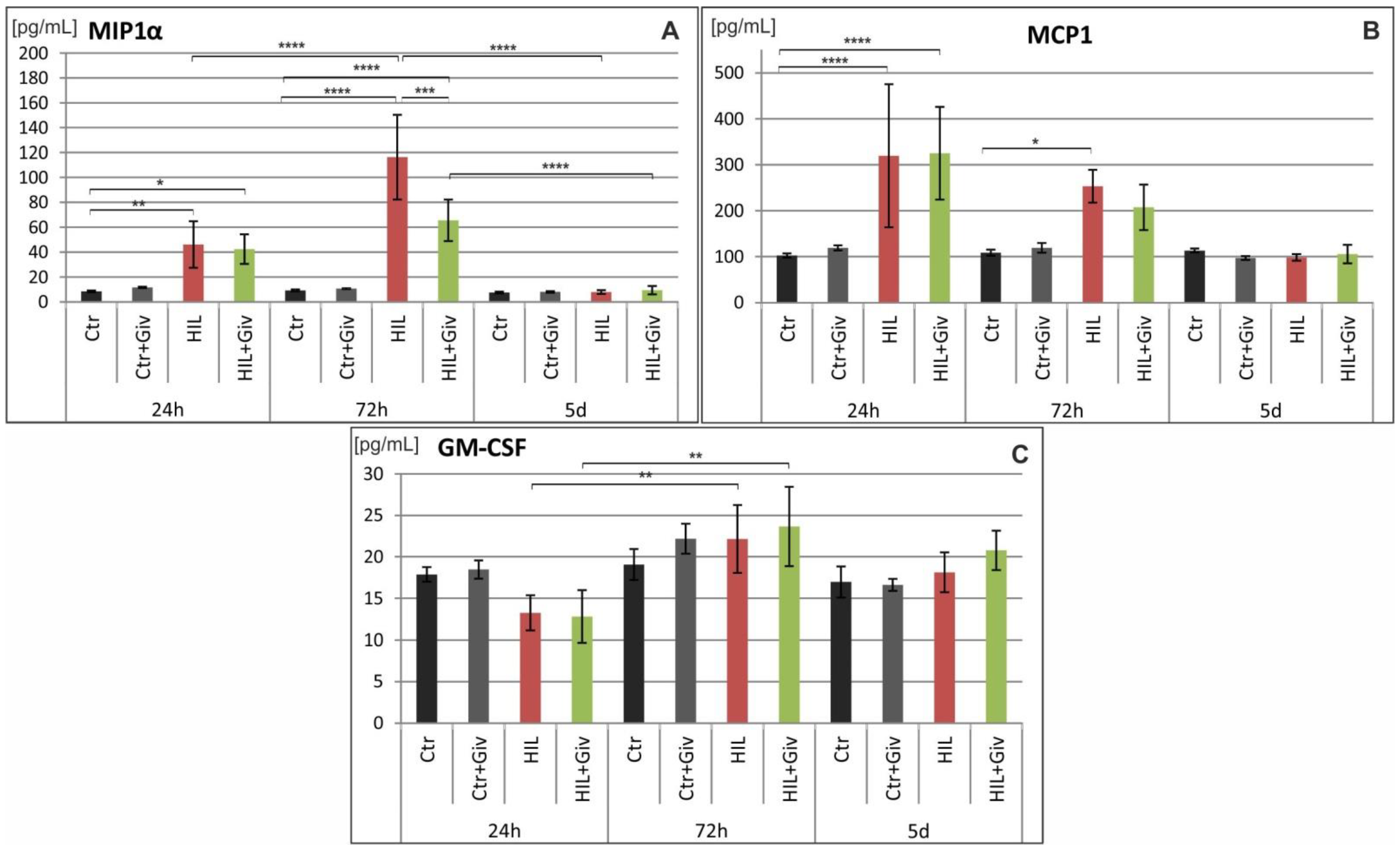
2.5. The Effect of Givinostat on the Levels of Cytokines and Chemokines in Rat Brains after HI
3. Discussion
4. Materials and Methods
4.1. Primary Glial Cell Culture (In Vitro Experiments)
4.2. Experimental Neonatal Hypoxia-Ischemia (In Vivo Experiments)
4.3. Drug Treatment
4.4. Tissue Preparation
4.5. Quantitative Polymerase Chain Reaction (qPCR)
4.6. Immunohistochemical Staining
4.7. Evaluation of Cytokine and Chemokine Expression after Hypoxia-Ischemia (Luminex)
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The Role of Inflammation in Perinatal Brain Injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; McCullough, L.D. Inflammatory Responses in Hypoxic Ischemic Encephalopathy. Acta Pharmacol. Sin. 2013, 34, 1121–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor, S.; Rocha-Ferreira, E.; Rahim, A.; Hagberg, H.; Edwards, D. New Possibilities for Neuroprotection in Neonatal Hypoxic-Ischemic Encephalopathy. Eur. J. Pediatr. 2022, 181, 875–887. [Google Scholar] [CrossRef]
- Ziemka-Nalecz, M.; Jaworska, J.; Zalewska, T. Insights Into the Neuroinflammatory Responses After Neonatal Hypoxia-Ischemia. J. Neuropathol. Exp. Neurol. 2017, 76, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Rowe, M.; Ren, M.; Hong, J.-S.; Chen, P.-S.; Chuang, D.-M. Histone Deacetylase Inhibitors Exhibit Anti-Inflammatory and Neuroprotective Effects in a Rat Permanent Ischemic Model of Stroke: Multiple Mechanisms of Action. J. Pharmacol. Exp. Ther. 2007, 321, 892–901. [Google Scholar] [CrossRef]
- Kim, H.J.; Leeds, P.; Chuang, D.-M. The HDAC Inhibitor, Sodium Butyrate, Stimulates Neurogenesis in the Ischemic Brain. J. Neurochem. 2009, 110, 1226–1240. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Chopp, M.; Kassis, H.; Jia, L.F.; Hozeska-Solgot, A.; Zhang, R.L.; Chen, C.; Cui, Y.S.; Zhang, Z.G. Valproic Acid Increases White Matter Repair and Neurogenesis after Stroke. Neuroscience 2012, 220, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Leng, Y.; Jeong, M.; Leeds, P.R.; Chuang, D.-M. Valproic Acid Reduces Brain Damage Induced by Transient Focal Cerebral Ischemia in Rats: Potential Roles of Histone Deacetylase Inhibition and Heat Shock Protein Induction. J. Neurochem. 2004, 89, 1358–1367. [Google Scholar] [CrossRef]
- Ziemka-Nalecz, M.; Jaworska, J.; Sypecka, J.; Polowy, R.; Filipkowski, R.K.; Zalewska, T. Sodium Butyrate, a Histone Deacetylase Inhibitor, Exhibits Neuroprotective/Neurogenic Effects in a Rat Model of Neonatal Hypoxia-Ischemia. Mol. Neurobiol. 2017, 54, 5300–5318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleiss, B.; Nilsson, M.K.L.; Blomgren, K.; Mallard, C. Neuroprotection by the Histone Deacetylase Inhibitor Trichostatin A in a Model of Lipopolysaccharide-Sensitised Neonatal Hypoxic-Ischaemic Brain Injury. J. Neuroinflamm. 2012, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Vojinovic, J.; Damjanov, N. HDAC Inhibition in Rheumatoid Arthritis and Juvenile Idiopathic Arthritis. Mol. Med. 2011, 17, 397–403. [Google Scholar] [CrossRef]
- Jaworska, J.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Potential Neuroprotective Role of a Histone Deacetylase Inhibitor, Sodium Butyrate, after Neonatal Hypoxia-Ischemia. J. Neuroinflamm. 2017, 14, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Yang, G.; Yang, Y.; Ding, F.; Hu, F. Protective Effects of Histone Deacetylase Inhibition by Scriptaid on Brain Injury in Neonatal Rat Models of Cerebral Ischemia and Hypoxia. Int. J. Clin. Exp. Pathol. 2020, 13, 179–191. [Google Scholar] [PubMed]
- Leoni, F.; Fossati, G.; Lewis, E.C.; Lee, J.-K.; Porro, G.; Pagani, P.; Modena, D.; Moras, M.L.; Pozzi, P.; Reznikov, L.L.; et al. The Histone Deacetylase Inhibitor ITF2357 Reduces Production of Pro-Inflammatory Cytokines in Vitro and Systemic Inflammation in Vivo. Mol. Med. 2005, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Carta, S.; Tassi, S.; Semino, C.; Fossati, G.; Mascagni, P.; Dinarello, C.A.; Rubartelli, A. Histone Deacetylase Inhibitors Prevent Exocytosis of Interleukin-1beta-Containing Secretory Lysosomes: Role of Microtubules. Blood 2006, 108, 1618–1626. [Google Scholar] [CrossRef] [Green Version]
- Golay, J.; Cuppini, L.; Leoni, F.; Micò, C.; Barbui, V.; Domenghini, M.; Lombardi, L.; Neri, A.; Barbui, A.M.; Salvi, A.; et al. The Histone Deacetylase Inhibitor ITF2357 Has Anti-Leukemic Activity in Vitro and in Vivo and Inhibits IL-6 and VEGF Production by Stromal Cells. Leukemia 2007, 21, 1892–1900. [Google Scholar] [CrossRef]
- Shein, N.A.; Grigoriadis, N.; Alexandrovich, A.G.; Simeonidou, C.; Lourbopoulos, A.; Polyzoidou, E.; Trembovler, V.; Mascagni, P.; Dinarello, C.A.; Shohami, E. Histone Deacetylase Inhibitor ITF2357 Is Neuroprotective, Improves Functional Recovery, and Induces Glial Apoptosis Following Experimental Traumatic Brain Injury. FASEB J. 2009, 23, 4266–4275. [Google Scholar] [CrossRef] [Green Version]
- Shein, N.A.; Shohami, E. Histone Deacetylase Inhibitors as Therapeutic Agents for Acute Central Nervous System Injuries. Mol. Med. 2011, 17, 448–456. [Google Scholar] [CrossRef]
- Suuronen, T.; Huuskonen, J.; Pihlaja, R.; Kyrylenko, S.; Salminen, A. Regulation of Microglial Inflammatory Response by Histone Deacetylase Inhibitors. J. Neurochem. 2003, 87, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, H.; Gressens, P.; Mallard, C. Inflammation during Fetal and Neonatal Life: Implications for Neurologic and Neuropsychiatric Disease in Children and Adults. Ann. Neurol. 2012, 71, 444–457. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Vannucci, S.J. Perinatal Hypoxic-Ischemic Brain Damage: Evolution of an Animal Model. Dev. Neurosci. 2005, 27, 81–86. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshiko, M.; Arnoux, I.; Avignone, E.; Yamamoto, N.; Audinat, E. Deficiency of the Microglial Receptor CX3CR1 Impairs Postnatal Functional Development of Thalamocortical Synapses in the Barrel Cortex. J. Neurosci. 2012, 32, 15106–15111. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Paolicelli, R.C.; Sforazzini, F.; Weinhard, L.; Bolasco, G.; Pagani, F.; Vyssotski, A.L.; Bifone, A.; Gozzi, A.; Ragozzino, D.; et al. Deficient Neuron-Microglia Signaling Results in Impaired Functional Brain Connectivity and Social Behavior. Nat. Neurosci. 2014, 17, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Mallard, C.; Tremblay, M.-E.; Vexler, Z.S. Microglia and Neonatal Brain Injury. Neuroscience 2019, 405, 68–76. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V Cortical Neurons Require Microglial Support for Survival during Postnatal Development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia Regulate the Number of Neural Precursor Cells in the Developing Cerebral Cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef] [Green Version]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalancette-Hébert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective Ablation of Proliferating Microglial Cells Exacerbates Ischemic Injury in the Brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustino, J.V.; Wang, X.; Johnson, C.E.; Klibanov, A.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Microglial Cells Contribute to Endogenous Brain Defenses after Acute Neonatal Focal Stroke. J. Neurosci. 2011, 31, 12992–13001. [Google Scholar] [CrossRef] [Green Version]
- Varnum, M.M.; Ikezu, T. The Classification of Microglial Activation Phenotypes on Neurodegeneration and Regeneration in Alzheimer’s Disease Brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266. [Google Scholar] [CrossRef]
- Bonestroo, H.J.C.; Nijboer, C.H.A.; van Velthoven, C.T.J.; Kavelaars, A.; Hack, C.E.; van Bel, F.; Heijnen, C.J. Cerebral and Hepatic Inflammatory Response after Neonatal Hypoxia-Ischemia in Newborn Rats. Dev. Neurosci. 2013, 35, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, K.; Llovera, G.; Recasens, M.; Chertoff, M.; Giménez-Llort, L.; Gonzalez, B.; Acarin, L. Temporal Expression of Cytokines and Signal Transducer and Activator of Transcription Factor 3 Activation after Neonatal Hypoxia/Ischemia in Mice. Dev. Neurosci. 2013, 35, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Zalewska, T.; Sypecka, J.; Ziemka-Nalecz, M. Effect of the HDAC Inhibitor, Sodium Butyrate, on Neurogenesis in a Rat Model of Neonatal Hypoxia-Ischemia: Potential Mechanism of Action. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M.; Priller, J. Microglia and Brain Macrophages in the Molecular Age: From Origin to Neuropsychiatric Disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia-Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Eyo, U.B.; Wu, L.-J. Bidirectional Microglia-Neuron Communication in the Healthy Brain. Neural Plast 2013, 2013, 456857. [Google Scholar] [CrossRef]
- Sheridan, G.K.; Murphy, K.J. Neuron-Glia Crosstalk in Health and Disease: Fractalkine and CX3CR1 Take Centre Stage. Open Biol. 2013, 3, 130181. [Google Scholar] [CrossRef] [Green Version]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W.G.M. Neuronal “On” and “Off” Signals Control Microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial Physiology: Unique Stimuli, Specialized Responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Nishiyori, A.; Minami, M.; Ohtani, Y.; Takami, S.; Yamamoto, J.; Kawaguchi, N.; Kume, T.; Akaike, A.; Satoh, M. Localization of Fractalkine and CX3CR1 MRNAs in Rat Brain: Does Fractalkine Play a Role in Signaling from Neuron to Microglia? FEBS Lett. 1998, 429, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Maciejewski-Lenoir, D.; Chen, S.; Feng, L.; Maki, R.; Bacon, K.B. Characterization of Fractalkine in Rat Brain Cells: Migratory and Activation Signals for CX3CR-1-Expressing Microglia. J. Immunol. 1999, 163, 1628–1635. [Google Scholar]
- Zujovic, V.; Benavides, J.; Vigé, X.; Carter, C.; Taupin, V. Fractalkine Modulates TNF-Alpha Secretion and Neurotoxicity Induced by Microglial Activation. Glia 2000, 29, 305–315. [Google Scholar] [CrossRef]
- Mizuno, T.; Kawanokuchi, J.; Numata, K.; Suzumura, A. Production and Neuroprotective Functions of Fractalkine in the Central Nervous System. Brain Res. 2003, 979, 65–70. [Google Scholar] [CrossRef]
- Lyons, A.; Lynch, A.M.; Downer, E.J.; Hanley, R.; O’Sullivan, J.B.; Smith, A.; Lynch, M.A. Fractalkine-Induced Activation of the Phosphatidylinositol-3 Kinase Pathway Attentuates Microglial Activation in Vivo and in Vitro. J. Neurochem. 2009, 110, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, S.; Perego, C.; Ortolano, F.; De Simoni, M.-G. CX3CR1 Deficiency Induces an Early Protective Inflammatory Environment in Ischemic Mice. Glia 2013, 61, 827–842. [Google Scholar] [CrossRef]
- Tang, Z.; Gan, Y.; Liu, Q.; Yin, J.-X.; Liu, Q.; Shi, J.; Shi, F.-D. CX3CR1 Deficiency Suppresses Activation and Neurotoxicity of Microglia/Macrophage in Experimental Ischemic Stroke. J. Neuroinflamm. 2014, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Cipriani, R.; Villa, P.; Chece, G.; Lauro, C.; Paladini, A.; Micotti, E.; Perego, C.; De Simoni, M.-G.; Fredholm, B.B.; Eusebi, F.; et al. CX3CL1 Is Neuroprotective in Permanent Focal Cerebral Ischemia in Rodents. J. Neurosci. 2011, 31, 16327–16335. [Google Scholar] [CrossRef] [Green Version]
- Dénes, A.; Ferenczi, S.; Halász, J.; Környei, Z.; Kovács, K.J. Role of CX3CR1 (Fractalkine Receptor) in Brain Damage and Inflammation Induced by Focal Cerebral Ischemia in Mouse. J. Cereb. Blood Flow Metab. 2008, 28, 1707–1721. [Google Scholar] [CrossRef] [Green Version]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of Microglial Neurotoxicity by the Fractalkine Receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of Tau Pathology by the Microglial Fractalkine Receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertollini, C.; Ragozzino, D.; Gross, C.; Limatola, C.; Eusebi, F. Fractalkine/CX3CL1 Depresses Central Synaptic Transmission in Mouse Hippocampal Slices. Neuropharmacology 2006, 51, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Ragozzino, D.; Di Angelantonio, S.; Trettel, F.; Bertollini, C.; Maggi, L.; Gross, C.; Charo, I.F.; Limatola, C.; Eusebi, F. Chemokine Fractalkine/CX3CL1 Negatively Modulates Active Glutamatergic Synapses in Rat Hippocampal Neurons. J. Neurosci. 2006, 26, 10488–10498. [Google Scholar] [CrossRef] [PubMed]
- Lauro, C.; Chece, G.; Monaco, L.; Antonangeli, F.; Peruzzi, G.; Rinaldo, S.; Paone, A.; Cutruzzolà, F.; Limatola, C. Fractalkine Modulates Microglia Metabolism in Brain Ischemia. Front. Cell. Neurosci. 2019, 13, 414. [Google Scholar] [CrossRef]
- Limatola, C.; Lauro, C.; Catalano, M.; Ciotti, M.T.; Bertollini, C.; Di Angelantonio, S.; Ragozzino, D.; Eusebi, F. Chemokine CX3CL1 Protects Rat Hippocampal Neurons against Glutamate-Mediated Excitotoxicity. J. Neuroimmunol. 2005, 166, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Deiva, K.; Geeraerts, T.; Salim, H.; Leclerc, P.; Héry, C.; Hugel, B.; Freyssinet, J.-M.; Tardieu, M. Fractalkine Reduces N-Methyl-d-Aspartate-Induced Calcium Flux and Apoptosis in Human Neurons through Extracellular Signal-Regulated Kinase Activation. Eur. J. Neurosci. 2004, 20, 3222–3232. [Google Scholar] [CrossRef]
- Ivacko, J.; Szaflarski, J.; Malinak, C.; Flory, C.; Warren, J.S.; Silverstein, F.S. Hypoxic-Ischemic Injury Induces Monocyte Chemoattractant Protein-1 Expression in Neonatal Rat Brain. J. Cereb. Blood Flow Metab. 1997, 17, 759–770. [Google Scholar] [CrossRef]
- Bona, E.; Andersson, A.L.; Blomgren, K.; Gilland, E.; Puka-Sundvall, M.; Gustafson, K.; Hagberg, H. Chemokine and Inflammatory Cell Response to Hypoxia-Ischemia in Immature Rats. Pediatr. Res. 1999, 45, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barks, J.D.; Schielke, G.P.; Silverstein, F.S. Attenuation of Hypoxia-Ischemia-Induced Monocyte Chemoattractant Protein-1 Expression in Brain of Neonatal Mice Deficient in Interleukin-1 Converting Enzyme. Brain Res. Mol. Brain Res. 2001, 90, 57–67. [Google Scholar] [CrossRef]
- Cowell, R.M.; Xu, H.; Galasso, J.M.; Silverstein, F.S. Hypoxic-Ischemic Injury Induces Macrophage Inflammatory Protein-1alpha Expression in Immature Rat Brain. Stroke 2002, 33, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Hallenbeck, J.M.; Ruetzler, C.; Bol, D.; Thomas, K.; Berman, N.E.J.; Vogel, S.N. Overexpression of Monocyte Chemoattractant Protein 1 in the Brain Exacerbates Ischemic Brain Injury and Is Associated with Recruitment of Inflammatory Cells. J. Cereb. Blood Flow Metab. 2003, 23, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Hughes, P.M.; Allegrini, P.R.; Rudin, M.; Perry, V.H.; Mir, A.K.; Wiessner, C. Monocyte Chemoattractant Protein-1 Deficiency Is Protective in a Murine Stroke Model. J. Cereb. Blood Flow Metab. 2002, 22, 308–317. [Google Scholar] [CrossRef] [Green Version]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Absence of the Chemokine Receptor CCR2 Protects against Cerebral Ischemia/Reperfusion Injury in Mice. Stroke 2007, 38, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Galasso, J.M.; Miller, M.J.; Cowell, R.M.; Harrison, J.K.; Warren, J.S.; Silverstein, F.S. Acute Excitotoxic Injury Induces Expression of Monocyte Chemoattractant Protein-1 and Its Receptor, CCR2, in Neonatal Rat Brain. Exp. Neurol. 2000, 165, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Galasso, J.M.; Liu, Y.; Szaflarski, J.; Warren, J.S.; Silverstein, F.S. Monocyte Chemoattractant Protein-1 Is a Mediator of Acute Excitotoxic Injury in Neonatal Rat Brain. Neuroscience 2000, 101, 737–744. [Google Scholar] [CrossRef]
- Stanimirovic, D.; Satoh, K. Inflammatory Mediators of Cerebral Endothelium: A Role in Ischemic Brain Inflammation. Brain Pathol. 2000, 10, 113–126. [Google Scholar] [CrossRef]
- Sinn, D.-I.; Kim, S.-J.; Chu, K.; Jung, K.-H.; Lee, S.-T.; Song, E.-C.; Kim, J.-M.; Park, D.-K.; Kun Lee, S.; Kim, M.; et al. Valproic Acid-Mediated Neuroprotection in Intracerebral Hemorrhage via Histone Deacetylase Inhibition and Transcriptional Activation. Neurobiol. Dis. 2007, 26, 464–472. [Google Scholar] [CrossRef]
- Dammann, O.; O’Shea, T.M. Cytokines and Perinatal Brain Damage. Clin. Perinatol. 2008, 35, 643–663. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-J.; Liu, W.; Wang, J.-L.; Zhang, Y.; Zhao, D.-J.; Wang, T.-J.; Li, Y.-Y. The Role of TNF-α, IL-6, IL-10, and GDNF in Neuronal Apoptosis in Neonatal Rat with Hypoxic-Ischemic Encephalopathy. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 905–909. [Google Scholar] [PubMed]
- Szaflarski, J.; Burtrum, D.; Silverstein, F.S. Cerebral Hypoxia-Ischemia Stimulates Cytokine Gene Expression in Perinatal Rats. Stroke 1995, 26, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fossati, G.; Marchetti, C.; Modena, D.; Pozzi, P.; Reznikov, L.L.; Moras, M.L.; Azam, T.; Abbate, A.; Mascagni, P.; et al. Specific Inhibition of Histone Deacetylase 8 Reduces Gene Expression and Production of Proinflammatory Cytokines in Vitro and in Vivo. J. Biol. Chem. 2015, 290, 2368–2378. [Google Scholar] [CrossRef] [Green Version]
- Faraco, G.; Pittelli, M.; Cavone, L.; Fossati, S.; Porcu, M.; Mascagni, P.; Fossati, G.; Moroni, F.; Chiarugi, A. Histone Deacetylase (HDAC) Inhibitors Reduce the Glial Inflammatory Response in Vitro and in Vivo. Neurobiol. Dis. 2009, 36, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.E.; Vannucci, R.C.; Brierley, J.B. The Influence of Immaturity on Hypoxic-Ischemic Brain Damage in the Rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawelec, P.; Sypecka, J.; Zalewska, T.; Ziemka-Nalecz, M. Analysis of Givinostat/ITF2357 Treatment in a Rat Model of Neonatal Hypoxic-Ischemic Brain Damage. Int. J. Mol. Sci. 2022, 23, 8287. https://doi.org/10.3390/ijms23158287
Pawelec P, Sypecka J, Zalewska T, Ziemka-Nalecz M. Analysis of Givinostat/ITF2357 Treatment in a Rat Model of Neonatal Hypoxic-Ischemic Brain Damage. International Journal of Molecular Sciences. 2022; 23(15):8287. https://doi.org/10.3390/ijms23158287
Chicago/Turabian StylePawelec, Paulina, Joanna Sypecka, Teresa Zalewska, and Malgorzata Ziemka-Nalecz. 2022. "Analysis of Givinostat/ITF2357 Treatment in a Rat Model of Neonatal Hypoxic-Ischemic Brain Damage" International Journal of Molecular Sciences 23, no. 15: 8287. https://doi.org/10.3390/ijms23158287