
Pseudomonas Aeruginosa Lung Infection Subverts Lymphocytic Responses through IL-23 and IL-22 Post-Transcriptional Regulation

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
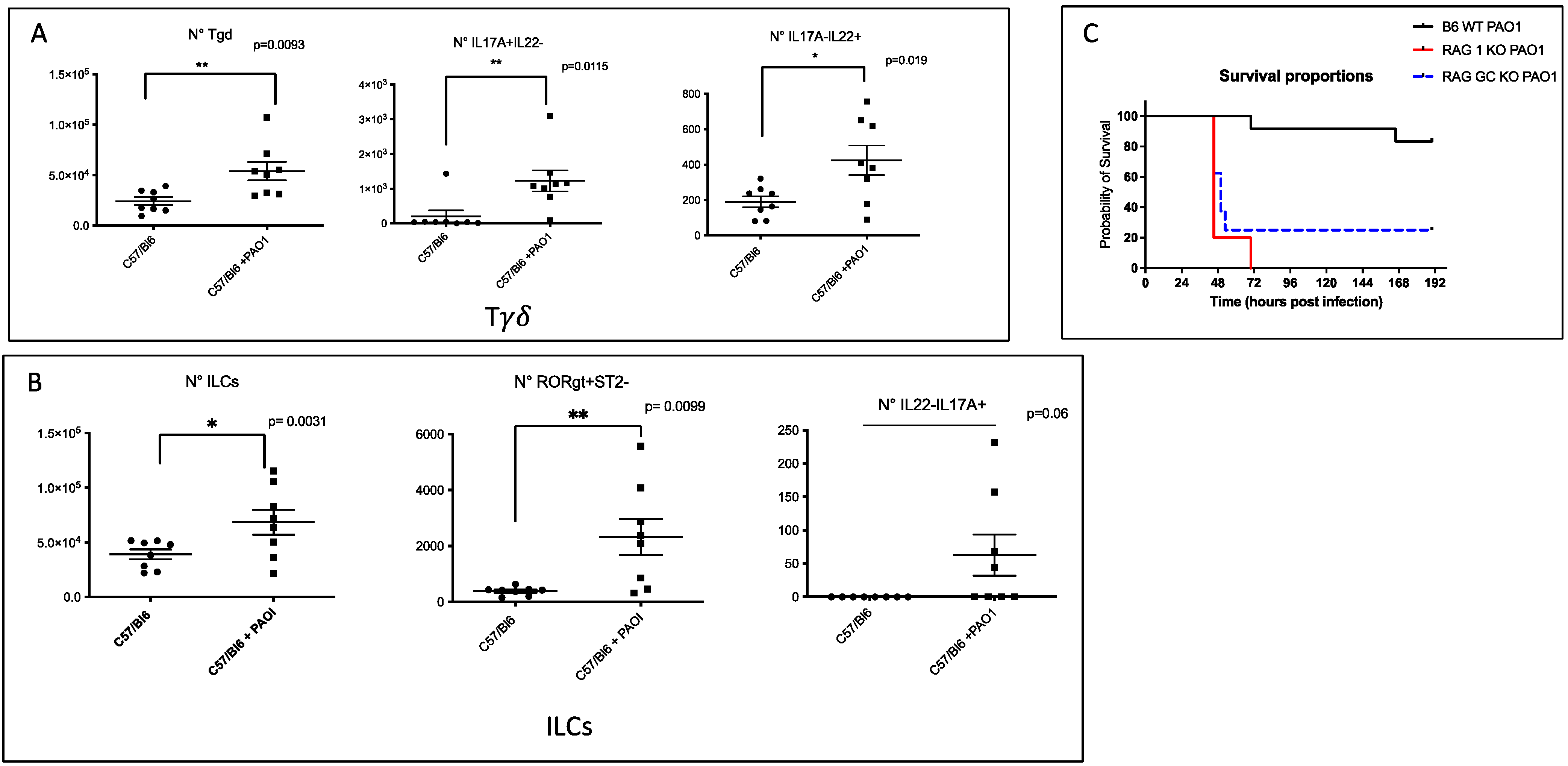
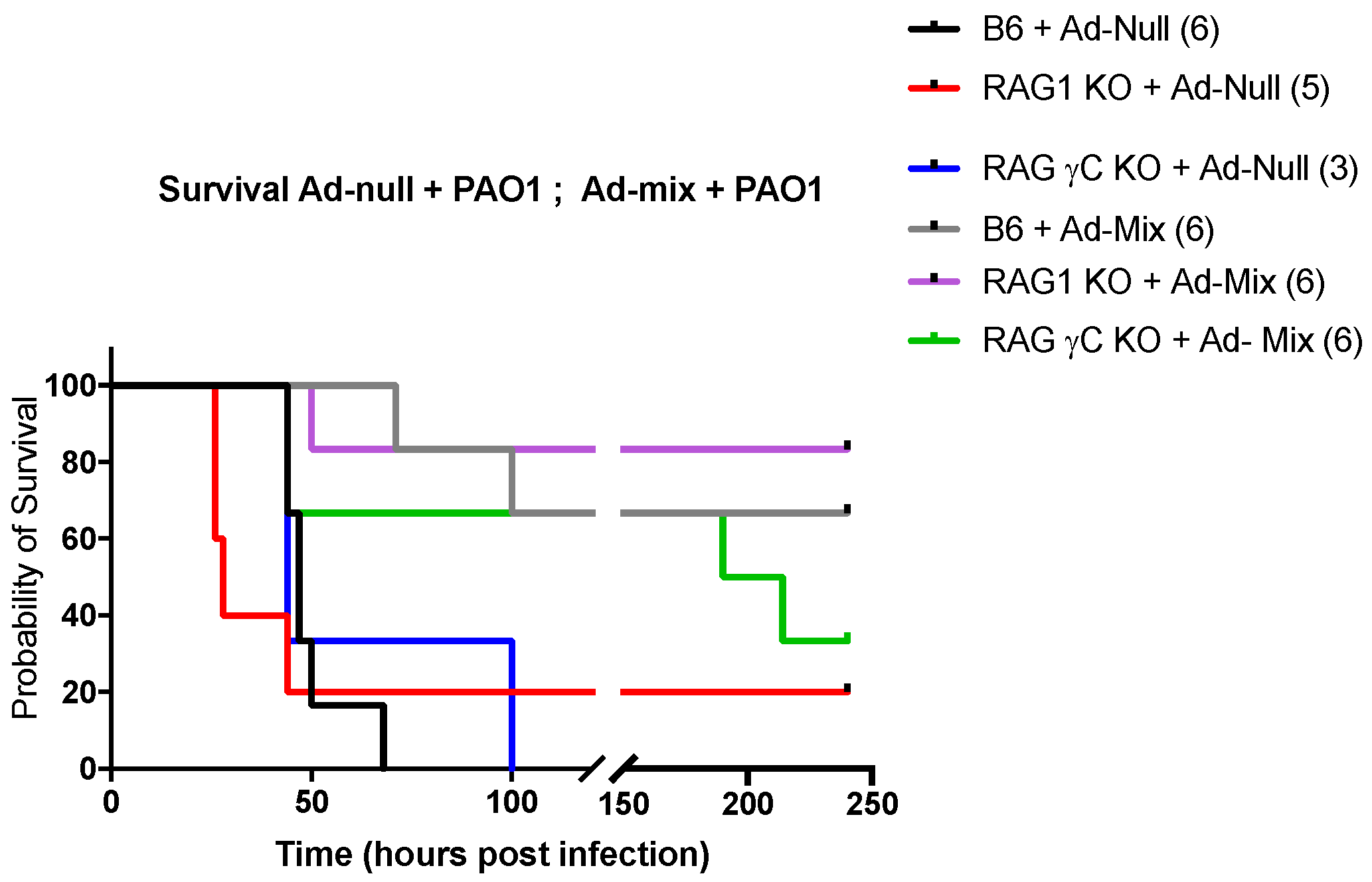
2.1. IL-17+, IL-22+ Tγδ and IL-17+ ILCs Increase Post Pseudomonas Aeruginosa Lung Infection, and Lymphocytes Are Important to Modulate Lung Resistance to Pseudomonas Aeruginosa Infections
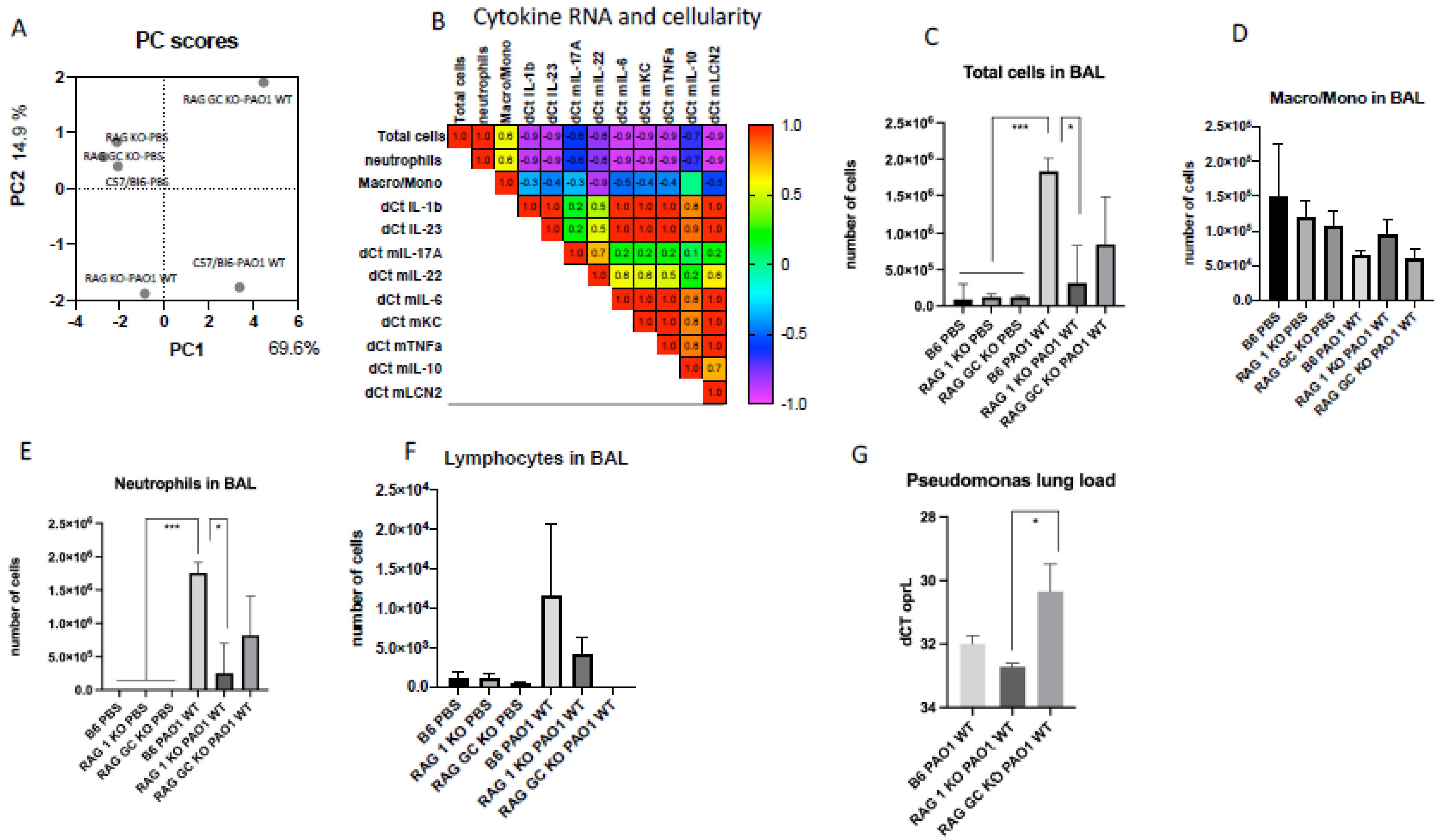
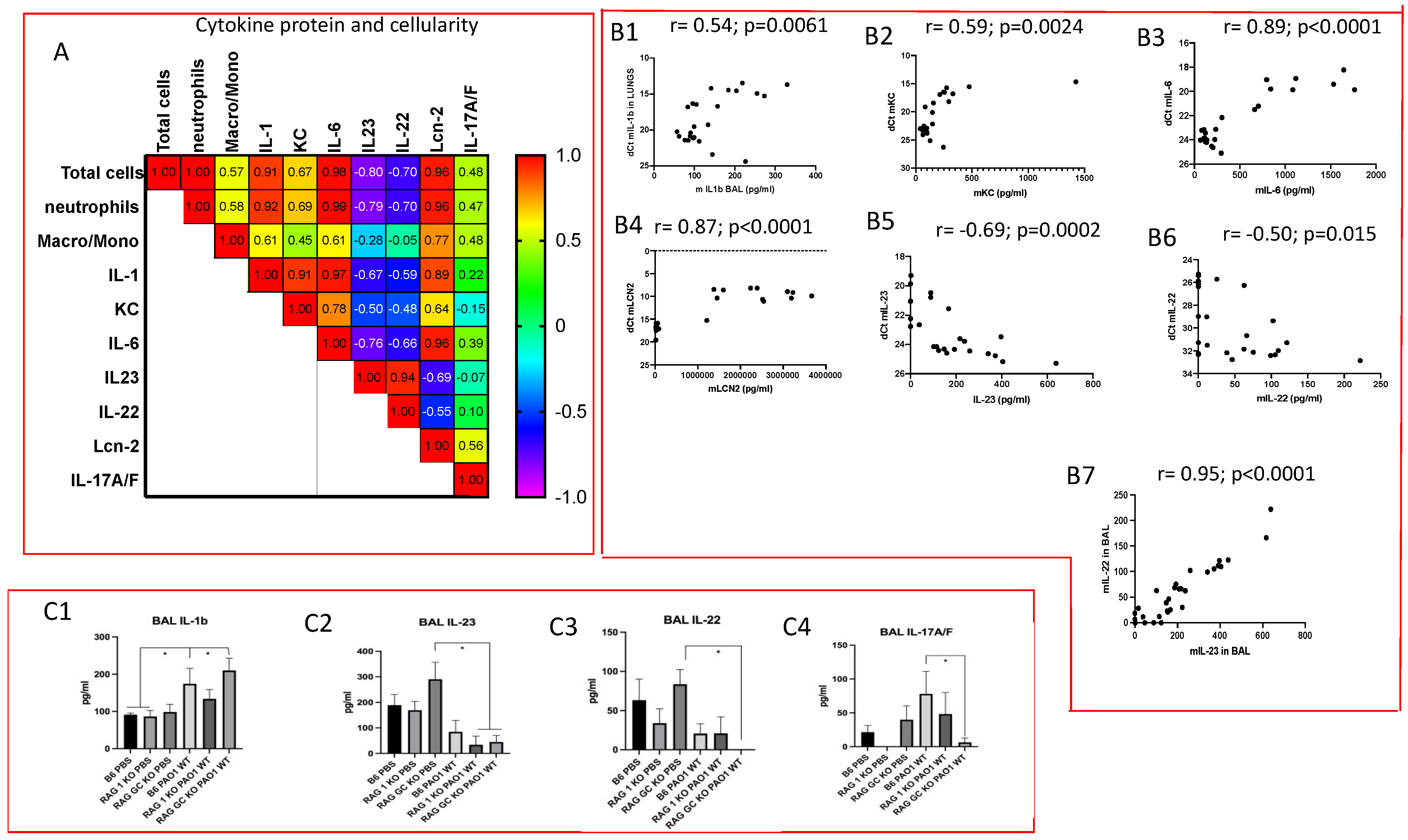
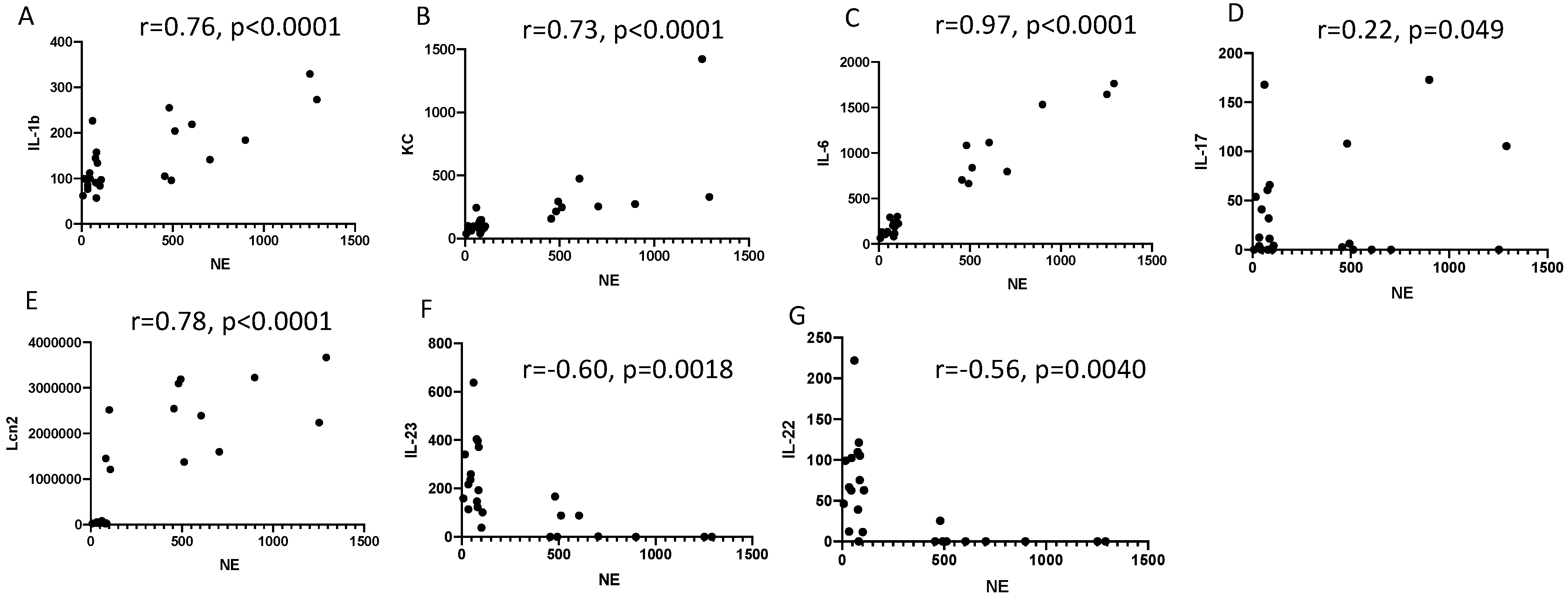
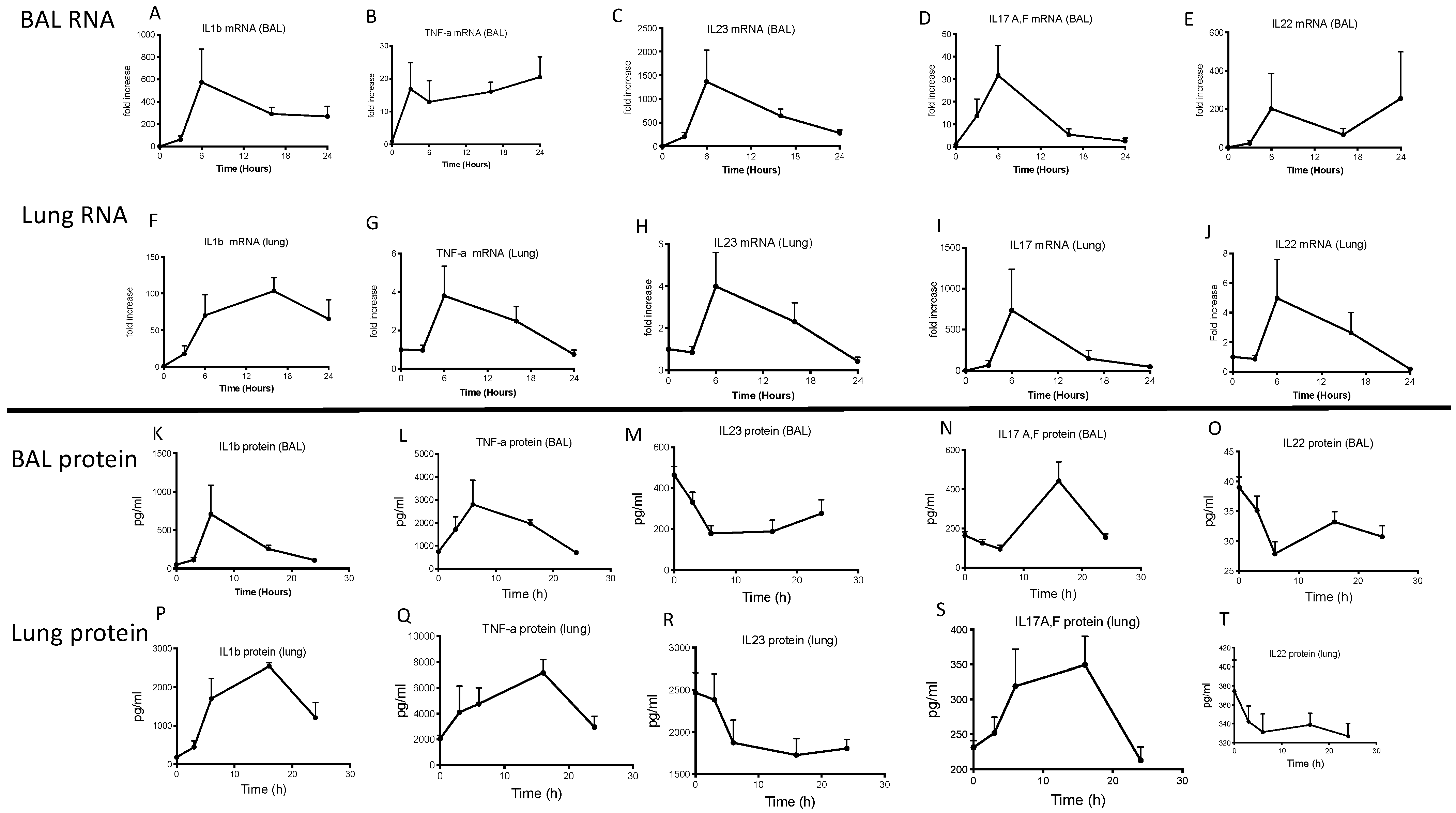
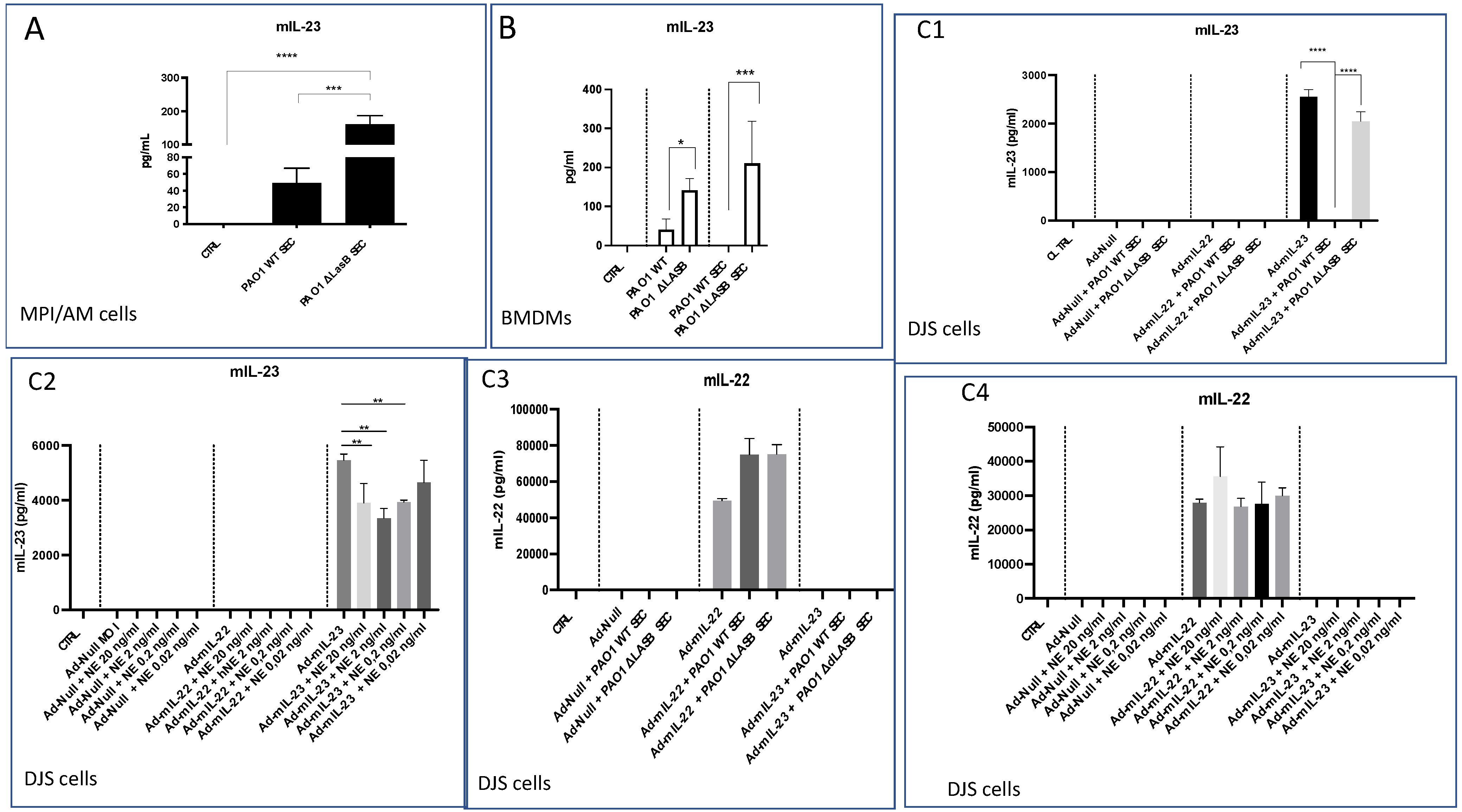
2.2. Lung IL-23 and IL-22 Protein Accumulation Is Down-Regulated Post PAO1 -Infection, Revealing a Post-Transcriptional Regulation
2.3. In Vivo Over-Expression of IL-23 Alone Does Not Rescue Mice from a Lethal PAO1 Infection
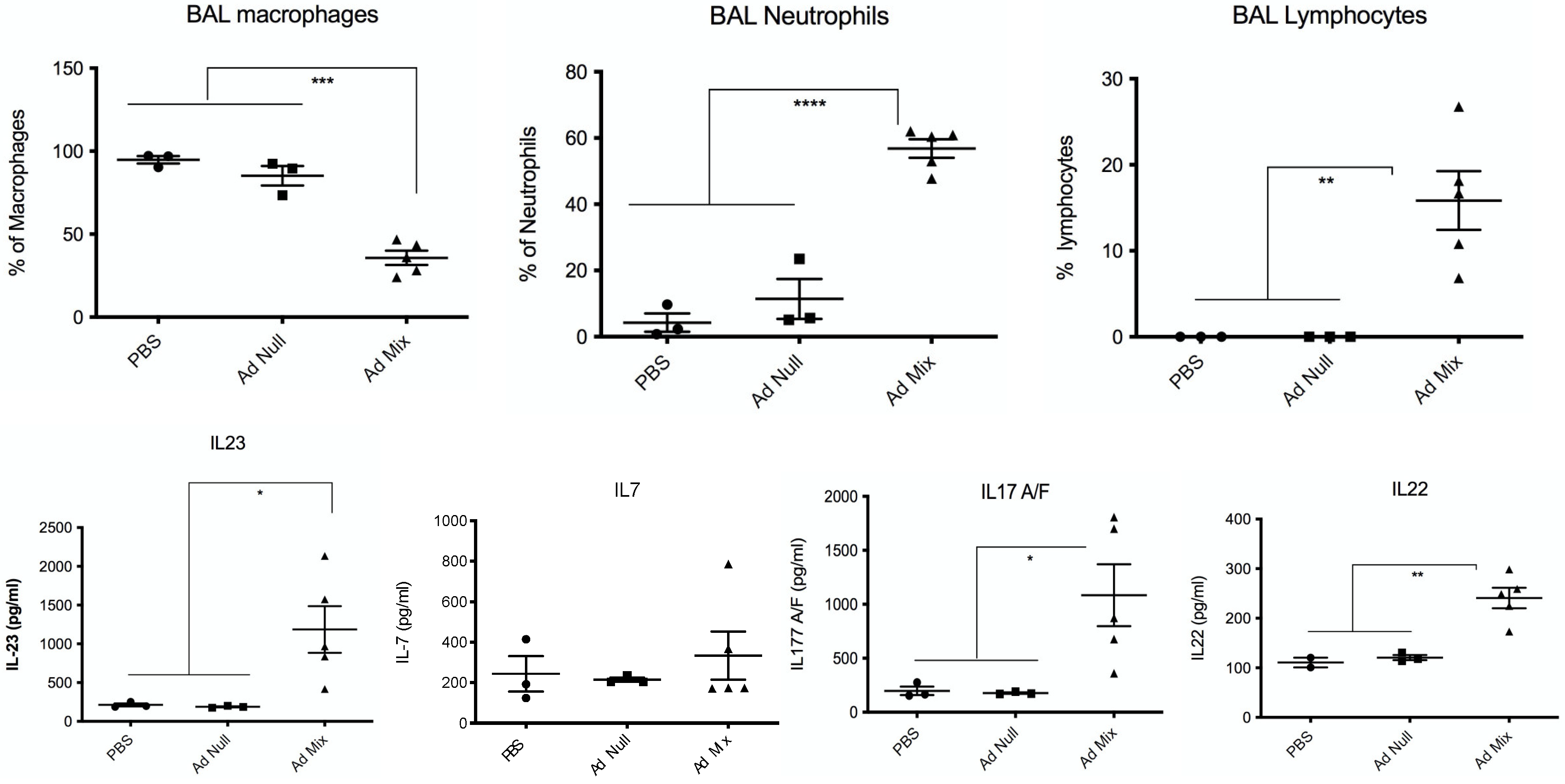
2.4. In Vivo Over-Expression of an IL-23, IL-1β and IL-7 Cocktail Protects against PAO1 Lethal Infection
3. Discussions
4. Materials and Methods
4.1. Recombinant Proteins and ELISA Assays
4.2. Adenovirus Constructs and Adenovirus Infection
4.3. Pseudomonas Aeruginosa O1 Strain, Secretome Production, and Bacterial Load Measurement
4.4. Cells, Cell Cultures, and Protocols
4.4.1. Primary Alveolar Macrophages
4.4.2. Bone Marrow-Derived Macrophage Generation
4.4.3. Cell Lines
4.5. Animals Procedures
4.6. Flow Cytometry Analysis
4.7. RNA Extraction, Reverse Transcription, and qPCR
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edelman, A.; Sallenave, J.-M. Cystic fibrosis, a multi-systemic mucosal disease: 25 years after the discovery of CFTR. Int. J. Biochem. Cell Biol. 2014, 52, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Sallenave, J.-M. Phagocytic and signaling innate immune receptors: Are they dysregulated in cystic fibrosis in the fight against Pseudomonas aeruginosa? Int. J. Biochem. Cell Biol. 2014, 52, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Bastaert, F.; Kheir, S.; Saint-Criq, V.; Villeret, B.; Dang, P.M.; El-Benna, J.; Sirard, J.C.; Voulhoux, R.; Sallenave, J.M. Pseudomonas aeruginosa LasB Subverts Alveolar Macrophage Activity by Interfering With Bacterial Killing Through Downregulation of Innate Immune Defense, Reactive Oxygen Species Generation, and Complement Activation. Front. Immunol. 2018, 9, 1675. [Google Scholar] [CrossRef] [PubMed]
- Saint-Criq, V.; Villeret, B.; Bastaert, F.; Kheir, S.; Hatton, A.; Cazes, A.; Xing, Z.; Sermet-Gaudelus, I.; Garcia-Verdugo, I.; Edelman, A.; et al. Pseudomonas aeruginosa LasB protease impairs innate immunity in mice and humans by targeting a lung epithelial cystic fibrosis transmembrane regulator–IL-6–antimicrobial–repair pathway. Thorax 2018, 73, 49–61. [Google Scholar] [CrossRef]
- Beatty, A.L.; Malloy, J.L.; Wright, J.R. Pseudomonas aeruginosa degrades pulmonary surfactant and increases conversion in vitro. Am. J. Respir. Cell Mol. Biol. 2005, 32, 128–134. [Google Scholar] [CrossRef]
- Kuang, Z.; Hao, Y.; Walling, B.E.; Jeffries, J.L.; Ohman, D.E.; Lau, G.W. Pseudomonas aeruginosa elastase provides an escape from phagocytosis by degrading the pulmonary surfactant protein-A. PLoS ONE 2011, 6, e27091. [Google Scholar] [CrossRef]
- Beaufort, N.; Corvazier, E.; Mlanaoindrou, S.; de Bentzmann, S.; Pidard, D. Disruption of the endothelial barrier by proteases from the bacterial pathogen Pseudomonas aeruginosa: Implication of matrilysis and receptor cleavage. PLoS ONE 2013, 8, e75708. [Google Scholar] [CrossRef]
- Ruffin, M.; Brochiero, E. Repair Process Impairment by Pseudomonas aeruginosa in Epithelial Tissues: Major Features and Potential Therapeutic Avenues. Front. Cell Infect. Microbiol. 2019, 9, 182. [Google Scholar] [CrossRef]
- Aujla, S.J.; Chan, Y.R.; Zheng, M.; Fei, M.; Askew, D.J.; Pociask, D.A.; Reinhart, T.A.; McAllister, F.; Edeal, J.; Gaus, K.; et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008, 14, 275–281. [Google Scholar] [CrossRef]
- Bayes, H.K.; Ritchie, N.D.; Evans, T.J. Interleukin-17 Is Required for Control of Chronic Lung Infection Caused by Pseudomonas aeruginosa. Infect. Immun. 2016, 84, 3507–3516. [Google Scholar] [CrossRef]
- Dubin, P.J.; Martz, A.; Eisenstatt, J.R.; Fox, M.D.; Logar, A.; Kolls, J.K. Interleukin-23-mediated inflammation in Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 2012, 80, 398–409. [Google Scholar] [CrossRef]
- Liu, J.; Qu, H.; Li, Q.; Ye, L.; Ma, G.; Wan, H. The responses of γδ T-cells against acute Pseudomonas aeruginosa pulmonary infection in mice via interleukin-17. Pathog. Dis. 2013, 68, 44–51. [Google Scholar] [CrossRef]
- Omar, T.; Ziltener, P.; Chamberlain, E.; Cheng, Z.; Johnston, B. Mice Lacking γδ T Cells Exhibit Impaired Clearance of Pseudomonas aeruginosa Lung Infection and Excessive Production of Inflammatory Cytokines. Infect. Immun. 2020, 88, e00171-20. [Google Scholar] [CrossRef]
- Smith, D.J.; Geoffrey, R.H.; Scott, C.B.; David, W.R. Reduced mucosal associated invariant T-cells are associated with increased disease severity and Pseudomonas aeruginosa infection in cystic fibrosis. PLoS ONE 2014, 9, e109891. [Google Scholar] [CrossRef]
- Benoit, P.; Sigounas, V.Y.; Thompson, J.L.; van Rooijen, N.; Poynter, M.E.; Wargo, M.J.; Boyson, J.E. The role of CD1d-restricted NKT cells in the clearance of Pseudomonas aeruginosa from the lung is dependent on the host genetic background. Infect. Immun. 2015, 83, 2557–2565. [Google Scholar] [CrossRef]
- Kinjo, T.; Nakamatsu, M.; Nakasone, C.; Yamamoto, N.; Kinjo, Y.; Miyagi, K.; Uezu, K.; Nakamura, K.; Higa, F.; Tateyama, M.; et al. NKT cells play a limited role in the neutrophilic inflammatory responses and host defense to pulmonary infection with Pseudomonas aeruginosa. Microbes Infect. 2006, 8, 2679–2685. [Google Scholar] [CrossRef]
- Morihara, K.; Tsuzuki, H.; Harada, M.; Iwata, T. Purification of human plasma alpha 1-proteinase inhibitor and its inactivation by Pseudomonas aeruginosa elastase. J. Biochem. 1984, 95, 795–804. [Google Scholar] [CrossRef]
- Hong, Y.Q.; Ghebrehiwet, B. Effect of Pseudomonas aeruginosa elastase and alkaline protease on serum complement and isolated components C1q and C3. Clin. Immunol. Immunopathol. 1992, 62, 133–138. [Google Scholar] [CrossRef]
- Azghani, A.O. Pseudomonas aeruginosa and epithelial permeability: Role of virulence factors elastase and exotoxin A. Am. J. Respir. Cell Mol. Biol. 1996, 15, 132–140. [Google Scholar] [CrossRef]
- Alcorn, J.F.; Wright, J.R. Degradation of pulmonary surfactant protein D by Pseudomonas aeruginosa elastase abrogates innate immune function. J. Biol. Chem. 2004, 279, 30871–30879. [Google Scholar] [CrossRef]
- Leduc, D.; Beaufort, N.; de Bentzmann, S.; Rousselle, J.C.; Namane, A.; Chignard, M.; Pidard, D. The Pseudomonas aeruginosa LasB metalloproteinase regulates the human urokinase-type plasminogen activator receptor through domain-specific endoproteolysis. Infect. Immun. 2007, 75, 3848–3858. [Google Scholar] [CrossRef]
- Potempa, J.; Pike, R.N. Corruption of innate immunity by bacterial proteases. J. Innate. Immun. 2009, 1, 70–87. [Google Scholar] [CrossRef]
- Ruffin, M.; Bilodeau, C.; Maillé, E.; LaFayette, S.L.; McKay, G.A.; Thu Ngan Trinh, N.; Beaudoin, T.; Desrosiers, M.Y.; Rousseau, S.; Nguyen, D.; et al. Quorum-sensing inhibition abrogates the deleterious impact of Pseudomonas aeruginosa on airway epithelial repair. FASEB J. 2016, 30, 3011–3025. [Google Scholar] [CrossRef]
- Magdaleno, S.M.; Wang, G.; Jackson, K.J.; Ray, M.K.; Welty, S.; Costa, R.H.; DeMayo, F.J. Interferon-gamma regulation of Clara cell gene expression: In vivo and in vitro. Am. J. Physiol. 1997, 272, L1142–L1151. [Google Scholar] [CrossRef]
- Sun, J.; LaRock, D.L.; Skowronski, E.A.; Kimmey, J.M.; Olson, J.; Jiang, Z.; O’Donoghue, A.J.; Nizet, V.; LaRock, C.N. The Pseudomonas aeruginosa protease LasB directly activates IL-1β. EBioMedicine 2020, 60, 102984. [Google Scholar] [CrossRef]
- Descamps, D.; Le Gars, M.; Balloy, V.; Barbier, D.; Maschalidi, S.; Tohme, M.; Chignard, M.; Ramphal, R.; Manoury, B.; Sallenave, J. Toll-like receptor 5 (TLR5), IL-1β secretion, and asparagine endopeptidase are critical factors for alveolar macrophage phagocytosis and bacterial killing. Proc. Natl. Acad. Sci. USA 2012, 5, 1619–1624. [Google Scholar] [CrossRef]
- Reiniger, N.; Lee, M.M.; Coleman, F.T.; Ray, C.; Golan, D.E.; Pier, G.B. Resistance to Pseudomonas aeruginosa chronic lung infection requires cystic fibrosis transmembrane conductance regulator-modulated interleukin-1 (IL-1) release and signaling through the IL-1 receptor. Infect. Immun. 2007, 75, 1598–1608. [Google Scholar] [CrossRef]
- Bekiaris, V.; Šedý, J.R.; Ware, C.F. Mixing Signals: Molecular Turn Ons and Turn Offs for Innate γδ T-Cells. Front. Immunol. 2014, 5, 654. [Google Scholar] [CrossRef]
- Michel, M.L.; Pang, D.J.; Haque, S.F.; Potocnik, A.J.; Pennington, D.J.; Hayday, A.C. Interleukin 7 (IL-7) selectively promotes mouse and human IL-17-producing γδ cells. Proc. Natl. Acad. Sci. USA 2012, 109, 17549–17554. [Google Scholar] [CrossRef]
- Hassane, M.; Jouan, Y.; Creusat, F.; Soulard, D.; Boisseau, C.; Gonzalez, L.; Patin, E.C.; Heuzé-Vourc’h, N.; Sirard, J.C.; Faveeuw, C.; et al. Interleukin-7 protects against bacterial respiratory infection by promoting IL-17A-producing innate T-cell response. Mucosal Immunol. 2020, 13, 128–139. [Google Scholar] [CrossRef]
- Willyard, C. The drug-resistant bacteria that pose the greatest health threats. Nat. News 2017, 543, 15. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Flume, P.A.; Biner, R.F.; Downey, D.G.; Brown, C.; Jain, M.; Fischer, R.; De Boeck, K.; Sawicki, G.S.; Chang, P.; Paz-Diaz, H.; et al. Long-term safety and efficacy of tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years or older who are homozygous or heterozygous for Phe508del CFTR (EXTEND): An open-label extension study. Lancet Respir. Med. 2021, 9, 733–746. [Google Scholar] [CrossRef]
- Bals, R.; Hiemstra, P.S. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Leiva-Juárez, M.M.; Kolls, J.K.; Evans, S.E. Lung epithelial cells: Therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol. 2018, 11, 21–34. [Google Scholar] [CrossRef]
- Cleaver, J.O.; You, D.; Michaud, D.R.; Guzmán Pruneda, F.A.; Leiva Juarez, M.M.; Zhang, J.; Weill, P.M.; Adachi, R.; Gong, L.; Moghaddam, S.J.; et al. Lung epithelial cells are essential effectors of inducible resistance to pneumonia. Mucosal Immunol. 2014, 7, 78–88. [Google Scholar] [CrossRef]
- Kheir, S.; Villeret, B.; Garcia-Verdugo, I.; Sallenave, J.M. IL-6-elafin genetically modified macrophages as a lung immunotherapeutic strategy against Pseudomonas aeruginosa infections. Mol. Ther. 2022, 30, 355–369. [Google Scholar] [CrossRef]
- Van Maele, L.; Carnoy, C.; Cayet, D.; Ivanov, S.; Porte, R.; Deruy, E.; Chabalgoity, J.A.; Renauld, J.C.; Eberl, G.; Benecke, A.G.; et al. Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J. Infect. Dis. 2014, 210, 493–503. [Google Scholar] [CrossRef]
- Van Maele, L.; Carnoy, C.; Cayet, D.; Songhet, P.; Dumoutier, L.; Ferrero, I.; Janot, L.; Erard, F.; Bertout, J.; Leger, H.; et al. TLR5 signaling stimulates the innate production of IL-17 and IL-22 by CD3(neg)CD127+ immune cells in spleen and mucosa. J. Immunol. 2010, 185, 1177–1185. [Google Scholar] [CrossRef]
- Guillon, A.; Brea, D.; Luczka, E.; Hervé, V.; Hasanat, S.; Thorey, C.; Pérez-Cruz, M.; Hordeaux, J.; Mankikian, J.; Gosset, P.; et al. Inactivation of the interleukin-22 pathway in the airways of cystic fibrosis patients. Cytokine 2019, 113, 470–474. [Google Scholar] [CrossRef]
- Sutton, C.E.; Stephen, J.L.; Cheryl, M.S.; Corinna, F.B.; Lavelle, C.E.; Kingston, H.G.M. Inter leukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef]
- Sun, R.; Abraham, C. IL-23 promotes antimicrobial pathways in human macrophages, which are reduced with the IBD-protective IL23R R381Q variant. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 673–697. [Google Scholar] [CrossRef]
- Sun, R.; Hedl, M.; Abraham, C. IL23 induces IL23R recycling and amplifies innate receptor-induced signaling and cytokines in human macrophages, and the IBD-protective IL23R R381Q variant modulates these outcomes. Gut 2020, 69, 264–273. [Google Scholar] [CrossRef]
- Wolf, K.; Schulz, C.; Riegger, G.A.; Pfeifer, M. Tumour necrosis factor-alpha induced CD70 and interleukin-7R mRNA expression in BEAS-2B cells. Eur. Respir. J. 2002, 20, 369–375. [Google Scholar] [CrossRef]
- Ponte, R.; Rancez, M.; Figueiredo-Morgado, S.; Dutrieux, J.; Fabre-Mersseman, V.; Charmeteau-de-Muylder, B.; Guilbert, T.; Routy, J.P.; Cheynier, R.; Couëdel-Courteille, A. Acute Simian Immunodeficiency Virus Infection Triggers Early and Transient Interleukin-7 Production in the Gut, Leading to Enhanced Local Chemokine Expression and Intestinal Immune Cell Homing. Front. Immunol. 2017, 8, 588. [Google Scholar] [CrossRef]
- Logerot, S.; Figueiredo-Morgado, S.; Charmeteau-de-Muylder, B.; Sandouk, A.; Drillet-Dangeard, A.S.; Bomsel, M.; Bourgault-Villada, I.; Couëdel-Courteille, A.; Cheynier, R.; Rancez, M. IL-7-Adjuvanted Vaginal Vaccine elicits strong mucosal immune responses in non-human primates. Front. Immunol. 2021, 12, 614115. [Google Scholar] [CrossRef]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef]
- Bugaut, H.; Aractingi, S. Major Role of the IL17/23 Axis in Psoriasis Supports the Development of New Targeted Therapies. Front. Immunol. 2021, 12, 621956. [Google Scholar] [CrossRef]
- Bett, A.J.; Haddara, W.; Prevec, L.; Graham, F.L. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. USA 1994, 91, 8802–8806. [Google Scholar] [CrossRef]
- Happel, K.I.; Lockhart, E.A.; Mason, C.M.; Porretta, E.; Keoshkerian, E.; Odden, A.R.; Nelson, S.; Ramsay, A.J. Pulmonary interleukin-23 gene delivery increases local T-cell immunity and controls growth of Mycobacterium tuberculosis in the lungs. Infect. Immun. 2005, 73, 5782–5788. [Google Scholar] [CrossRef]
- Le Gall, F.; Le Berre, R.; Rosec, S.; Hardy, J.; Gouriou, S.; Boisramé-Gastrin, S.; Vallet, S.; Rault, G.; Payan, C.; Héry-Arnaud, G. Proposal of a quantitative PCR-based protocol for an optimal Pseudomonas aeruginosa detection in patients with cystic fibrosis. BMC Microbiol. 2013, 13, 143. [Google Scholar] [CrossRef]
- Villeret, B.; Solhonne, B.; Straube, M.; Lemaire, F.; Cazes, A.; Garcia-Verdugo, I.; Sallenave, J.M. Influenza A Virus pre-Infection exacerbates Pseudomonas aeruginosa-mediated Lung Damage through Increased MMP-9 Expression, Decreased Elafin Production and Tissue Resilience. Front. Immunol. 2020, 11, 117. [Google Scholar] [CrossRef]
- Fejer, G.; Wegner, M.D.; Györy, I.; Cohen, I.; Engelhard, P.; Voronov, E.; Manke, T.; Ruzsics, Z.; Dölken, L.; Prazeres da Costa, O.; et al. Nontransformed, GM-CSF-dependent macrophage lines are a unique model to study tissue macrophage functions. Proc. Natl. Acad. Sci. USA 2013, 110, 2191–2198. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villeret, B.; Ghinnagow, R.; Kheir, S.; Born-Bony, M.; Kolls, J.K.; Garcia-Verdugo, I.; Sallenave, J.-M. Pseudomonas Aeruginosa Lung Infection Subverts Lymphocytic Responses through IL-23 and IL-22 Post-Transcriptional Regulation. Int. J. Mol. Sci. 2022, 23, 8427. https://doi.org/10.3390/ijms23158427
Villeret B, Ghinnagow R, Kheir S, Born-Bony M, Kolls JK, Garcia-Verdugo I, Sallenave J-M. Pseudomonas Aeruginosa Lung Infection Subverts Lymphocytic Responses through IL-23 and IL-22 Post-Transcriptional Regulation. International Journal of Molecular Sciences. 2022; 23(15):8427. https://doi.org/10.3390/ijms23158427
Chicago/Turabian StyleVilleret, Bérengère, Reem Ghinnagow, Saadé Kheir, Maëlys Born-Bony, Jay K. Kolls, Ignacio Garcia-Verdugo, and Jean-Michel Sallenave. 2022. "Pseudomonas Aeruginosa Lung Infection Subverts Lymphocytic Responses through IL-23 and IL-22 Post-Transcriptional Regulation" International Journal of Molecular Sciences 23, no. 15: 8427. https://doi.org/10.3390/ijms23158427
APA StyleVilleret, B., Ghinnagow, R., Kheir, S., Born-Bony, M., Kolls, J. K., Garcia-Verdugo, I., & Sallenave, J.-M. (2022). Pseudomonas Aeruginosa Lung Infection Subverts Lymphocytic Responses through IL-23 and IL-22 Post-Transcriptional Regulation. International Journal of Molecular Sciences, 23(15), 8427. https://doi.org/10.3390/ijms23158427