
Emodin Sensitizes Cervical Cancer Cells to Vinblastine by Inducing Apoptosis and Mitotic Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Emodin and Vinblastine Decrease Cell Viability
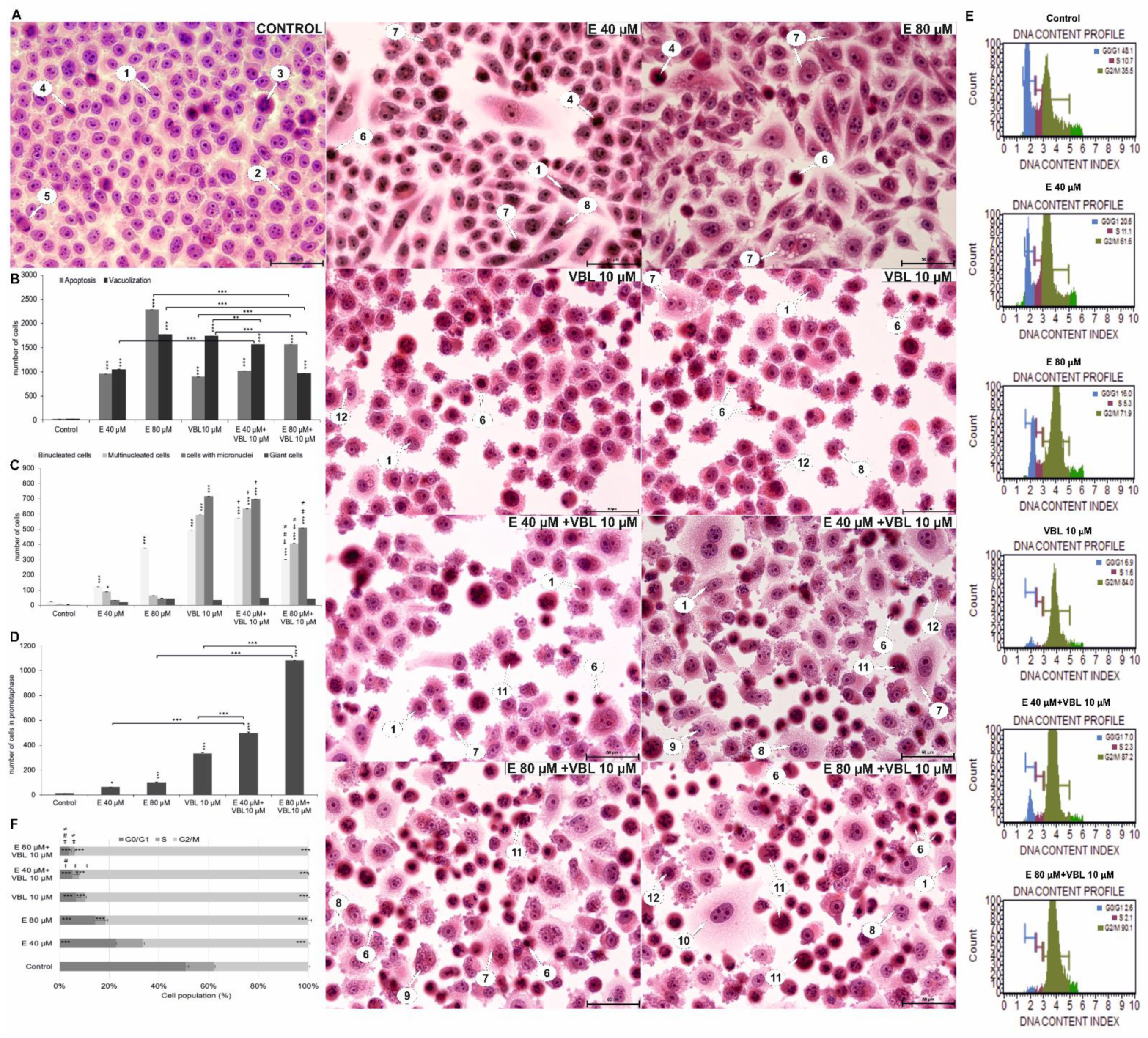
2.2. Emodin and Vinblastine Induce Cell Apoptosis by Activating Caspase 3/7
2.3. Emodin and Vinblastine Inactivate the Bcl-2 Protein
2.4. Emodin and Vinblastine Induce Mitotic Death
2.5. Emodin and Vinblastine Induce Phosphorylation of ATM Kinase
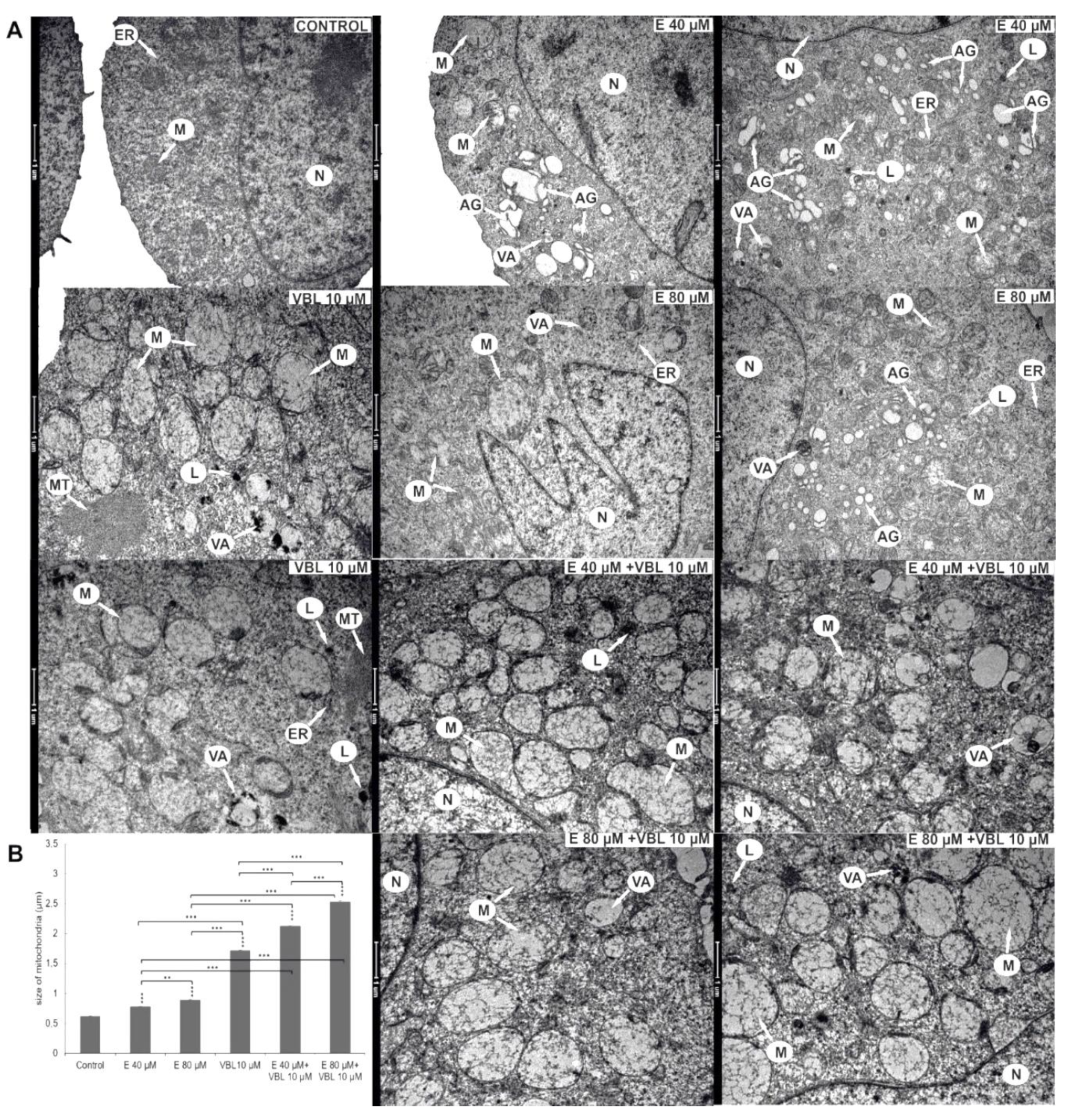
2.6. Emodin and Vinblastine Induced Ultrastructural Changes in Cervical Cancer Cells
2.7. Emodin and Vinblastine Induce ROS Production and Changes in Mitochondria in HeLa Cells
2.8. Emodin and Vinblastine Induce Morphological Changes in HeLa Cells
2.9. Emodin and Vinblastine Disrupt the Cell Cycle
2.10. Emodin and Vinblastine Reorganize the Cytoskeleton
3. Discussion
4. Materials and Methods
4.1. In Vitro Culture Conditions
4.2. Assessment of Cell Viability
4.3. Detection of Apoptosis
4.4. Caspase 3/7 Activity Test
4.5. Assessment of Bcl-2 Protein Phosphorylation
4.6. Measurement of the Reactive Oxygen Species Generation
4.7. Measurement of the Mitochondrial Membrane Potential (Δψm)
4.8. Microscopic Evaluation of Changes in Mitochondrial Membrane Potential
4.9. DAPI Staining
4.10. DNA Damage Assessment
4.11. Assessment of Ultrastructural Changes
4.12. Assessment of Morphological Changes
4.13. Cell Cycle Analysis
4.14. Fluorescent Labeling of F-Actin
4.15. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitra, T.; Elangovan, S. Cervical cancer development, chemoresistance, and therapy: A snapshot of involvement of microRNA. Mol. Cell. Biochem. 2021, 476, 4363–4385. [Google Scholar] [CrossRef] [PubMed]
- Sarenac, T.; Mikov, M. Cervical Cancer, Different Treatments and Importance of Bile Acids as Therapeutic Agents in This Disease. Front. Pharmacol. 2019, 10, 484. [Google Scholar] [CrossRef] [Green Version]
- Sathyanarayanan, A.; Chandrasekaran, K.S.; Karunagaran, D. microRNA-145 modulates epithelial-mesenchymal transition and suppresses proliferation, migration and invasion by targeting SIP1 in human cervical cancer cells. Cell. Oncol. 2017, 40, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Asiaf, A.; Ahmad, S.T.; Mohammad, S.O.; Zargar, M.A. Review of the current knowledge on the epidemiology, pathogenesis, and prevention of human papillomavirus infection. Eur. J. Cancer Prev. Off. J. Eur. Cancer Prev. Organ. 2014, 23, 206–224. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almagro, L.; Fernandez-Perez, F.; Pedreno, M.A. Indole alkaloids from Catharanthus roseus: Bioproduction and their effect on human health. Molecules 2015, 20, 2973–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Xu, Z. Indole Alkaloids with Potential Anticancer Activity. Curr. Top. Med. Chem. 2020, 20, 1938–1949. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.L.; Huang, W.; Zhu, H.P.; Peng, C.; Zhao, Q.; Han, B. Advances in indole-containing alkaloids as potential anticancer agents by regulating autophagy. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 149, 112827. [Google Scholar] [CrossRef]
- Selimovic, D.; Badura, H.E.; El-Khattouti, A.; Soell, M.; Porzig, B.B.; Spernger, A.; Ghanjati, F.; Santourlidis, S.; Haikel, Y.; Hassan, M. Vinblastine-induced apoptosis of melanoma cells is mediated by Ras homologous A protein (Rho A) via mitochondrial and non-mitochondrial-dependent mechanisms. Apoptosis Int. J. Program. Cell Death 2013, 18, 980–997. [Google Scholar] [CrossRef]
- Upreti, M.; Lyle, C.S.; Skaug, B.; Du, L.; Chambers, T.C. Vinblastine-induced apoptosis is mediated by discrete alterations in subcellular location, oligomeric structure, and activation status of specific Bcl-2 family members. J. Biol. Chem. 2006, 281, 15941–15950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.T.; Huang, Y.W.; Yang, C.H.; Huang, K.S. Drug delivery systems and combination therapy by using vinca alkaloids. Curr. Top. Med. Chem. 2015, 15, 1491–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zdioruk, M.; Want, A.; Mietelska-Porowska, A.; Laskowska-Kaszub, K.; Wojsiat, J.; Klejman, A.; Uzarowska, E.; Koza, P.; Olejniczak, S.; Pikul, S.; et al. A New Inhibitor of Tubulin Polymerization Kills Multiple Cancer Cell Types and Reveals p21-Mediated Mechanism Determining Cell Death after Mitotic Catastrophe. Cancers 2020, 12, 2161. [Google Scholar] [CrossRef]
- Wang, D.; Wang, X.H.; Yu, X.; Cao, F.; Cai, X.; Chen, P.; Li, M.; Feng, Y.; Li, H.; Wang, X. Pharmacokinetics of Anthraquinones from Medicinal Plants. Front. Pharmacol. 2021, 12, 638993. [Google Scholar] [CrossRef] [PubMed]
- van Gorkom, B.A.; de Vries, E.G.; Karrenbeld, A.; Kleibeuker, J.H. Review article: Anthranoid laxatives and their potential carcinogenic effects. Aliment. Pharmacol. Ther. 1999, 13, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, D.; Li, L.; Yang, S.; Du, G.; Lu, Y. Anti-inflammatory Effects and Mechanisms of Rhein, an Anthraquinone Compound, and Its Applications in Treating Arthritis: A Review. Nat. Prod. Bioprospect. 2020, 10, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Kemegne, G.A.; Mkounga, P.; Essia Ngang, J.J.; Sado Kamdem, S.L.; Nkengfack, A.E. Antimicrobial structure activity relationship of five anthraquinones of emodine type isolated from Vismia laurentii. BMC Microbiol. 2017, 17, 41. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Singh, A.; Samanta, S.K.; Singha Roy, A. Naturally occurring anthraquinones as potential inhibitors of SARS-CoV-2 main protease: An integrated computational study. Biologia 2022, 77, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, G.K.; Yadav, A.; Mandal, P.; Tripathi, A.; Das, M. Immunomodulatory potential of Rhein, an anthraquinone moiety of Cassia occidentalis seeds. Toxicol. Lett. 2016, 245, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Wang, C.; Li, D.; Hou, H. Novel anthraquinone compounds as anticancer agents and their potential mechanism. Future Med. Chem. 2020, 12, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chu, S.; Liu, Y.; Chen, N. Neuroprotective Effects of Anthraquinones from Rhubarb in Central Nervous System Diseases. Evid.-Based Complement. Altern. Med. Ecam 2019, 2019, 3790728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.K.; Noh, E.M.; Moon, S.J.; Kim, J.M.; Kwon, K.B.; Park, B.H.; You, Y.O.; Hwang, B.M.; Kim, H.J.; Kim, B.S.; et al. Emodin suppresses inflammatory responses and joint destruction in collagen-induced arthritic mice. Rheumatology 2013, 52, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.W.; Shim, D.W.; Shin, W.Y.; Heo, K.H.; Kwak, S.B.; Sim, E.J.; Jeong, J.H.; Kang, T.B.; Lee, K.H. Anti-inflammatory effect of emodin via attenuation of NLRP3 inflammasome activation. Int. J. Mol. Sci. 2015, 16, 8102–8109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Wen, K.; Caruso, F.; Belli, S. Emodin Scavenging of Superoxide Radical Includes pi-pi Interaction. X-ray Crystal Structure, Hydrodynamic Voltammetry and Theoretical Studies. Antioxidants 2020, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trybus, W.; Krol, T.; Trybus, E.; Stachurska, A.; Krol, G.; Kopacz-Bednarska, A. Emodin Induces Death in Human Cervical Cancer Cells Through Mitotic Catastrophe. Anticancer Res. 2019, 39, 679–686. [Google Scholar] [CrossRef]
- Trybus, W.; Krol, T.; Trybus, E.; Kopacz-Bednarska, A.; Krol, G.; Karpowicz, E. Changes in the Lysosomal System of Cervical Cancer Cells Induced by Emodin Action. Anticancer Res. 2017, 37, 6087–6096. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.C.; Chung, J.G. Anticancer potential of emodin. Biomedicine 2012, 2, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.; Zhang, X.; Wang, L.; Li, X.; Xie, T.; Zhao, J.; Yan, J.; Wang, L.; Ye, H.; Jiao, S.; et al. Herb-sourced emodin inhibits angiogenesis of breast cancer by targeting VEGFA transcription. Theranostics 2020, 10, 6839–6853. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, P.; Mao, H.; Wanga, A.; Zhang, X. Emodin sensitizes paclitaxel-resistant human ovarian cancer cells to paclitaxel-induced apoptosis in vitro. Oncol. Rep. 2009, 21, 1605–1610. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.L.; Bu, H.; Li, H.; Chen, H.; Guo, H.C.; Wang, Z.H.; Tong, H.F.; Ni, Z.L.; Liu, H.B.; Lin, S.Z. Emodin reverses gemcitabine resistance in pancreatic cancer cells via the mitochondrial apoptosis pathway in vitro. Int. J. Oncol. 2012, 40, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Lee, Y.M.; Oh, T.I.; Shin, D.H.; Kim, G.H.; Kan, S.Y.; Kang, H.; Kim, J.H.; Kim, B.M.; Yim, W.J.; et al. Emodin Sensitizes Hepatocellular Carcinoma Cells to the Anti-Cancer Effect of Sorafenib through Suppression of Cholesterol Metabolism. Int. J. Mol. Sci. 2018, 19, 3127. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Lu, H.; Wang, S.; Chen, B.; Liu, Z.; Ke, X.; Liu, T.; Fu, J. The anthraquinone derivative Emodin inhibits angiogenesis and metastasis through downregulating Runx2 activity in breast cancer. Int. J. Oncol. 2015, 46, 1619–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chen, H.; Chen, J.; Hong, Z.; Liao, Y.; Zhang, Q.; Tong, H. Emodin sensitizes human pancreatic cancer cells to EGFR inhibitor through suppressing Stat3 signaling pathway. Cancer Manag. Res. 2019, 11, 8463–8473. [Google Scholar] [CrossRef] [Green Version]
- Watroly, M.N.; Sekar, M.; Fuloria, S.; Gan, S.H.; Jeyabalan, S.; Wu, Y.S.; Subramaniyan, V.; Sathasivam, K.V.; Ravi, S.; Mat Rani, N.N.I.; et al. Chemistry, Biosynthesis, Physicochemical and Biological Properties of Rubiadin: A Promising Natural Anthraquinone for New Drug Discovery and Development. Drug Des. Dev. Ther. 2021, 15, 4527–4549. [Google Scholar] [CrossRef] [PubMed]
- Huertas, D.; Soler, M.; Moreto, J.; Villanueva, A.; Martinez, A.; Vidal, A.; Charlton, M.; Moffat, D.; Patel, S.; McDermott, J.; et al. Antitumor activity of a small-molecule inhibitor of the histone kinase Haspin. Oncogene 2012, 31, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, Z.; Zhang, J. Emodin enhances antitumor effect of paclitaxel on human non-small-cell lung cancer cells in vitro and in vivo. Drug Des. Dev. Ther. 2019, 13, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Bastians, H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2007, 10, 162–181. [Google Scholar] [CrossRef] [PubMed]
- Horbay, R.; Stoika, R. Giant cell formation: The way to cell death or cell survival? Cent. Eur. J. Biol. 2011, 6, 675–684. [Google Scholar] [CrossRef]
- Calvino, E.; Tejedor, M.C.; Sancho, P.; Herraez, A.; Diez, J.C. JNK and NFkappaB dependence of apoptosis induced by vinblastine in human acute promyelocytic leukaemia cells. Cell Biochem. Funct. 2015, 33, 211–219. [Google Scholar] [CrossRef]
- Kuboki, M.; Ito, A.; Simizu, S.; Umezawa, K. Activation of apoptosis by caspase-3-dependent specific RelB cleavage in anticancer agent-treated cancer cells: Involvement of positive feedback mechanism. Biochem. Biophys. Res. Commun. 2015, 456, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Król, T.; Schmidt, A.; Kołątaj, A.; Witek, B. Vinblastine-induced autophagy in mouse liver. Comp. Biochem. Physiol. C Toxicol. Pharm. 1994, 107, 165–169. [Google Scholar] [CrossRef]
- Munafo, D.B.; Colombo, M.I. A novel assay to study autophagy: Regulation of autophagosome vacuole size by amino acid deprivation. J. Cell Sci. 2001, 114, 3619–3629. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, W.W.; Sun, X.; Qian, D.; Tang, D.D.; Zhang, L.L.; Li, M.Y.; Wang, L.Y.; Wu, C.J.; Peng, W. The versatile emodin: A natural easily acquired anthraquinone possesses promising anticancer properties against a variety of cancers. Int. J. Biol. Sci. 2022, 18, 3498–3527. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Ni, B.; Fu, J.; Yin, X.; You, L.; Leng, X.; Liang, X.; Ni, J. Emodin induces apoptosis in human hepatocellular carcinoma HepaRG cells via the mitochondrial caspasedependent pathway. Oncol. Rep. 2018, 40, 1985–1993. [Google Scholar] [CrossRef]
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, M.A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anti-Cancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef]
- Gadde, S.; Heald, R. Mechanisms and molecules of the mitotic spindle. Curr. Biol. CB 2004, 14, R797–R805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Egea, G.; Serra-Peinado, C.; Gavilan, M.; Rios, R. Cytoskeleton and Golgi-apparatus interactions: A two-way road of function and structure. Cell Health Cytoskelet. 2015, 7, 37–54. [Google Scholar] [CrossRef] [Green Version]
- Thyberg, J.; Moskalewski, S. Role of microtubules in the organization of the Golgi complex. Exp. Cell Res. 1999, 246, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Trybus, W.; Krol, T.; Trybus, E.; Stachurska, A.; Kopacz-Bednarska, A.; Krol, G. Induction of Mitotic Catastrophe in Human Cervical Cancer Cells After Administration of Aloe-emodin. Anticancer Res. 2018, 38, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Portugal, J.; Mansilla, S.; Bataller, M. Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr. Pharm. Des. 2010, 16, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Valent, A.; Raslova, H.; Yakushijin, K.; Horne, D.; Feunteun, J.; Lenoir, G.; Medema, R.; et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004, 23, 4362–4370. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.J.; Kaufman, G.P.; Mavis, C.; Czuczman, M.S.; Hernandez-Ilizaliturri, F.J. Mitotic catastrophe and cell cycle arrest are alternative cell death pathways executed by bortezomib in rituximab resistant B-cell lymphoma cells. Oncotarget 2017, 8, 12741–12753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maskey, D.; Yousefi, S.; Schmid, I.; Zlobec, I.; Perren, A.; Friis, R.; Simon, H.U. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat. Commun. 2013, 4, 2130. [Google Scholar] [CrossRef] [Green Version]
- Geriyol, P.; Basavanneppa, H.B.; Dhananjaya, B.L. Protecting effect of caffeine against vinblastine (an anticancer drug) induced genotoxicity in mice. Drug Chem. Toxicol. 2015, 38, 188–195. [Google Scholar] [CrossRef]
- Mhaidat, N.M.; Alzoubi, K.H.; Khabour, O.F.; Alawneh, K.Z.; Raffee, L.A.; Alsatari, E.S.; Hussein, E.I.; Bani-Hani, K.E. Assessment of genotoxicity of vincristine, vinblastine and vinorelbine in human cultured lymphocytes: A comparative study. Balk. J. Med. Genet. BJMG 2016, 19, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Trybus, W.; Krol, T.; Trybus, E.; Stachurska, A.; Krol, G. The potential antitumor effect of chrysophanol in relation to cervical cancer cells. J. Cell. Biochem. 2021, 122, 639–652. [Google Scholar] [CrossRef]
- Trybus, W.; Król, T.; Trybus, E.; Stachurska, A. Physcion Induces Potential Anticancer Effects in Cervical Cancer Cells. Cells 2021, 10, 2029. [Google Scholar] [CrossRef]
- Mueller, S.O.; Stopper, H. Characterization of the genotoxicity of anthraquinones in mammalian cells. Biochim. Biophys. Acta 1999, 1428, 406–414. [Google Scholar] [CrossRef]
- Lin, X.H.; Wan, H.Y.; Zhang, Y.F.; Chen, J.H. Studies of the interaction between Aloe-emodin and DNA and preparation of DNA biosensor for detection of PML-RARalpha fusion gene in acute promyelocytic leukemia. Talanta 2008, 74, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.F.; Lai, T.Y.; Hsia, T.C.; Tang, Y.J.; Yang, J.S.; Chiang, J.H.; Lu, C.C.; Liu, C.M.; Wang, H.L.; Chung, J.G. Danthron induces DNA damage and inhibits DNA repair gene expressions in GBM 8401 human brain glioblastoma multiforms cells. Neurochem. Res. 2010, 35, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.T.; Silva, G.; Pungartnik, C.; Brendel, M. Study of DNA-emodin interaction by FTIR and UV-vis spectroscopy. J. Photochem. Photobiol. B Biol. 2012, 111, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Stagni, V.; Cirotti, C.; Barila, D. Ataxia-Telangiectasia Mutated Kinase in the Control of Oxidative Stress, Mitochondria, and Autophagy in Cancer: A Maestro With a Large Orchestra. Front. Oncol. 2018, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Kamsler, A.; Daily, D.; Hochman, A.; Stern, N.; Shiloh, Y.; Rotman, G.; Barzilai, A. Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from Atm-deficient mice. Cancer Res. 2001, 61, 1849–1854. [Google Scholar] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navrkalova, V.; Kafkova, L.R.; Divoky, V.; Pospisilova, S. Oxidative stress as a therapeutic perspective for ATM-deficient chronic lymphocytic leukemia patients. Haematologica 2015, 100, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.; Goldstine, J.V.; Gatti, R.A. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum. Mol. Genet. 2007, 16, 2154–2164. [Google Scholar] [CrossRef]
- Krol, T. Activity of lysosomal system in mouse liver after taxol administration. Gen. Pharmacol. 1998, 30, 239–243. [Google Scholar] [CrossRef]
- Wu, Z.Z.; Zhang, G.; Long, M.; Wang, H.B.; Song, G.B.; Cai, S.X. Comparison of the viscoelastic properties of normal hepatocytes and hepatocellular carcinoma cells under cytoskeletal perturbation. Biorheology 2000, 37, 279–290. [Google Scholar] [PubMed]
- Smith, C.D.; Mooberry, S.L.; Zhang, X.; Helt, A.M. A sensitive assay for taxol and other microtubule-stabilizing agent. Cancer Lett. 1994, 79, 213–219. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trybus, W.; Trybus, E.; Król, T. Emodin Sensitizes Cervical Cancer Cells to Vinblastine by Inducing Apoptosis and Mitotic Death. Int. J. Mol. Sci. 2022, 23, 8510. https://doi.org/10.3390/ijms23158510
Trybus W, Trybus E, Król T. Emodin Sensitizes Cervical Cancer Cells to Vinblastine by Inducing Apoptosis and Mitotic Death. International Journal of Molecular Sciences. 2022; 23(15):8510. https://doi.org/10.3390/ijms23158510
Chicago/Turabian StyleTrybus, Wojciech, Ewa Trybus, and Teodora Król. 2022. "Emodin Sensitizes Cervical Cancer Cells to Vinblastine by Inducing Apoptosis and Mitotic Death" International Journal of Molecular Sciences 23, no. 15: 8510. https://doi.org/10.3390/ijms23158510
APA StyleTrybus, W., Trybus, E., & Król, T. (2022). Emodin Sensitizes Cervical Cancer Cells to Vinblastine by Inducing Apoptosis and Mitotic Death. International Journal of Molecular Sciences, 23(15), 8510. https://doi.org/10.3390/ijms23158510