
Wound Healing Impairment in Type 2 Diabetes Model of Leptin-Deficient Mice—A Mechanistic Systematic Review


Abstract
:1. Introduction
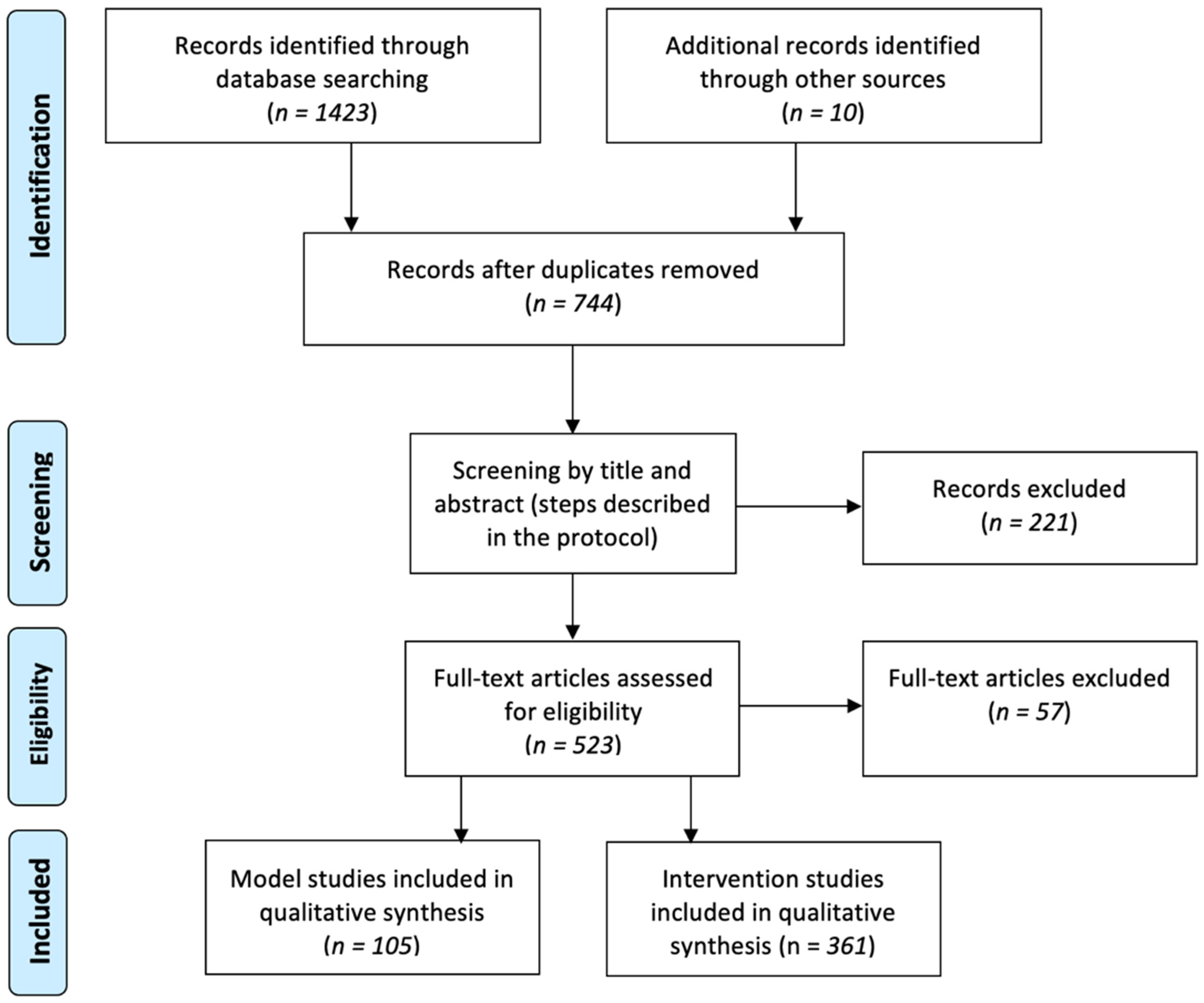
2. Methods
2.1. Study Selection
2.2. Data Extraction and Analyses
3. Results and Discussion
3.1. Literature Review
3.1.1. Phenotype and Wound Healing Dynamics
3.1.2. Growth Factors
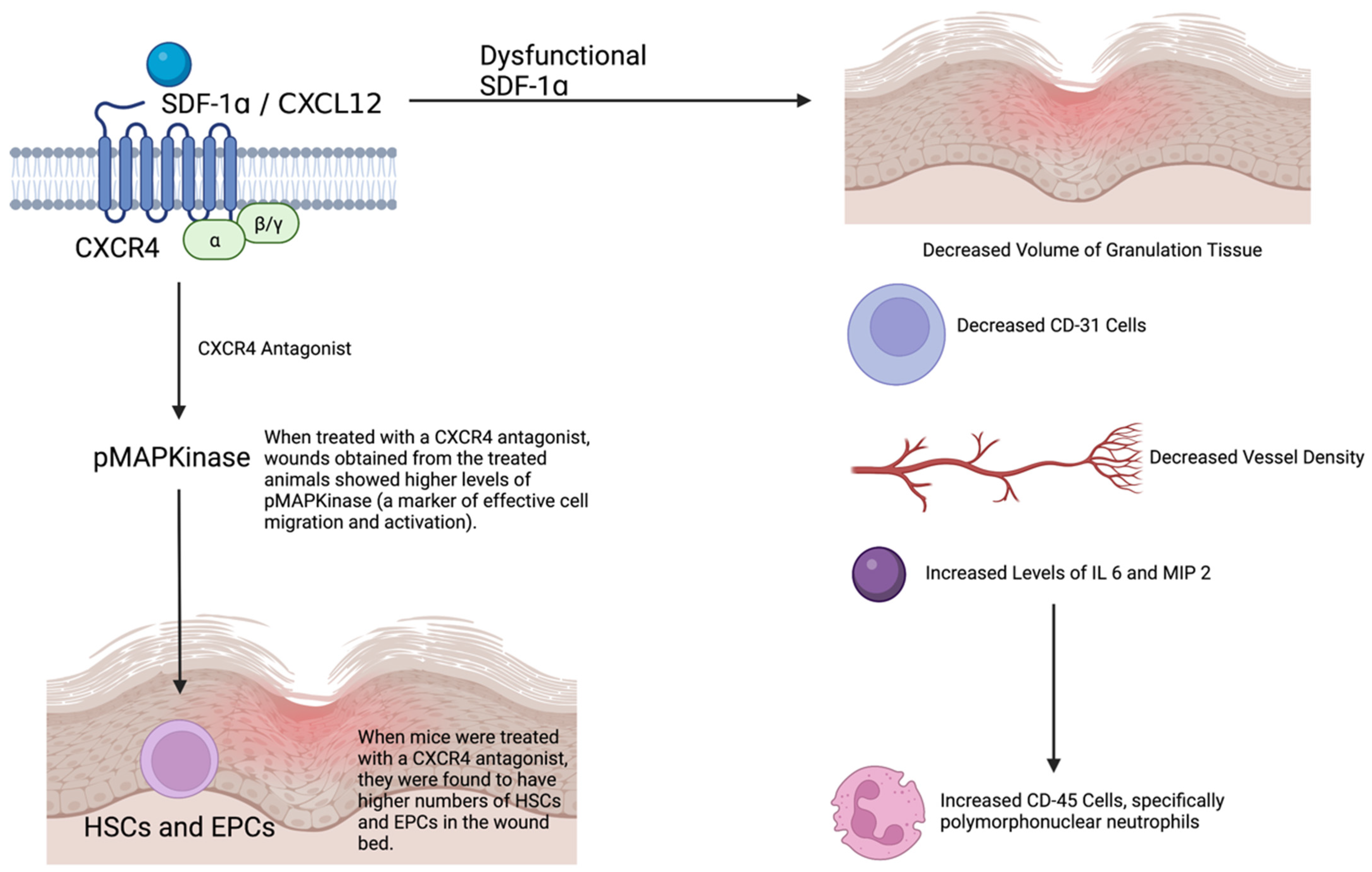
3.1.3. Angiogenesis
3.1.4. Cytokines and Immunological Apparatus
3.1.5. Macrophages
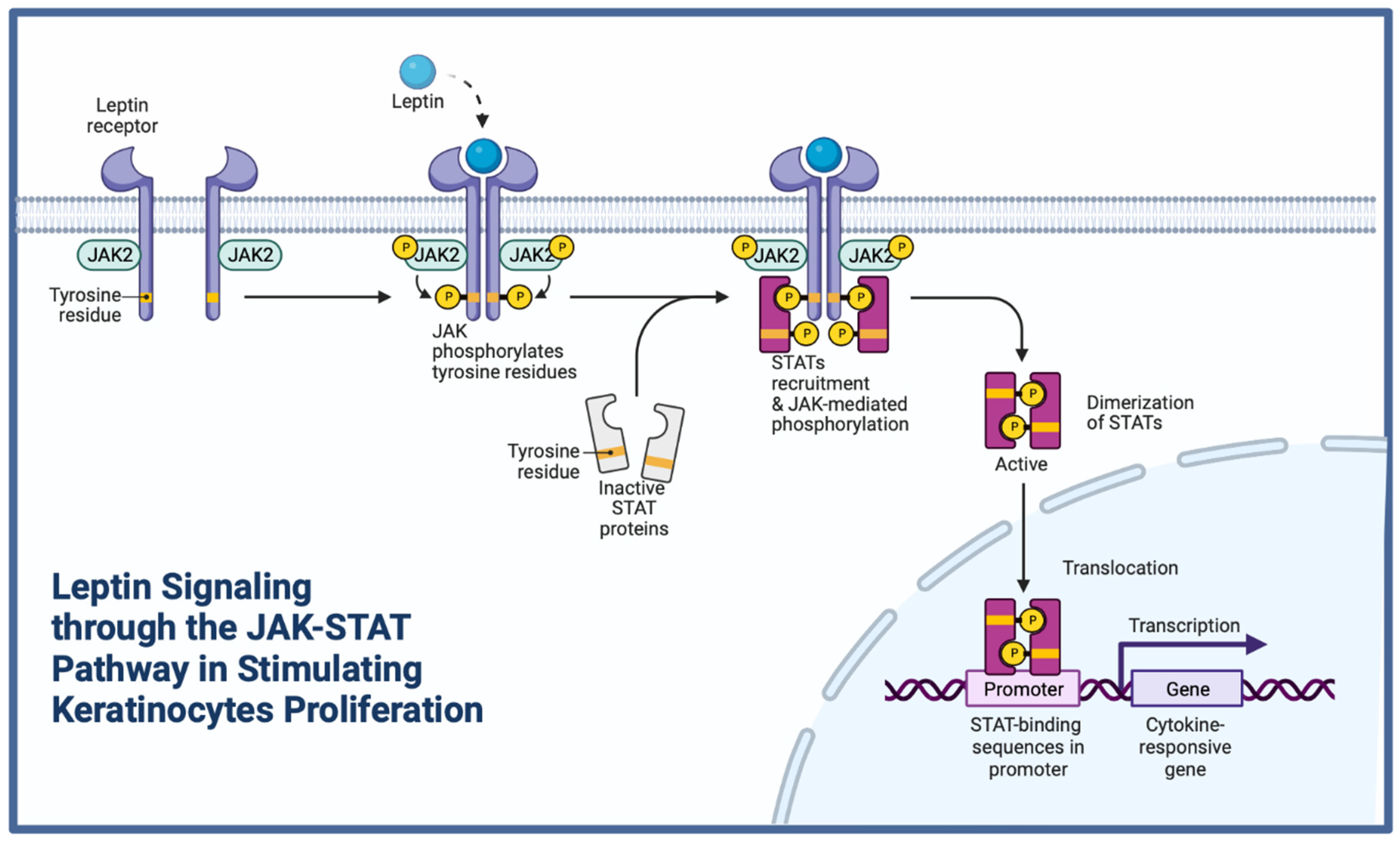
3.1.6. Leptin
3.1.7. Insulin
3.1.8. Advanced Glycation End-Products (AGEs)
3.1.9. Fibroblasts and Extracellular Matrix (ECM)
3.1.10. Apoptosis and Autophagy
3.1.11. Stem Cells (Bone Marrow)
3.1.12. Non-Coding RNAs
3.1.13. Bacterial Burden and Biofilm
3.1.14. Models of Chronic Diabetic Wounds
3.1.15. Other Studies
3.1.16. Interventions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intervention | In Vivo Studies | Most Advanced Clinical Trial | Reference |
|---|---|---|---|
| rPDGF-BB (Becaplermin) | [118,119,120,121,122,123,124] | Phase 3 | [125] |
| Collagen application (scaffold) | [126,127] | Phase 4 | [128] |
| Chitosan Gel Application | [129,130,131] | Phase 2 | [132] |
| bFGF | [119,133,134] | Phase 3 | [135] |
| VEGF, HIF1-α | [136,137,138,139] | Phase 2 | [140] |
| Negative Pressure Wound Therapy | [141] | Phase 2 | [142] |
| Cold Plasma Therapy | [143,144] | Phase 2 | [145] |
| Topical Insulin | [111,146] | Phase 2 | [147] |
| Low Magnitude High-Frequency Vibration Platform | [148] | Phase 2 | [149] |
| Nitric Oxide Releasing Patch | [150,151,152] | Phase 3 | [153] |
3.2. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Boulton, A.J.; Vileikyte, L.; Ragnarson-Tennvall, G.; Apelqvist, J. The global burden of diabetic foot disease. Lancet 2005, 366, 1719–1724. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Uivaraseanu, B.; Bungau, S.; Tit, D.M.; Fratila, O.; Rus, M.; Maghiar, T.A.; Maghiar, O.; Pantis, C.; Vesa, C.M.; Zaha, D.C. Clinical, Pathological and Microbiological Evaluation of Diabetic Foot Syndrome. Medicina 2020, 56, 380. [Google Scholar] [CrossRef]
- Edmonds, M.; Manu, C.; Vas, P. The current burden of diabetic foot disease. J. Clin. Orthop. Trauma 2021, 17, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummel, K.P.; Coleman, D.L.; Lane, P.W. The influence of genetic background on expression of mutations at the diabetes locus in the mouse. I. C57BL-KsJ and C57BL-6J strains. Biochem. Genet. 1972, 7, 1–13. [Google Scholar] [CrossRef]
- Coleman, D.L. Obese and diabetes: Two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 1978, 14, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Senter, L.H.; Legrand, E.K.; Laemmerhirt, K.E.; Kiorpes, T.C. Assessment of full-thickness wounds in the genetically diabetic mouse for suitability as a wound healing model. Wound Repair Regen. 1995, 3, 351–358. [Google Scholar] [CrossRef]
- Brem, H.; Tomic-Canic, M.; Entero, H.; Hanflik, A.M.; Wang, V.M.; Fallon, J.T.; Ehrlich, H.P. The synergism of age and db/db genotype impairs wound healing. Exp. Gerontol. 2007, 42, 523–531. [Google Scholar] [CrossRef]
- Sullivan, S.R.; Underwood, R.A.; Gibran, N.S.; Sigle, R.O.; Usui, M.L.; Carter, W.G.; Olerud, J.E. Validation of a model for the study of multiple wounds in the diabetic mouse (db/db). Plast. Reconstr. Surg. 2004, 113, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.C.; Kryger, Z.B.; Buck, D.W., II; De La Garza, M.; Galiano, R.D.; Mustoe, T.A. Limitations of the db/db mouse in translational wound healing research: Is the NONcNZO10 polygenic mouse model superior? Wound Repair Regen. 2010, 18, 605–613. [Google Scholar] [CrossRef]
- Berdal, M.; Jenssen, T. No association between glycemia and wound healing in an experimental db/db mouse model. ISRN Endocrinol. 2013, 2013, 307925. [Google Scholar] [CrossRef] [PubMed]
- Michaels, J.T.; Churgin, S.S.; Blechman, K.M.; Greives, M.R.; Aarabi, S.; Galiano, R.D.; Gurtner, G.C. db/db mice exhibit severe wound-healing impairments compared with other murine diabetic strains in a silicone-splinted excisional wound model. Wound Repair. Regen. 2007, 15, 665–670. [Google Scholar] [CrossRef]
- Tkalcević, V.I.; Cuzić, S.; Parnham, M.J.; Pasalić, I.; Brajsa, K. Differential evaluation of excisional non-occluded wound healing in db/db mice. Toxicol. Pathol. 2009, 37, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.T.; McKeever, C.C.; Vonu, P.; Patterson, C.; Liu, P.Y. Dynamic Histological Events and Molecular Changes in Excisional Wound Healing of Diabetic DB/DB Mice. J. Surg. Res. 2019, 238, 186–197. [Google Scholar] [CrossRef]
- Gnyawali, S.C.; Sinha, M.; El Masry, M.S.; Wulff, B.; Ghatak, S.; Soto-Gonzalez, F.; Wilgus, T.A.; Roy, S.; Sen, C.K. High resolution ultrasound imaging for repeated measure of wound tissue morphometry, biomechanics and hemodynamics under fetal, adult and diabetic conditions. PLoS ONE 2020, 15, e0241831. [Google Scholar] [CrossRef]
- Trousdale, R.K.; Jacobs, S.; Simhaee, D.A.; Wu, J.K.; Lustbader, J.W. Wound closure and metabolic parameter variability in a db/db mouse model for diabetic ulcers. J. Surg. Res. 2009, 151, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Goren, I.; Kämpfer, H.; Podda, M.; Pfeilschifter, J.; Frank, S. Leptin and wound inflammation in diabetic ob/ob mice: Differential regulation of neutrophil and macrophage influx and a potential role for the scab as a sink for inflammatory cells and mediators. Diabetes 2003, 52, 2821–2832. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Breeden, M.; Hübner, G.; Greenhalgh, D.G.; Longaker, M.T. Induction of keratinocyte growth factor expression is reduced and delayed during wound healing in the genetically diabetic mouse. J. Investig. Dermatol. 1994, 103, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Beer, H.D.; Longaker, M.T.; Werner, S. Reduced expression of PDGF and PDGF receptors during impaired wound healing. J. Investig. Dermatol. 1997, 109, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.L.; Kane, C.D.; Chernausek, S.D.; Greenhalgh, D.G. Differential expression and localization of insulin-like growth factors I and II in cutaneous wounds of diabetic and nondiabetic mice. Am. J. Pathol. 1997, 151, 715–724. [Google Scholar] [PubMed]
- Botusan, I.R.; Sunkari, V.G.; Savu, O.; Catrina, A.I.; Grunler, J.; Lindberg, S.; Pereira, T.; Yla-Herttuala, S.; Poellinger, L.; Brismar, K.; et al. Stabilization of HIF-1alpha is critical to improve wound healing in diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19426–19431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, S.; Hübner, G.; Breier, G.; Longaker, M.T.; Greenhalgh, D.G.; Werner, S. Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. J. Biol. Chem. 1995, 270, 12607–12613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, W.W.; Yang, C.; Dai, Z.Y.; Che, D.; Feng, J.; Mao, Y.L.; Cheng, R.; Wang, Z.X.; He, X.M.; Zhou, T.; et al. High Levels of Pigment Epithelium-Derived Factor in Diabetes Impair Wound Healing Through Suppression of Wnt Signaling. Diabetes 2015, 64, 1407–1419. [Google Scholar] [CrossRef] [Green Version]
- Kämpfer, H.; Pfeilschifter, J.; Frank, S. Expressional regulation of angiopoietin-1 and -2 and the tie-1 and -2 receptor tyrosine kinases during cutaneous wound healing: A comparative study of normal and impaired repair. Lab. Investig. 2001, 81, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, K.; Asai, J.; Li, M.; Thorne, T.; Losordo, D.W.; D’Amore, P.A. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am. J. Pathol. 2007, 170, 1178–1191. [Google Scholar] [CrossRef] [Green Version]
- Goren, I.; Müller, E.; Schiefelbein, D.; Gutwein, P.; Seitz, O.; Pfeilschifter, J.; Frank, S. Akt1 controls insulin-driven VEGF biosynthesis from keratinocytes: Implications for normal and diabetes-impaired skin repair in mice. J. Investig. Dermatol. 2009, 129, 752–764. [Google Scholar] [CrossRef] [Green Version]
- Zins, S.R.; Amare, M.F.; Tadaki, D.K.; Elster, E.A.; Davis, T.A. Comparative analysis of angiogenic gene expression in normal and impaired wound healing in diabetic mice: Effects of extracorporeal shock wave therapy. Angiogenesis 2010, 13, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Kotlinowski, J.; Grochot-Przeczek, A.; Taha, H.; Kozakowska, M.; Pilecki, B.; Skrzypek, K.; Bartelik, A.; Derlacz, R.; Horrevoets, A.J.G.; Pap, A.; et al. PPAR gamma activation but not PPAR gamma haplodeficiency affects proangiogenic potential of endothelial cells and bone marrow-derived progenitors. Cardiovasc. Diabetol. 2014, 13, 150. [Google Scholar] [CrossRef] [Green Version]
- Langer, S.; Beescho, C.; Ring, A.; Dorfmann, O.; Steinau, H.U.; Spindler, N. A new in vivo model using a dorsal skinfold chamber to investigate microcirculation and angiogenesis in diabetic wounds. GMS Interdiscip. Plast. Reconstr. Surg. Dgpw 2016, 5, Doc09. [Google Scholar] [CrossRef] [PubMed]
- Okonkwo, U.A.; Chen, L.; Ma, D.; Haywood, V.A.; Barakat, M.; Urao, N.; Dipietro, L.A. Compromised angiogenesis and vascular Integrity in impaired diabetic wound healing. PLoS ONE 2020, 15, e0231962. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J.; Jin, D.P.; Buck, D.W., II; Galiano, R.D.; Mustoe, T.A. Impaired response of mature adipocytes of diabetic mice to hypoxia. Exp. Cell Res. 2011, 317, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Sinha, M.; Pal, D.; Tabasum, S.; Gnyawali, S.C.; Khona, D.; Sarkar, S.; Mohanty, S.K.; Soto-Gonzalez, F.; Khanna, S.; et al. Cutaneous Epithelial to Mesenchymal Transition Activator ZEB1 Regulates Wound Angiogenesis and Closure in a Glycemic Status-Dependent Manner. Diabetes 2019, 68, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Kämpfer, H.; Paulukat, J.; Mühl, H.; Wetzler, C.; Pfeilschifter, J.; Frank, S. Lack of interferon-gamma production despite the presence of interleukin-18 during cutaneous wound healing. Mol. Med. 2000, 6, 1016–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzler, C.; Kämpfer, H.; Stallmeyer, B.; Pfeilschifter, J.; Frank, S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: Prolonged persistence of neutrophils and macrophages during the late phase of repair. J. Investig. Dermatol. 2000, 115, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goren, I.; Kämpfer, H.; Müller, E.; Schiefelbein, D.; Pfeilschifter, J.; Frank, S. Oncostatin M expression is functionally connected to neutrophils in the early inflammatory phase of skin repair: Implications for normal and diabetes-impaired wounds. J. Investig. Dermatol. 2006, 126, 628–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zykova, S.N.; Jenssen, T.G.; Berdal, M.; Olsen, R.; Myklebust, R.; Seljelid, R. Altered cytokine and nitric oxide secretion in vitro by macrophages from diabetic type II-like db/db mice. Diabetes 2000, 49, 1451–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Desta, T.; He, H.; Graves, D.T. Diabetes alters the response to bacteria by enhancing fibroblast apoptosis. Endocrinology 2004, 145, 2997–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.I.; Kim, K.H.; Lau, L.F. The matricellular protein CCN1 mediates neutrophil efferocytosis in cutaneous wound healing. Nat. Commun. 2015, 6, 7386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawaya, A.P.; Stone, R.C.; Brooks, S.R.; Pastar, I.; Jozic, I.; Hasneen, K.; O’Neill, K.; Mehdizadeh, S.; Head, C.R.; Strbo, N.; et al. Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat. Commun. 2020, 11, 4678. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.R.; Mills, R.E.; Costanzo, A.E.; Jameson, J.M. Gammadelta T cells are reduced and rendered unresponsive by hyperglycemia and chronic TNFalpha in mouse models of obesity and metabolic disease. PLoS ONE 2010, 5, e11422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.R.; Costanzo, A.E.; Jameson, J.M. Dysfunctional gamma delta T Cells Contribute to Impaired Keratinocyte Homeostasis in Mouse Models of Obesity. J. Investig. Dermatol. 2011, 131, 2409–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurmann, C.; Seitz, O.; Sader, R.; Pfeilschifter, J.; Goren, I.; Frank, S. Role of wound macrophages in skin flap loss or survival in an experimental diabetes model. Br. J. Surg. 2010, 97, 1437–1451. [Google Scholar] [CrossRef] [PubMed]
- Kampfer, H.; Schmidt, R.; Geisslinger, G.; Pfeilschifter, J.; Frank, S. Wound inflammation in diabetic ob/ob mice—Functional coupling of prostaglandin biosynthesis to cyclooxygenase-1 activity in diabetes-impaired wound healing. Diabetes 2005, 54, 1543–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, S.; Seitz, O.; Schürmann, C.; Hermes, N.; Müller, E.; Pfeilschifter, J.; Goren, I. Wound healing in mice with high-fat diet- or ob gene-induced diabetes-obesity syndromes: A comparative study. Exp. Diabetes Res. 2010, 2010, 476969. [Google Scholar] [CrossRef] [Green Version]
- Schürmann, C.; Goren, I.; Linke, A.; Pfeilschifter, J.; Frank, S. Deregulated unfolded protein response in chronic wounds of diabetic ob/ob mice: A potential connection to inflammatory and angiogenic disorders in diabetes-impaired wound healing. Biochem. Biophys. Res. Commun. 2014, 446, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Mirza, R.; Koh, T.J. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 2011, 56, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Mirza, R.E.; Fang, M.M.; Ennis, W.J.; Koh, T.J. Blocking Interleukin-1 beta Induces a Healing-Associated Wound Macrophage Phenotype and Improves Healing in Type 2 Diabetes. Diabetes 2013, 62, 2579–2587. [Google Scholar] [CrossRef] [Green Version]
- Rodero, M.P.; Hodgson, S.S.; Hollier, B.; Combadiere, C.; Khosrotehrani, K. Reduced Il17a expression distinguishes a Ly6c(lo)MHCII(hi) macrophage population promoting wound healing. J. Investig. Dermatol. 2013, 133, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Goren, I.; Pfeilschifter, J.; Frank, S. Uptake of Neutrophil-Derived Ym1 Protein Distinguishes Wound Macrophages in the Absence of Interleukin-4 Signaling in Murine Wound Healing. Am. J. Pathol. 2014, 184, 3249–3261. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Tie, G.; Wang, S.; Tutto, A.; DeMarco, N.; Khair, L.; Fazzio, T.G.; Messina, L.M. Diabetes impairs wound healing by Dnmt1-dependent dysregulation of hematopoietic stem cells differentiation towards macrophages. Nat. Commun. 2018, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, S.; Jayaraman, V.; Huelsmann, E.J.; Bonish, B.; Burgad, D.; Sivaramakrishnan, G.; Qin, S.S.; DiPietro, L.A.; Zloza, A.; Zhang, C.X.; et al. Pro-Inflammatory Chemokine CCL2 (MCP-1) Promotes Healing in Diabetic Wounds by Restoring the Macrophage Response. PLoS ONE 2014, 9, e91574. [Google Scholar] [CrossRef] [Green Version]
- Guest, C.B.; Chakour, K.S.; Freund, G.G. Macropinocytosis is decreased in diabetic mouse macrophages and is regulated by AMPK. BMC Immunol. 2008, 9, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, R.E.; Fang, M.M.; Weinheimer-Haus, E.M.; Ennis, W.J.; Koh, T.J. Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes 2014, 63, 1103–1114. [Google Scholar] [CrossRef] [Green Version]
- Stallmeyer, B.; Kämpfer, H.; Podda, M.; Kaufmann, R.; Pfeilschifter, J.; Frank, S. A novel keratinocyte mitogen: Regulation of leptin and its functional receptor in skin repair. J. Investig. Dermatol. 2001, 117, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, L.M.; Li, J.; Liu, Y.; Zhang, C.Y.; Zhang, Y.J.; Zen, K. Protein Tyrosine Phosphatase 1B Impairs Diabetic Wound Healing Through Vascular Endothelial Growth Factor Receptor 2 Dephosphorylation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goren, I.; Pfeilschifter, J.; Frank, S. Determination of leptin signaling pathways in human and murine keratinocytes. Biochem. Biophys. Res. Commun. 2003, 303, 1080–1085. [Google Scholar] [CrossRef]
- Ring, B.D.; Scully, S.; Davis, C.R.; Baker, M.B.; Cullen, M.J.; Pelleymounter, M.A.; Danilenko, D.M. Systemically and topically administered leptin both accelerate wound healing in diabetic ob/ob mice. Endocrinology 2000, 141, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Gao, F.; Li, C.Z.; Xue, Y.M. Expression of leptin and its long-form receptor in the marginal cutaneous tissues of diabetic foot ulcers. Acta Diabetol. 2012, 49, S205–S214. [Google Scholar] [CrossRef]
- Kämpfer, H.; Pfeilschifter, J.; Frank, S. Expression and activity of arginase isoenzymes during normal and diabetes-impaired skin repair. J. Investig. Dermatol. 2003, 121, 1544–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodson, W.H., III; Hunt, T.K. Wound collagen accumulation in obese hyperglycemic mice. Diabetes 1986, 35, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.A.; Saghizadeh, M.; Ong, J.M.; Bosch, R.J.; Deem, R.; Simsolo, R.B. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J. Clin. Investig. 1995, 95, 2111–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goren, I.; Muller, E.; Pfeilschifter, J.; Frank, S. Severely impaired insulin signaling in chronic wounds of diabetic ob/ob mice—A potential role of tumor necrosis factor-alpha. Am. J. Pathol. 2006, 168, 765–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.Q.; Lee, S.; Wilson, H.; Seeger, M.; Iordanov, H.; Gatla, N.; Whittington, A.; Bach, D.; Lu, J.Y.; Paller, A.S. Ganglioside GM3 depletion reverses impaired wound healing in diabetic mice by activating IGF-1 and insulin receptors. J. Investig. Dermatol. 2014, 134, 1446–1455. [Google Scholar] [CrossRef] [Green Version]
- Peppa, M.; Zhang, J.G.; Cai, W.J.; Brem, H.; Vlassara, H. Wound healing in diabetic db/db((+/+)) mice is regulated by dietary content in glycotoxins under stable hyperglycemia. Diabetes 2002, 51, A17–A18. [Google Scholar]
- Ji, X.Y.; Chen, Y.; Ye, G.H.; Dong, M.W.; Lin, K.Z.; Han, J.G.; Feng, X.P.; Li, X.B.; Yu, L.S.; Fan, Y.Y. Detection of RAGE expression and its application to diabetic wound age estimation. Int. J. Leg. Med. 2017, 131, 691–698. [Google Scholar] [CrossRef]
- Kim, J.H.; Yoon, N.Y.; Kim, D.H.; Jung, M.; Jun, M.; Park, H.Y.; Chung, C.H.; Lee, K.; Kim, S.; Park, C.S.; et al. Impaired permeability and antimicrobial barriers in type 2 diabetes skin are linked to increased serum levels of advanced glycation end-product. Exp. Dermatol. 2018, 27, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Goova, M.T.; Li, J.; Kislinger, T.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Nowygrod, S.; Wolf, B.M.; Caliste, X.; Yan, S.F.; et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am. J. Pathol. 2001, 159, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Lerman, O.Z.; Galiano, R.D.; Armour, M.; Levine, J.P.; Gurtner, G.C. Cellular dysfunction in the diabetic fibroblast—Impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am. J. Pathol. 2003, 162, 303–312. [Google Scholar] [CrossRef]
- Liu, R.K.; Bal, H.S.; Desta, T.; Behl, Y.; Graves, D.T. Tumor necrosis factor-alpha mediates diabetes-enhanced apoptosis of matrix-producing cells and impairs diabetic healing. Am. J. Pathol. 2006, 168, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, M.F.; Li, J.; Chehab, L.; Desta, T.; Chino, T.; Krothpali, N.; Behl, Y.; Alikhani, M.; Yang, J.; Braasch, C.; et al. Impaired wound healing in mouse models of diabetes is mediated by TNF-α dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia 2010, 53, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Chen, B.; Kao, H.K.; Murphy, G.; Orgill, D.P.; Guo, L.F. Lack of FGF-7 Further Delays Cutaneous Wound Healing in Diabetic Mice. Plast. Reconstr. Surg. 2011, 128, 673E–684E. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.; Yu, J.; Um, J.; Dubon, M.J.; Park, K.S. Substance P modulates properties of normal and diabetic dermal fibroblasts. Tissue Eng. Regen. Med. 2016, 13, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Choi, S.; Um, J.; Park, K.S. Reduced Expression of YAP in Dermal Fibroblasts is Associated with Impaired Wound Healing in Type 2 Diabetic Mice. Tissue Eng. Regen. Med. 2017, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Al-Mashat, H.A.; Kandru, S.; Liu, R.K.; Behl, Y.; Desta, T.; Graves, D.T. Diabetes enhances mRNA levels of proapoptotic genes and caspase activity, which contribute to impaired healing. Diabetes 2006, 55, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.Y.; Lin, C.; Xu, P.; Wu, S.; Fu, X.J.; Xia, W.D.; Yao, M. AGEs Induced Autophagy Impairs Cutaneous Wound Healing via Stimulating Macrophage Polarization to M1 in Diabetes. Sci. Rep. 2016, 6, 36416. [Google Scholar] [CrossRef]
- Brown, D.L.; Kao, W.W.; Greenhalgh, D.G. Apoptosis down-regulates inflammation under the advancing epithelial wound edge: Delayed patterns in diabetes and improvement with topical growth factors. Surgery 1997, 121, 372–380. [Google Scholar] [CrossRef]
- Shin, L.; Peterson, D.A. Impaired therapeutic capacity of autologous stem cells in a model of type 2 diabetes. Stem Cells Transl. Med. 2012, 1, 125–135. [Google Scholar] [CrossRef]
- Fiorina, P.; Pietramaggiori, G.; Scherer, S.S.; Jurewicz, M.; Mathews, J.C.; Vergani, A.; Thomas, G.; Orsenigo, E.; Staudacher, C.; La Rosa, S.; et al. The mobilization and effect of endogenous bone marrow progenitor cells in diabetic wound healing. Cell Transpl. 2010, 19, 1369–1381. [Google Scholar] [CrossRef]
- Tepper, O.M.; Carr, J.; Allen, R.J., Jr.; Chang, C.C.; Lin, C.D.; Tanaka, R.; Gupta, S.M.; Levine, J.P.; Saadeh, P.B.; Warren, S.M. Decreased circulating progenitor cell number and failed mechanisms of stromal cell-derived factor-1alpha mediated bone marrow mobilization impair diabetic tissue repair. Diabetes 2010, 59, 1974–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermudez, D.M.; Xu, J.W.; Herdrich, B.J.; Radu, A.; Mitchell, M.E.; Liechty, K.W. Inhibition of stromal cell-derived factor-1 alpha further impairs diabetic wound healing. J. Vasc. Surg. 2011, 53, 774–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Yang, X.; Zhang, Z.; Song, J.; Guan, Y.F.; Zou, D.J.; Miao, C.Y. Depletion of NAD pool contributes to impairment of endothelial progenitor cell mobilization in diabetes. Metab. Clin. Exp. 2016, 65, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Barman, P.K.; Urao, N.; Koh, T.J. Diabetes induces myeloid bias in bone marrow progenitors associated with enhanced wound macrophage accumulation and impaired healing. J. Pathol. 2019, 249, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, F.F.; Zhang, X.Y.; Coppin, E.; Vasamsetti, S.B.; Modugu, G.; Schloss, M.J.; Rohde, D.; McAlpine, C.S.; Iwamoto, Y.; Libby, P.; et al. Bone Marrow Endothelial Cells Regulate Myelopoiesis in Diabetes Mellitus. Circulation 2020, 142, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Li, H.N.; O’Meara, M.; Zhang, X.; Zhang, K.Z.; Seyoum, B.; Yi, Z.P.; Kaufman, R.J.; Monks, T.J.; Wang, J.M. Ameliorating Methylglyoxal-Induced Progenitor Cell Dysfunction for Tissue Repair in Diabetes. Diabetes 2019, 68, 1287–1302. [Google Scholar] [CrossRef]
- Wang, J.M.; Tao, J.; Chen, D.D.; Cai, J.J.; Irani, K.; Wang, Q.D.; Yuan, H.; Chen, A.F. MicroRNA miR-27b Rescues Bone Marrow-Derived Angiogenic Cell Function and Accelerates Wound Healing in Type 2 Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icli, B.; Nabzdyk, C.S.; Lujan-Hernandez, J.; Cahill, M.; Auster, M.E.; Wara, A.K.M.; Sun, X.H.; Ozdemir, D.; Giatsidis, G.; Orgill, D.P.; et al. Regulation of impaired angiogenesis in diabetic dermal wound healing by microRNA-26a. J. Mol. Cell. Cardiol. 2016, 91, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, D.Q.; Wang, A.X.; Chu, T.B.; Lohcharoenkal, W.; Zheng, X.W.; Grunler, J.; Narayanan, S.; Eliasson, S.; Herter, E.K.; et al. MicroRNA-132 with Therapeutic Potential in Chronic Wounds. J. Investig. Dermatol. 2017, 137, 2630–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icli, B.; Wu, W.; Ozdemir, D.; Li, H.; Cheng, H.S.; Haemmig, S.; Liu, X.; Giatsidis, G.; Avci, S.N.; Lee, N.; et al. MicroRNA-615-5p Regulates Angiogenesis and Tissue Repair by Targeting AKT/eNOS (Protein Kinase B/Endothelial Nitric Oxide Synthase) Signaling in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1458–1474. [Google Scholar] [CrossRef]
- Grice, E.A.; Snitkin, E.S.; Yockey, L.J.; Bermudez, D.M.; Liechty, K.W.; Segre, J.A.; Sequencing, N.C. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc. Natl. Acad. Sci. USA 2010, 107, 14799–14804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, G.A.; Zhao, A.G.; Usui, M.; Underwood, R.A.; Nguyen, H.; Beyenal, H.; Pulcini, E.D.; Hunt, A.A.; Bernstein, H.C.; Fleckman, P.; et al. Microsensor and transcriptomic signatures of oxygen depletion in biofilms associated with chronic wounds. Wound Repair Regen. 2016, 24, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Desmet, C.M.; Lafosse, A.; Vériter, S.; Porporato, P.E.; Sonveaux, P.; Dufrane, D.; Levêque, P.; Gallez, B. Application of Electron Paramagnetic Resonance (EPR) Oximetry to Monitor Oxygen in Wounds in Diabetic Models. PLoS ONE 2015, 10, e0144914. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Rich, J.; Hanses, F.; Lee, J.C. Defects in Innate Immunity Predispose C57BL/6J-Lepr(db)/Lepr(db) Mice to Infection by Staphylococcus aureus. Infect. Immun. 2009, 77, 1008–1014. [Google Scholar] [CrossRef] [Green Version]
- Naguib, G.; Al-Mashat, H.; Desta, T.; Graves, D.T. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J. Investig. Dermatol. 2004, 123, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Bauerle, E.A.; Usui, M.L.; Underwood, R.A.; Singh, P.K.; James, G.A.; Stewart, P.S.; Olerud, J.E.; Fleckman, P. Characterization of biofilm infected wounds in diabetic mice. J. Investig. Dermatol. 2010, 130, S2. [Google Scholar] [CrossRef]
- Zhao, G.; Hochwalt, P.C.; Usui, M.L.; Underwood, R.A.; Singh, P.K.; James, G.A.; Stewart, P.S.; Fleckman, P.; Olerud, J.E. Delayed wound healing in diabetic (db/db) mice with Pseudomonas aeruginosa biofilm challenge: A model for the study of chronic wounds. Wound Repair Regen. 2010, 18, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Usui, M.L.; Underwood, R.A.; Singh, P.K.; James, G.A.; Stewart, P.S.; Fleckman, P.; Olerud, J.E. Time course study of delayed wound healing in a biofilm-challenged diabetic mouse model. Wound Repair Regen. 2012, 20, 342–352. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.N.; Li, R.R.; Cheng, C.; Xu, J.Y.; Jin, C.X.; Gao, F.R.; Wang, J.; Zhang, J.P.; Zhang, J.F.; Wang, H.; et al. Pseudomonas aeruginosa infection alters the macrophage phenotype switching process during wound healing in diabetic mice. Cell Biol. Int. 2018, 42, 877–889. [Google Scholar] [CrossRef]
- Dhall, S.; Do, D.C.; Garcia, M.; Kim, J.; Mirebrahim, S.H.; Lyubovitsky, J.; Lonardi, S.; Nothnagel, E.A.; Schiller, N.; Martins-Green, M. Generating and reversing chronic wounds in diabetic mice by manipulating wound redox parameters. J. Diabetes Res. 2014, 2014, 562625. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Martins-Green, M. Protocol to create chronic wounds in diabetic mice. J. Vis. Exp. 2019, 2019, e57656. [Google Scholar] [CrossRef] [PubMed]
- Panayi, A.C.; Endo, Y.; Karvar, M.; Sensharma, P.; Haug, V.; Fu, S.Q.; Mi, B.B.; An, Y.; Orgill, D.P. Low mortality oxidative stress murine chronic wound model. BMJ Open Diabetes Res. Care 2020, 8, e001221. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Teixeira, L.B.C.; Raghunathan, V.K.; Covert, J.; Dubielzig, R.R.; Isseroff, R.R.; Schurr, M.; Abbott, N.L.; McAnulty, J.; Murphy, C.J. Full-thickness splinted skin wound healing models in db/db and heterozygous mice: Implications for wound healing impairment. Wound Repair Regen. 2014, 22, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Spenny, M.L.; Muangman, P.; Sullivan, S.R.; Bunnett, N.W.; Ansel, J.C.; Olerud, J.E.; Gibran, N.S. Neutral endopeptidase inhibition in diabetic wound repair. Wound Repair Regen. 2002, 10, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Schiefelbein, D.; Goren, I.; Fisslthaler, B.; Schmidt, H.; Geisslinger, G.; Pfeilschifter, J.; Frank, S. Biphasic regulation of HMG-CoA reductase expression and activity during wound healing and its functional role in the control of keratinocyte angiogenic and proliferative responses. J. Biol. Chem. 2008, 283, 15479–15490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medicherla, S.; Wadsworth, S.; Cullen, B.; Silcock, D.; Ma, J.Y.; Mangadu, R.; Kerr, I.; Chakravarty, S.; Luedtke, G.L.; Dugar, S.; et al. p38 MAPK inhibition reduces diabetes-induced impairment of wound healing. Diabetes Metab. Syndr. Obes. 2009, 2, 91–100. [Google Scholar]
- Pietramaggiori, G.; Scherer, S.S.; Alperovich, M.; Chen, B.; Orgill, D.P.; Wagers, A.J. Improved cutaneous healing in diabetic mice exposed to healthy peripheral circulation. J. Investig. Dermatol. 2009, 129, 2265–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurmann, C.; Schmidt, N.; Seitz, O.; Pfeilschifter, J.; Frank, S. Angiogenic response pattern during normal and impaired skin flap re-integration in mice: A comparative study. J. Cranio-Maxillofac. Surg. 2014, 42, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.M.; Coffman, B.; McGuire, P.G.; Howdieshell, T.R. Myocutaneous revascularization following graded ischemia in lean and obese mice. Diabetes Metab. Syndr. Obes. 2016, 9, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terao, M.; Murota, H.; Kimura, A.; Kato, A.; Ishikawa, A.; Igawa, K.; Miyoshi, E.; Katayama, I. 11 beta-Hydroxysteroid Dehydrogenase-1 Is a Novel Regulator of Skin Homeostasis and a Candidate Target for Promoting Tissue Repair. PLoS ONE 2011, 6, e25039. [Google Scholar] [CrossRef]
- Brazel, C.B.; Simon, J.C.; Tuckermann, J.P.; Saalbach, A. Inhibition of 11β-HSD1 expression by insulin in skin: Impact for diabetic wound healing. J. Clin. Med. 2020, 9, 3878. [Google Scholar] [CrossRef]
- Shibata, S.; Tada, Y.; Asano, Y.; Hau, C.S.; Kato, T.; Saeki, H.; Yamauchi, T.; Kubota, N.; Kadowaki, T.; Sato, S. Adiponectin regulates cutaneous wound healing by promoting keratinocyte proliferation and migration via the ERK signaling pathway. J. Immunol. 2012, 189, 3231–3241. [Google Scholar] [CrossRef] [Green Version]
- Sood, R.F.; Gu, H.W.; Djukovic, D.; Deng, L.L.; Ga, M.; Muffley, L.A.; Raftery, D.; Hocking, A.M. Targeted metabolic profiling of wounds in diabetic and nondiabetic mice. Wound Repair Regen. 2015, 23, 423–434. [Google Scholar] [CrossRef]
- Zhao, H.C.; Chen, J.C.; Chai, J.C.; Zhang, Y.C.; Yu, C.; Pan, Z.; Gao, P.; Zong, C.; Guan, Q.B.; Fu, Y.Q.; et al. Cytochrome P450 (CYP) epoxygenases as potential targets in the management of impaired diabetic wound healing. Lab. Investig. 2017, 97, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Wieman, T.J.; Smiell, J.M.; Su, Y. Efficacy and Safely of a Topical Gel Formulation of Recombinant Human Platelet-Derived Growth Factor-BB (Becaplermin) in Patients with Chronic Neuropathic Diabetic Ulcers: A phase III randomized placebo-controlled double-blind study. Diabetes Care 1998, 21, 822–827. [Google Scholar] [CrossRef]
- Papanas, N.; Maltezos, E. Benefit-risk assessment of becaplermin in the treatment of diabetic foot ulcers. Drug Saf. 2010, 33, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Ziyadeh, N.; Fife, D.; Walker, A.M.; Wilkinson, G.S.; Seeger, J.D. A matched cohort study of the risk of cancer in users of becaplermin. Adv. Ski. Wound Care 2011, 24, 31–39. [Google Scholar] [CrossRef]
- Greenhalgh, D.G.; Sprugel, K.H.; Murray, M.J.; Ross, R. PDGF and FGF stimulate wound healing in the genetically diabetic mouse. Am. J. Pathol. 1990, 136, 1235–1246. [Google Scholar]
- Albertson, S.; Hummel, R.P., III; Breeden, M.; Greenhalgh, D.G. PDGF and FGF reverse the healing impairment in protein-malnourished diabetic mice. Surgery 1993, 114, 368–372; discussion 372–373. [Google Scholar]
- Brown, R.L.; Breeden, M.P.; Greenhalgh, D.G. PDGF and TGF-alpha act synergistically to improve wound healing in the genetically diabetic mouse. J. Surg. Res. 1994, 56, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Kiritsy, C.P.; Antoniades, H.N.; Carlson, M.R.; Beaulieu, M.T.; D’Andrea, M.; Lynch, S.E. Combination of platelet-derived growth factor-BB and insulin-like growth factor-I is more effective than platelet-derived growth factor-BB alone in stimulating complete healing of full-thickness wounds in older diabetic mice. Wound Repair Regen. 1995, 3, 340–350. [Google Scholar] [CrossRef]
- Breitbart, A.S.; Laser, J.; Parrett, B.; Porti, D.; Grant, R.T.; Grande, D.A.; Mason, J.M. Accelerated diabetic wound healing using cultured dermal fibroblasts retrovirally transduced with the platelet-derived growth factor B gene. Ann. Plast. Surg. 2003, 51, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Keswani, S.G.; Katz, A.B.; Lim, F.Y.; Zoltick, P.; Radu, A.; Alaee, D.; Herlyn, M.; Crombleholme, T.M. Adenoviral mediated gene transfer of PDGF-B enhances wound healing in type I and type II diabetic wounds. Wound Repair Regen. 2004, 12, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Conejero, J.A.; Mason, J.M.; Parrett, B.M.; Wear-Maggitti, K.D.; Grant, R.T.; Breitbart, A.S. Lentiviral transfection with the PDGF-B gene improves diabetic wound healing. Plast. Reconstr. Surg. 2005, 116, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Canadian Institute of Health Research. Study of Combined Topical Growth Factor and Protease Inhibitor in Chronic Wound Healing; McMaster University: Hamilton, ON, Canada, 2016; NCT02845466. [Google Scholar]
- Chikazu, D.; Taguchi, T.; Koyama, H.; Hikiji, H.; Fujihara, H.; Suenaga, H.; Saijo, H.; Mori, Y.; Seto, I.; Iino, M.; et al. Improvement in wound healing by a novel synthetic collagen-gel dressing in genetically diabetic mice. Asian J. Oral Maxillofac. Surg. 2010, 22, 61–67. [Google Scholar] [CrossRef]
- Kondo, S.; Niiyama, H.; Yu, A.; Kuroyanagi, Y. Evaluation of a wound dressing composed of hyaluronic acid and collagen sponge containing epidermal growth factor in diabetic mice. J. Biomater. Sci. Polym. Ed. 2012, 23, 1729–1740. [Google Scholar] [CrossRef]
- Steinberg, J. Use of INTEGRA™ Flowable Wound Matrix to Manage Diabetic Foot Ulcers; Georgetown University: Washington, DC, USA, 2010; NCT01108263. [Google Scholar]
- Murakami, K.; Aoki, H.; Nakamura, S.; Nakamura, S.; Takikawa, M.; Hanzawa, M.; Kishimoto, S.; Hattori, H.; Tanaka, Y.; Kiyosawa, T.; et al. Hydrogel blends of chitin/chitosan, fucoidan and alginate as healing-impaired wound dressings. Biomaterials 2010, 31, 83–90. [Google Scholar] [CrossRef]
- Yuan, Z.; Zakhaleva, J.; Ren, H.; Liu, J.; Chen, W.; Pan, Y. Noninvasive and high-resolution optical monitoring of healing of diabetic dermal excisional wounds implanted with biodegradable in situ gelable hydrogels. Tissue Eng. Part C Methods 2010, 16, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagibayashi, S.; Kishimoto, S.; Ishihara, M.; Murakami, K.; Aoki, H.; Takikawa, M.; Fujita, M.; Sekido, M.; Kiyosawa, T. Novel hydrocolloid-sheet as wound dressing to stimulate healing-impaired wound healing in diabetic db/db mice. Biomed. Mater. Eng. 2012, 22, 301–310. [Google Scholar] [CrossRef]
- Primex. Clinical Trial Evaluating the Safety and Efficacy of the Use of Chitosan Gel in Patients with Chronic Wounds; University of Ljubljana: Ljubljana, Slovenia, 2019; NCT04178525. [Google Scholar]
- Tsuboi, R.; Rifkin, D.B. Recombinant basic fibroblast growth factor stimulates wound healing in healing-impaired db/db mice. J. Exp. Med. 1990, 172, 245–251. [Google Scholar] [CrossRef]
- Tsuboi, R.; Shi, C.M.; Rifkin, D.B.; Ogawa, H. A wound healing model using healing-impaired diabetic mice. J. Dermatol. 1992, 19, 673–675. [Google Scholar] [CrossRef]
- Olympus Biotech Corporation. The TRAfermin in Neuropathic Diabetic Foot Ulcer Study—Southern Europe The TRANS-South Study; Olympus Biotech Corporation: Hopkinton, MA, USA, 2010; NCT01217463. [Google Scholar]
- Kirchner, L.M.; Meerbaum, S.O.; Gruber, B.S.; Knoll, A.K.; Bulgrin, J.; Taylor, R.A.; Schmidt, S.P. Effects of vascular endothelial growth factor on wound closure rates in the genetically diabetic mouse model. Wound Repair Regen. 2003, 11, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Galiano, R.D.; Tepper, O.M.; Pelo, C.R.; Bhatt, K.A.; Callaghan, M.; Bastidas, N.; Bunting, S.; Steinmetz, H.G.; Gurtner, G.C. Topical vascular endothelial growth factor accelerates diabetic wound healing through increased angiogenesis and by mobilizing and recruiting bone marrow-derived cells. Am. J. Pathol. 2004, 164, 1935–1947. [Google Scholar] [CrossRef] [Green Version]
- Mace, K.A.; Yu, D.H.; Paydar, K.Z.; Boudreau, N.; Young, D.M. Sustained expression of Hif-1alpha in the diabetic environment promotes angiogenesis and cutaneous wound repair. Wound Repair Regen. 2007, 15, 636–645. [Google Scholar] [CrossRef]
- Brem, H.; Kodra, A.; Golinko, M.S.; Entero, H.; Stojadinovic, O.; Wang, V.M.; Sheahan, C.M.; Weinberg, A.D.; Woo, S.L.; Ehrlich, H.P.; et al. Mechanism of sustained release of vascular endothelial growth factor in accelerating experimental diabetic healing. J. Investig. Dermatol. 2009, 129, 2275–2287. [Google Scholar] [CrossRef] [Green Version]
- Genentech, Inc. A Study to Assess the Effect of Topical Recombinant Human Vascular Endothelial Growth Factor for Induction of Healing of Diabetic Foot Ulcers; Genentech, Inc.: San Francisco, CA, USA, 2006; NCT00351767. [Google Scholar]
- Song, H.; Xu, Y.; Chang, W.; Zhuang, J.; Wu, X. Negative pressure wound therapy promotes wound healing by suppressing macrophage inflammation in diabetic ulcers. Regen. Med. 2020, 15, 2341–2349. [Google Scholar] [CrossRef]
- Ministry of Health and Wellnes. Negative Pressure Wound Therapy in Diabetic Wounds; University of Mauritius: Moka, Mauritius, 2021; NCT05041244. [Google Scholar]
- Pan, S.; Zhang, S.; Chen, H. Low temperature plasma promotes the healing of chronic wounds in diabetic mice. J. Phys. D Appl. Phys. 2020, 53, 185205. [Google Scholar] [CrossRef]
- He, R.; Li, Q.; Shen, W.; Wang, T.; Lu, H.; Lu, J.; Lu, F.; Luo, M.; Zhang, J.; Gao, H.; et al. The efficacy and safety of cold atmospheric plasma as a novel therapy for diabetic wound in vitro and in vivo. Int. Wound J. 2020, 17, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Tschoepe, D. Cold Plasma Therapy for Acceleration of Wound Healing in Diabetic Foot; Ruhr University of Bochum: Bochum, Germany, 2019; NCT04205942. [Google Scholar]
- Wei, Q.; Zhang, Z.; Luo, J.; Kong, J.; Ding, Y.; Chen, Y.; Wang, K. Insulin treatment enhances pseudomonas aeruginosa biofilm formation by increasing intracellular cyclic di-GMP levels, leading to chronic wound infection and delayed wound healing. Am. J. Transl. Res. 2019, 11, 3261–3279. [Google Scholar]
- University of Campinas. Topic Insulin Accelerates Wound Healing in Diabetes; University of Campinas: Sao Paolo, Brazil, 2011; NCT01295177. [Google Scholar]
- Roberts, R.E.; Bilgen, O.; Kineman, R.D.; Koh, T.J. Parameter-Dependency of Low-Intensity Vibration for Wound Healing in Diabetic Mice. Front. Bioeng. Biotechnol. 2021, 9, 654920. [Google Scholar] [CrossRef]
- King, S.K.K. Vibration Enhances Diabetic ULCER Healing; Chinese University of Hong Kong: Hong Kong, China, 2020; NCT04275804. [Google Scholar]
- Gonzalez, K.; Salem, K.M.; Methé, B.; Li, K.; Hong, G.; Tzeng, E. Impact of Dietary Tungsten and Topical Nitrite in Diabetic Wound Healing and the Composition of the Wound Microbiome. JVS-Vasc. Sci. 2020, 1, 257. [Google Scholar] [CrossRef]
- Sindler, A.L.; Cox-York, K.; Reese, L.; Bryan, N.S.; Seals, D.R.; Gentile, C.L. Oral nitrite therapy improves vascular function in diabetic mice. Diab. Vasc. Dis. Res. 2015, 12, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, R.; Finnen, M.J. The effects of topical treatment with acidified nitrite on wound healing in normal and diabetic mice. Nitric Oxide 2006, 15, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Fundación Cardiovascular de Colombia. Clinical Trial for the Treatment of Diabetic Foot Ulcers Using a Nitric Oxide Releasing Patch: PATHON; Fundación Cardiovascular de Colombia: Floridablanca, Colombia, 2007; NCT00428727. [Google Scholar]
- Rai, V.; Moellmer, R.; Agrawal, D.K. Clinically relevant experimental rodent models of diabetic foot ulcer. Mol. Cell Biochem. 2022, 477, 1239–1247. [Google Scholar] [CrossRef]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.O.; Leung, K.S.; Fung, K.P.; Lam, F.F.; Ng, E.S.; Lau, K.M.; Chow, S.K.; Cheung, W.H. The characterization of a full-thickness excision open foot wound model in n5-streptozotocin (STZ)-induced type 2 diabetic rats that mimics diabetic foot ulcer in terms of reduced blood circulation, higher C-reactive protein, elevated inflammation, and reduced cell proliferation. Exp. Anim. 2017, 66, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Nord, C.; Eriksson, M.; Dicker, A.; Eriksson, A.; Grong, E.; Ilegems, E.; Marvik, R.; Kulseng, B.; Berggren, P.O.; Gorzsas, A.; et al. Biochemical profiling of diabetes disease progression by multivariate vibrational microspectroscopy of the pancreas. Sci. Rep. 2017, 7, 6646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomer, H.D.; Trentin, A.G. Skin wound healing in humans and mice: Challenges in translational research. J. Dermatol. Sci. 2018, 90, 3–12. [Google Scholar] [CrossRef] [Green Version]

| Animal Model | Modification | Ref. |
|---|---|---|
| db/db (BKS) 11 weeks old, females | Pseudomonas aeruginosa biofilm challenge | [97,98] |
| db/db (C57BL/6)/db/db (BKS) 5–6 months old/db/db 11 weeks old, males | Increasing oxidative stress via blocking catalase and glutathione peroxidase | [100,101,102] |
| db/db (BKS) 12 weeks old, males | Wound splinting to prevent contraction | [103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stachura, A.; Khanna, I.; Krysiak, P.; Paskal, W.; Włodarski, P. Wound Healing Impairment in Type 2 Diabetes Model of Leptin-Deficient Mice—A Mechanistic Systematic Review. Int. J. Mol. Sci. 2022, 23, 8621. https://doi.org/10.3390/ijms23158621
Stachura A, Khanna I, Krysiak P, Paskal W, Włodarski P. Wound Healing Impairment in Type 2 Diabetes Model of Leptin-Deficient Mice—A Mechanistic Systematic Review. International Journal of Molecular Sciences. 2022; 23(15):8621. https://doi.org/10.3390/ijms23158621
Chicago/Turabian StyleStachura, Albert, Ishani Khanna, Piotr Krysiak, Wiktor Paskal, and Paweł Włodarski. 2022. "Wound Healing Impairment in Type 2 Diabetes Model of Leptin-Deficient Mice—A Mechanistic Systematic Review" International Journal of Molecular Sciences 23, no. 15: 8621. https://doi.org/10.3390/ijms23158621