
Platelet Derivatives and the Immunomodulation of Wound Healing

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Innate and Adaptive Immune Cells in Wound Healing
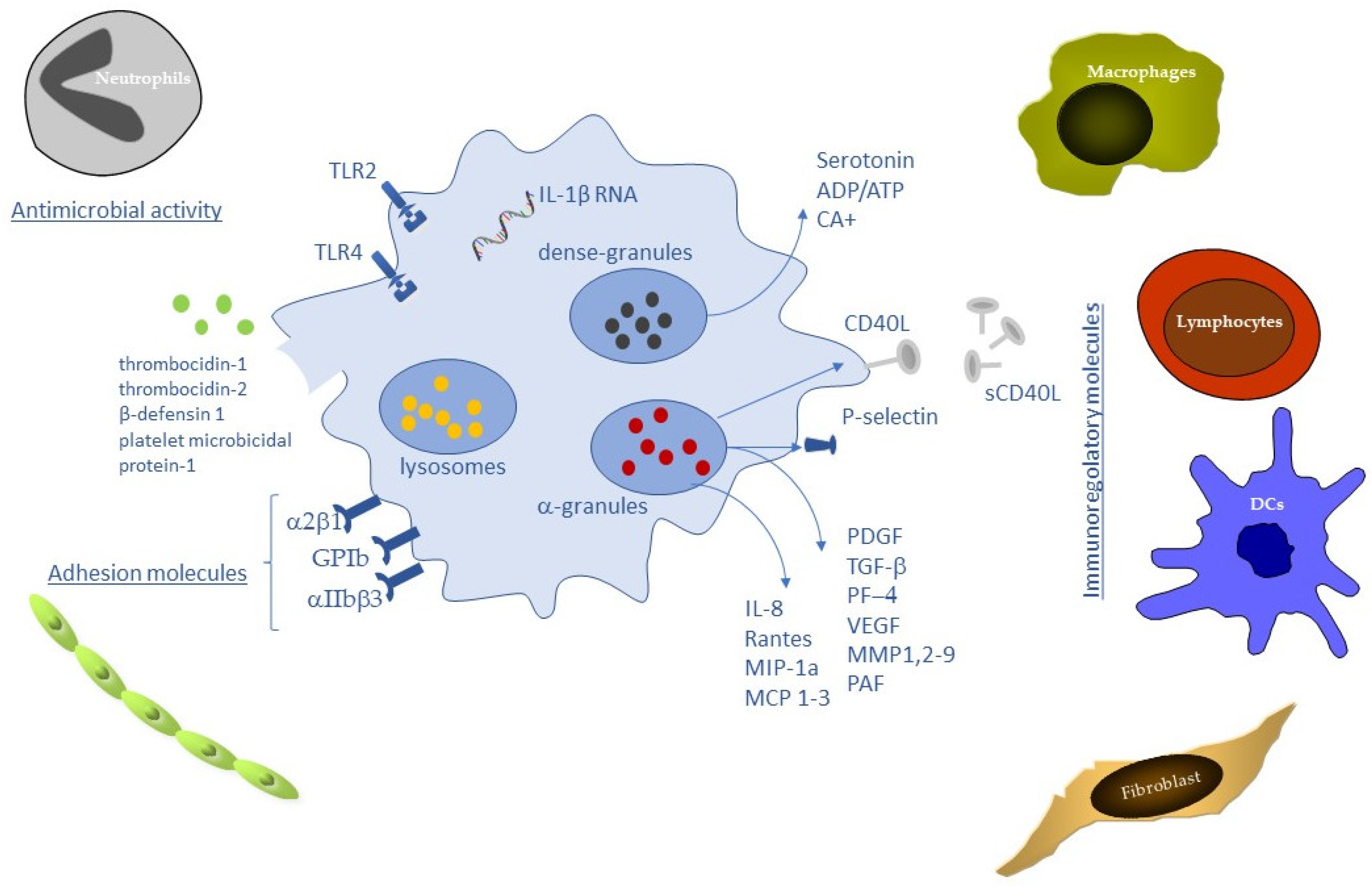
3. Platelet and the Immune Regulation of Wound Healing
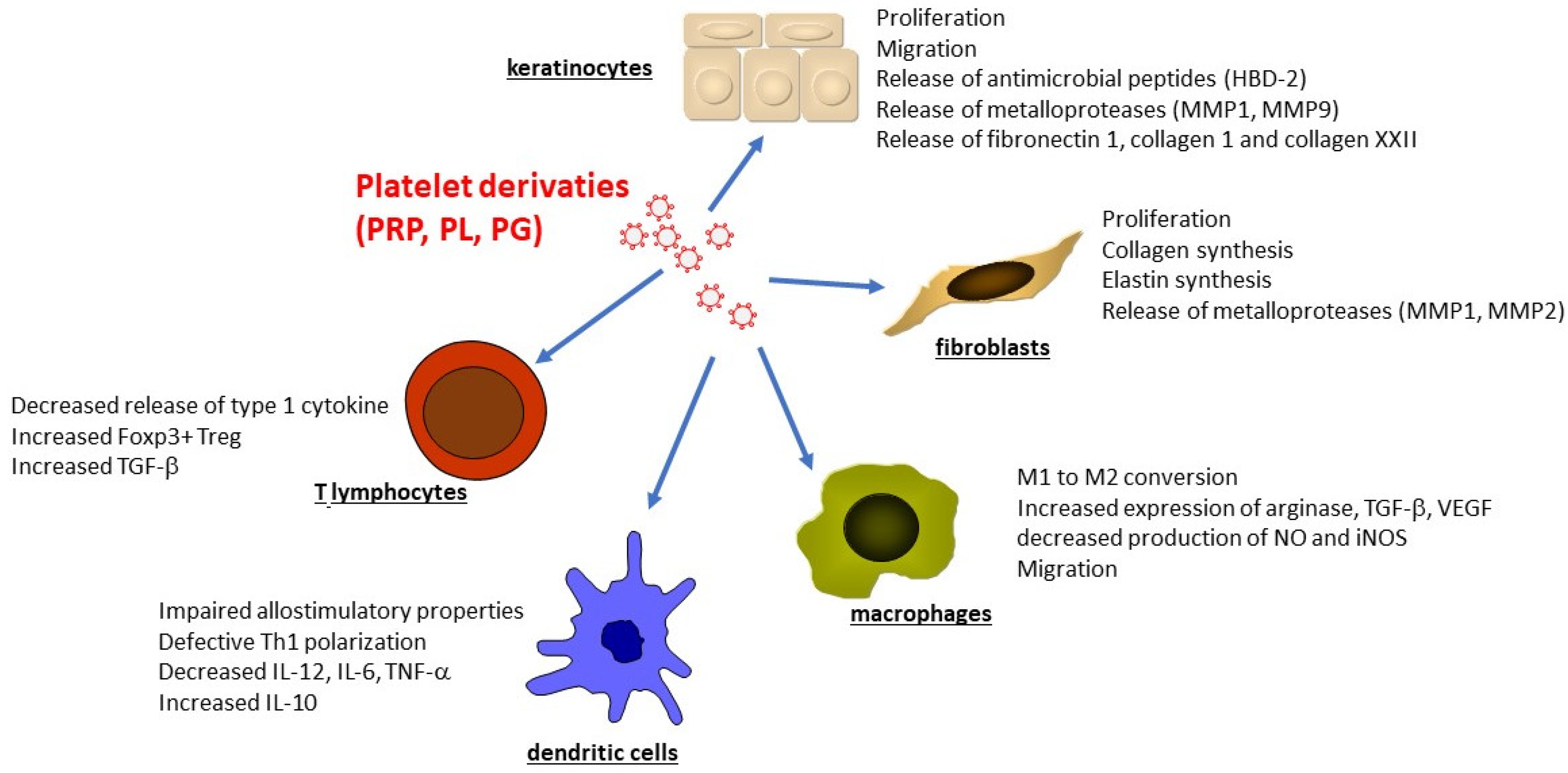
4. Platelet Derivates in Wound Healing
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound Repair and Regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Diegelmann, R.F. Wound Healing: An Overview of Acute, Fibrotic and Delayed Healing. Front. Biosci. 2004, 9, 283. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Ceilley, R. Chronic Wound Healing: A Review of Current Management and Treatments. Adv. Ther. 2017, 34, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janis, J.E.; Harrison, B. Wound Healing: Part I. Basic Science. Plast. Reconstr. Surg. 2014, 133, 199e–207e. [Google Scholar] [CrossRef]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound Healing: A Cellular Perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Mancuso, M.E.; Santagostino, E. Platelets: Much More than Bricks in a Breached Wall. Br. J. Haematol. 2017, 178, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Strbo, N.; Yin, N.; Stojadinovic, O. Innate and Adaptive Immune Responses in Wound Epithelialization. Adv. Wound Care 2014, 3, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Piipponen, M.; Li, D.; Landén, N.X. The Immune Functions of Keratinocytes in Skin Wound Healing. Int. J. Mol. Sci. 2020, 21, 8790. [Google Scholar] [CrossRef]
- Chen, L.; Guo, S.; Ranzer, M.J.; DiPietro, L.A. Toll-Like Receptor 4 Has an Essential Role in Early Skin Wound Healing. J. Investig. Dermatol. 2013, 133, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Takeda, K. Toll-like Receptor Signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Takeda, K. Toll-like Receptors in Innate Immunity. Int. Immunol. 2004, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, E.P.; Lu, H.; Jacobs, H.L.; Messina, C.G.M.; Bolsover, S.; Gabella, G.; Potma, E.O.; Warley, A.; Roes, J.; Segal, A.W. Killing Activity of Neutrophils Is Mediated through Activation of Proteases by K+ Flux. Nature 2002, 416, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.K.; Vivekanandan-Giri, A.; Tang, C.; Knight, J.S.; Mathew, A.; Padilla, R.L.; Gillespie, B.W.; Carmona-Rivera, C.; Liu, X.; Subramanian, V.; et al. Neutrophil Extracellular Trap-Derived Enzymes Oxidize High-Density Lipoprotein: An Additional Proatherogenic Mechanism in Systemic Lupus Erythematosus: NETs, HDL, and Lupus. Arthritis Rheumatol. 2014, 66, 2532–2544. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Lindbom, L. Phagocyte Partnership during the Onset and Resolution of Inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- Dovi, J.V.; He, L.-K.; DiPietro, L.A. Accelerated Wound Closure in Neutrophil-Depleted Mice. J. Leukoc. Biol. 2003, 73, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.L.; Holmes, G.R.; Bojarczuk, A.N.; Burgon, J.; Loynes, C.A.; Chimen, M.; Sawtell, A.K.; Hamza, B.; Willson, J.; Walmsley, S.R.; et al. A Zebrafish Compound Screen Reveals Modulation of Neutrophil Reverse Migration as an Anti-Inflammatory Mechanism. Sci. Transl. Med. 2014, 6, 225ra29. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Marneros, A.G. Macrophages Are Essential for the Early Wound Healing Response and the Formation of a Fibrovascular Scar. Am. J. Pathol. 2013, 182, 2407–2417. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Nair, M.G. Macrophages in Wound Healing: Activation and Plasticity. Immunol. Cell Biol. 2019, 97, 258–267. [Google Scholar] [CrossRef]
- Slauch, J.M. How Does the Oxidative Burst of Macrophages Kill Bacteria? Still an Open Question: How Do Phagocytic ROS Kill Bacteria? Mol. Microbiol. 2011, 80, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, L. The Impact of the Extracellular Matrix on Inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Müller, W.; Roers, A.; Eming, S.A. Differential Roles of Macrophages in Diverse Phases of Skin Repair. J. Int. 2010, 184, 3964–3977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and Functional Plasticity of Cells of Innate Immunity: Macrophages, Mast Cells and Neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibovich, S.J.; Polverini, P.J.; Shepard, H.M.; Wiseman, D.M.; Shively, V.; Nuseir, N. Macrophage-Induced Angiogenesis Is Mediated by Tumour Necrosis Factor-α. Nature 1987, 329, 630–632. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The Role of Myeloid Cells in the Promotion of Tumour Angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Willenborg, S.; Lucas, T.; van Loo, G.; Knipper, J.A.; Krieg, T.; Haase, I.; Brachvogel, B.; Hammerschmidt, M.; Nagy, A.; Ferrara, N.; et al. CCR2 Recruits an Inflammatory Macrophage Subpopulation Critical for Angiogenesis in Tissue Repair. Blood 2012, 120, 613–625. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, C.J.; Leibovich, S.J. Regulation of Macrophage Polarization and Wound Healing. Adv. Wound Care 2012, 1, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Boniakowski, A.E.; Kimball, A.S.; Jacobs, B.N.; Kunkel, S.L.; Gallagher, K.A. Macrophage-Mediated Inflammation in Normal and Diabetic Wound Healing. J. Immunol. 2017, 199, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Delavary, B.M.; van der Veer, W.M.; van Egmond, M.; Niessen, F.B.; Beelen, R.H.J. Macrophages in Skin Injury and Repair. Immunobiology 2011, 216, 753–762. [Google Scholar] [CrossRef]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glim, J.E.; Niessen, F.B.; Everts, V.; van Egmond, M.; Beelen, R.H.J. Platelet Derived Growth Factor-CC Secreted by M2 Macrophages Induces Alpha-Smooth Muscle Actin Expression by Dermal and Gingival Fibroblasts. Immunobiology 2013, 218, 924–929. [Google Scholar] [CrossRef]
- Wong, V.W.; Paterno, J.; Sorkin, M.; Glotzbach, J.P.; Levi, K.; Januszyk, M.; Rustad, K.C.; Longaker, M.T.; Gurtner, G.C. Mechanical Force Prolongs Acute Inflammation via T-cell-dependent Pathways during Scar Formation. FASEB J. 2011, 25, 4498–4510. [Google Scholar] [CrossRef]
- Satoh, T.; Nakagawa, K.; Sugihara, F.; Kuwahara, R.; Ashihara, M.; Yamane, F.; Minowa, Y.; Fukushima, K.; Ebina, I.; Yoshioka, Y.; et al. Identification of an Atypical Monocyte and Committed Progenitor Involved in Fibrosis. Nature 2017, 541, 96–101. [Google Scholar] [CrossRef]
- Toulon, A.; Breton, L.; Taylor, K.R.; Tenenhaus, M.; Bhavsar, D.; Lanigan, C.; Rudolph, R.; Jameson, J.; Havran, W.L. A Role for Human Skin–Resident T Cells in Wound Healing. J. Exp. Med. 2009, 206, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 Cells Represent a Distinct Human T Cell Subset Involved in Epidermal Immunity and Remodeling. J. Clin. Investig. 2009, 119, 3573–3585. [Google Scholar] [CrossRef] [Green Version]
- Avitabile, S.; Odorisio, T.; Madonna, S.; Eyerich, S.; Guerra, L.; Eyerich, K.; Zambruno, G.; Cavani, A.; Cianfarani, F. Interleukin-22 Promotes Wound Repair in Diabetes by Improving Keratinocyte Pro-Healing Functions. J. Investig. Dermatol. 2015, 135, 2862–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosbaum, A.; Prevel, N.; Truong, H.-A.; Mehta, P.; Ettinger, M.; Scharschmidt, T.C.; Ali, N.H.; Pauli, M.L.; Abbas, A.K.; Rosenblum, M.D. Cutting Edge: Regulatory T Cells Facilitate Cutaneous Wound Healing. J. Immunol. 2016, 196, 2010–2014. [Google Scholar] [CrossRef]
- Soloff, A.C.; Barratt-Boyes, S.M. Enemy at the Gates: Dendritic Cells and Immunity to Mucosal Pathogens. Cell Res. 2010, 20, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Saxena, M.; Bhardwaj, N. Dendritic Cell Subsets and Locations. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 348, pp. 1–68. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, Y.; Chen, L.; Xie, J.; Tang, J.; Zhao, J.; Shu, B.; Qi, S.; Chen, J.; Liang, G.; et al. Dendritic Epidermal T Cells Facilitate Wound Healing in Diabetic Mice. Am. J. Transl. Res. 2016, 8, 2375–2384. [Google Scholar] [PubMed]
- Keyes, B.E.; Liu, S.; Asare, A.; Naik, S.; Levorse, J.; Polak, L.; Lu, C.P.; Nikolova, M.; Pasolli, H.A.; Fuchs, E. Impaired Epidermal to Dendritic T Cell Signaling Slows Wound Repair in Aged Skin. Cell 2016, 167, 1323–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojadinovic, O.; Yin, N.; Lehmann, J.; Pastar, I.; Kirsner, R.S.; Tomic-Canic, M. Increased Number of Langerhans Cells in the Epidermis of Diabetic Foot Ulcers Correlates with Healing Outcome. Immunol. Res. 2013, 57, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Lamb, R.; Coles, M.C.; Bennett, C.L.; Ambler, C.A. Inducible Ablation of CD11c + Cells to Determine Their Role in Skin Wound Repair. Immunology 2021, 163, 105–111. [Google Scholar] [CrossRef]
- Gao, N.; Yin, J.; Yoon, G.S.; Mi, Q.-S.; Yu, F.-S.X. Dendritic Cell–Epithelium Interplay Is a Determinant Factor for Corneal Epithelial Wound Repair. Am. J. Pathol. 2011, 179, 2243–2253. [Google Scholar] [CrossRef]
- Giles, C. The Platelet Count and Mean Platelet Volume. Br. J. Haematol. 2008, 48, 31–37. [Google Scholar] [CrossRef]
- Savage, B.; Saldívar, E.; Ruggeri, Z.M. Initiation of Platelet Adhesion by Arrest onto Fibrinogen or Translocation on von Willebrand Factor. Cell 1996, 84, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The Final Steps of Integrin Activation: The End Game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Konstantinides, S.; Ware, J.; Marchese, P.; Almus-Jacobs, F.; Loskutoff, D.J.; Ruggeri, Z.M. Distinct Antithrombotic Consequences of Platelet Glycoprotein Ibα and VI Deficiency in a Mouse Model of Arterial Thrombosis. J. Thromb. Haemost. 2006, 4, 2014–2021. [Google Scholar] [CrossRef]
- Garraud, O.; Hamzeh-Cognasse, H.; Pozzetto, B.; Cavaillon, J.-M.; Cognasse, F. Bench-to-Bedside Review: Platelets and Active Immune Functions–New Clues for Immunopathology? Crit. Care 2013, 17, 236. [Google Scholar] [CrossRef] [Green Version]
- Assinger, A.; Schrottmaier, W.C.; Salzmann, M.; Rayes, J. Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front. Immunol. 2019, 10, 1687. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Koupenova, M.; Vitseva, O.; MacKay, C.R.; Beaulieu, L.M.; Benjamin, E.J.; Mick, E.; Kurt-Jones, E.A.; Ravid, K.; Freedman, J.E. Platelet-TLR7 Mediates Host Survival and Platelet Count during Viral Infection in the Absence of Platelet-Dependent Thrombosis. Blood 2014, 124, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Koessler, J.; Schuepferling, A.; Klingler, P.; Koessler, A.; Weber, K.; Boeck, M.; Kobsar, A. The Role of Proteasome Activity for Activating and Inhibitory Signalling in Human Platelets. Cell. Signal. 2019, 62, 109351. [Google Scholar] [CrossRef]
- Damien, P.; Cognasse, F.; Eyraud, M.-A.; Arthaud, C.-A.; Pozzetto, B.; Garraud, O.; Hamzeh-Cognasse, H. LPS Stimulation of Purified Human Platelets Is Partly Dependent on Plasma Soluble CD14 to Secrete Their Main Secreted Product, Soluble-CD40-Ligand. BMC Immunol. 2015, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Yeaman, M.R. Platelets: At the Nexus of Antimicrobial Defence. Nat. Rev. Microbiol. 2014, 12, 426–437. [Google Scholar] [CrossRef]
- Larsen, E.; Celi, A.; Gilbert, G.E.; Furie, B.C.; Erban, J.K.; Bonfanti, R.; Wagner, D.D.; Furie, B. PADGEM Protein: A Receptor That Mediates the Interaction of Activated Platelets with Neutrophils and Monocytes. Cell 1989, 59, 305–312. [Google Scholar] [CrossRef]
- De Luca, M.; Dunlop, L.C.; Andrews, R.K.; Flannery, J.V.; Ettling, R.; Cumming, D.A.; Veldman, G.M.; Berndt, M.C. A Novel Cobra Venom Metalloproteinase, Mocarhagin, Cleaves a 10-Amino Acid Peptide from the Mature N Terminus of P-Selectin Glycoprotein Ligand Receptor, PSGL-1, and Abolishes P-Selectin Binding. J. Biol. Chem. 1995, 270, 26734–26737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyrich, A.S.; Elstad, M.R.; McEver, R.P.; McIntyre, T.M.; Moore, K.L.; Morrissey, J.H.; Prescott, S.M.; Zimmerman, G.A. Activated Platelets Signal Chemokine Synthesis by Human Monocytes. J. Clin. Investig. 1996, 97, 1525–1534. [Google Scholar] [CrossRef] [Green Version]
- Deuel, T.F.; Senior, R.M.; Chang, D.; Griffin, G.L.; Heinrikson, R.L.; Kaiser, E.T. Platelet Factor 4 Is Chemotactic for Neutrophils and Monocytes. Proc. Natl. Acad. Sci. USA 1981, 78, 4584–4587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheuerer, B.; Ernst, M.; Dürrbaum-Landmann, I.; Fleischer, J.; Grage-Griebenow, E.; Brandt, E.; Flad, H.D.; Petersen, F. The CXC-Chemokine Platelet Factor 4 Promotes Monocyte Survival and Induces Monocyte Differentiation into Macrophages. Blood 2000, 95, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Mezger, M.; Nording, H.; Sauter, R.; Graf, T.; Heim, C.; von Bubnoff, N.; Ensminger, S.M.; Langer, H.F. Platelets and Immune Responses During Thromboinflammation. Front. Immunol. 2019, 10, 1731. [Google Scholar] [CrossRef] [PubMed]
- Seizer, P.; May, A.E. Platelets and Matrix Metalloproteinases. Thromb. Haemost. 2013, 110, 903–909. [Google Scholar] [CrossRef] [Green Version]
- Blumberg, N.; Spinelli, S.L.; Francis, C.W.; Taubman, M.B.; Phipps, R.P. The Platelet as an Immune Cell—CD40 Ligand and Transfusion Immunomodulation. Immunol. Res. 2009, 45, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Dewitte, A.; Tanga, A.; Villeneuve, J.; Lepreux, S.; Ouattara, A.; Desmoulière, A.; Combe, C.; Ripoche, J. New Frontiers for Platelet CD154. Exp. Hematol. Oncol. 2015, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Henn, V.; Slupsky, J.R.; Gräfe, M.; Anagnostopoulos, I.; Förster, R.; Müller-Berghaus, G.; Kroczek, R.A. CD40 Ligand on Activated Platelets Triggers an Inflammatory Reaction of Endothelial Cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef]
- Jin, Y.; Nonoyama, S.; Morio, T.; Imai, K.; Ochs, H.D.; Mizutani, S. Characterization of Soluble CD40 Ligand Released from Human Activated Platelets. J. Med. Dent. Sci. 2001, 48, 23–27. [Google Scholar]
- Lindmark, E.; Tenno, T.; Siegbahn, A. Role of Platelet P-Selectin and CD40 Ligand in the Induction of Monocytic Tissue Factor Expression. Arter. Thromb. Vasc. Biol. 2000, 20, 2322–2328. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, R.I.; Reichenbach, F.; Kraft, P.; Kumar, A.; Lescan, M.; Todt, F.; Göbel, K.; Hilgendorf, I.; Geisler, T.; Bauer, A.; et al. Platelets Induce Apoptosis via Membrane-Bound FasL. Blood 2015, 126, 1483–1493. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.T.; McIntyre, T.M. Lipopolysaccharide Signaling without a Nucleus: Kinase Cascades Stimulate Platelet Shedding of Proinflammatory IL-1β–Rich Microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef] [PubMed]
- Puhlmann, M.; Weinreich, D.M.; Farma, J.M.; Carroll, N.M.; Turner, E.M.; Alexander, H.R. Interleukin-1β Induced Vascular Permeability Is Dependent on Induction of Endothelial Tissue Factor (TF) Activity. J. Transl. Med. 2005, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Kasper, B.; Brandt, E.; Brandau, S.; Petersen, F. Platelet Factor 4 (CXC Chemokine Ligand 4) Differentially Regulates Respiratory Burst, Survival, and Cytokine Expression of Human Monocytes by Using Distinct Signaling Pathways. J. Immunol. 2007, 179, 2584–2591. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-ΚB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Passacquale, G.; Vamadevan, P.; Pereira, L.; Hamid, C.; Corrigall, V.; Ferro, A. Monocyte-Platelet Interaction Induces a Pro-Inflammatory Phenotype in Circulating Monocytes. PLoS ONE 2011, 6, e25595. [Google Scholar] [CrossRef] [Green Version]
- Scull, C.M.; Hays, W.D.; Fischer, T.H. Macrophage Pro-Inflammatory Cytokine Secretion Is Enhanced Following Interaction with Autologous Platelets. J. Inflamm. 2010, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carestia, A.; Mena, H.A.; Olexen, C.M.; Ortiz Wilczyñski, J.M.; Negrotto, S.; Errasti, A.E.; Gómez, R.M.; Jenne, C.N.; Carrera Silva, E.A.; Schattner, M. Platelets Promote Macrophage Polarization toward Pro-Inflammatory Phenotype and Increase Survival of Septic Mice. Cell Rep. 2019, 28, 896–908. [Google Scholar] [CrossRef] [Green Version]
- Gudbrandsdottir, S.; Hasselbalch, H.C.; Nielsen, C.H. Activated Platelets Enhance IL-10 Secretion and Reduce TNF-α Secretion by Monocytes. J. Immunol. 2013, 191, 4059–4067. [Google Scholar] [CrossRef] [Green Version]
- Linke, B.; Schreiber, Y.; Picard-Willems, B.; Slattery, P.; Nüsing, R.M.; Harder, S.; Geisslinger, G.; Scholich, K. Activated Platelets Induce an Anti-Inflammatory Response of Monocytes/Macrophages through Cross-Regulation of PGE2 and Cytokines. Mediat. Inflamm. 2017, 2017, 1463216. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Pan, K.; Chen, L.; Jin, K. Platelet Derived Vesicles Enhance the TGF-Beta Signaling Pathway of M1 Macrophage. Front. Endocrinol. 2022, 13, 868893. [Google Scholar] [CrossRef]
- Lax, S.; Rayes, J.; Wichaiyo, S.; Haining, E.J.; Lowe, K.; Grygielska, B.; Laloo, R.; Flodby, P.; Borok, Z.; Crandall, E.D.; et al. Platelet CLEC-2 Protects against Lung Injury via Effects of Its Ligand Podoplanin on Inflammatory Alveolar Macrophages in the Mouse. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 313, L1016–L1029. [Google Scholar] [CrossRef] [PubMed]
- Kissel, K.; Berber, S.; Nockher, A.; Santoso, S.; Bein, G.; Hackstein, H. Human Platelets Target Dendritic Cell Differentiation and Production of Proinflammatory Cytokines. Transfusion 2006, 46, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Saris, A.; Steuten, J.; Schrijver, D.P.; van Schijndel, G.; Zwaginga, J.J.; van Ham, S.M.; ten Brinke, A. Inhibition of Dendritic Cell Activation and Modulation of T Cell Polarization by the Platelet Secretome. Front. Immunol. 2021, 12, 631285. [Google Scholar] [CrossRef]
- Zamora, C.; Cantó, E.; Nieto, J.C.; Ortiz, M.A.; Diaz-Torné, C.; Diaz-Lopez, C.; Llobet, J.M.; Juarez, C.; Vidal, S. Functional Consequences of Platelet Binding to T Lymphocytes in Inflammation. J. Leukoc. Biol. 2013, 94, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Kühne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed Transport of Neutrophil-Derived Extracellular Vesicles Enables Platelet-Mediated Innate Immune Response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Deryckx, S.; Wang, J.-L.; Valentin, H.; Peronne, C.; de Vries, J.E.; Bernard, A.; Benveniste, J.; Thomas, Y. Immunoregulatory Functions of Paf-Acether. VI. Inhibition of T Cell Activation via CD3 and Potentiation of T Cell Activation via CD2. Int. Immunol. 1990, 2, 545–553. [Google Scholar] [CrossRef]
- Fleischer, J.; Grage-Griebenow, E.; Kasper, B.; Heine, H.; Ernst, M.; Brandt, E.; Flad, H.-D.; Petersen, F. Platelet Factor 4 Inhibits Proliferation and Cytokine Release of Activated Human T Cells. J. Immunol. 2002, 169, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Bacon, K.B.; Premack, B.A.; Gardner, P.; Schall, T.J. Activation of Dual T Cell Signaling Pathways by the Chemokine RANTES. Science 1995, 269, 1727–1730. [Google Scholar] [CrossRef]
- Acres, R.B.; Lamb, J.R.; Feldman, M. Effects of Platelet-Derived Growth Factor and Epidermal Growth Factor on Antigen-Induced Proliferation of Human T-Cell Lines. Immunology 1985, 54, 9–16. [Google Scholar]
- Elzey, B.D.; Schmidt, N.W.; Crist, S.A.; Kresowik, T.P.; Harty, J.T.; Nieswandt, B.; Ratliff, T.L. Platelet-Derived CD154 Enables T-Cell Priming and Protection against Listeria Monocytogenes Challenge. Blood 2008, 111, 3684–3691. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, N.; Zhu, L.; Ersoy, M.; Hermansson, A.; Hjemdahl, P.; Hu, H.; Hansson, G.K.; Li, N. Platelets Regulate CD4+ T-Cell Differentiation via Multiple Chemokines in Humans. Thromb. Haemost. 2011, 106, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Zamora, C.; Cantó, E.; Nieto, J.C.; Bardina, J.; Diaz-Torné, C.; Moya, P.; Magallares, B.; Ortiz, M.A.; Julià, G.; Juarez, C.; et al. Binding of Platelets to Lymphocytes: A Potential Anti-Inflammatory Therapy in Rheumatoid Arthritis. J. Immunol. 2017, 198, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Battaglia, M.; Lee, S.H.; Sun, Q.-H.; Aster, R.H.; Visentin, G.P. Platelet Factor 4 Differentially Modulates CD4+ CD25+ (Regulatory) versus CD4+ CD25− (Nonregulatory) T Cells. J. Immunol. 2005, 174, 2680–2686. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Huang, Z.; Stålesen, R.; Hansson, G.K.; Li, N. Platelets Provoke Distinct Dynamics of Immune Responses by Differentially Regulating CD4 + T-Cell Proliferation. J. Thromb. Haemost. 2014, 12, 1156–1165. [Google Scholar] [CrossRef]
- Gawaz, M.; Vogel, S. Platelets in Tissue Repair: Control of Apoptosis and Interactions with Regenerative Cells. Blood 2013, 122, 2550–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerletti, C.; De Gaetano, G.; Lorenzet, R. Platelet-Leukocyte Interactions: Multiple Links between Inflammation, Blood Coagulation and Vascular Risk. Mediterr. J. Hematol. Infect. Dis. 2010, 2, e2010023. [Google Scholar] [CrossRef]
- Suthar, M.; Gupta, S.; Bukhari, S.; Ponemone, V. Treatment of Chronic Non-Healing Ulcers Using Autologous Platelet Rich Plasma: A Case Series. J. Biomed. Sci. 2017, 24, 16. [Google Scholar] [CrossRef] [Green Version]
- Menchisheva, Y.; Mirzakulova, U.; Yui, R. Use of Platelet-rich Plasma to Facilitate Wound Healing. Int. Wound J. 2019, 16, 343–353. [Google Scholar] [CrossRef]
- Martinez-Zapata, M.J.; Martí-Carvajal, A.J.; Solà, I.; Expósito, J.A.; Bolíbar, I.; Rodríguez, L.; Garcia, J. Autologous Platelet-Rich Plasma for Treating Chronic Wounds. In Cochrane Database of Systematic Reviews; The Cochrane Collaboration, Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2012; p. CD006899. [Google Scholar] [CrossRef]
- Hirase, T.; Ruff, E.; Surani, S.; Ratnani, I. Topical Application of Platelet-Rich Plasma for Diabetic Foot Ulcers: A Systematic Review. World J. Diabetes 2018, 9, 172–179. [Google Scholar] [CrossRef]
- Alves, R.; Grimalt, R. A Review of Platelet-Rich Plasma: History, Biology, Mechanism of Action, and Classification. Skin Appendage Disord. 2018, 4, 18–24. [Google Scholar] [CrossRef]
- Knighton, D.R.; Ciresi, K.F.; Fiegel, V.D.; Austin, L.L.; Butler, E.L. Classification and Treatment of Chronic Nonhealing Wounds. Ann. Surg. 1986, 204, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, A.; El-Said, M.; Emile, S.; Youssef, M.; Khafagy, W.; Elshobaky, A. Randomized Controlled Trial on Autologous Platelet-Rich Plasma Versus Saline Dressing in Treatment of Non-Healing Diabetic Foot Ulcers. World J. Surg. 2020, 44, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Yaghoubi, Y.; Movassaghpour, A.; Shakouri, K.; Mehdizadeh, A.; Pishgahi, A.; Yousefi, M. Novel Therapeutic Approaches in Utilizing Platelet Lysate in Regenerative Medicine: Are We Ready for Clinical Use? J. Cell Physiol. 2019, 234, 17172–17186. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-H.; Lo, W.-C.; Hsu, W.-C.; Wei, H.-J.; Liu, H.-Y.; Lee, C.-H.; Tina Chen, S.-Y.; Shieh, Y.-H.; Williams, D.F.; Deng, W.-P. Synergistic Anabolic Actions of Hyaluronic Acid and Platelet-Rich Plasma on Cartilage Regeneration in Osteoarthritis Therapy. Biomaterials 2014, 35, 9599–9607. [Google Scholar] [CrossRef]
- Klaassen, M.A.; Pietrzak, W.S. Platelet-Rich Plasma Application and Heterotopic Bone Formation Following Total Hip Arthroplasty. J. Investig. Surg. 2011, 24, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Miron, R.J.; Fujioka-Kobayashi, M.; Bishara, M.; Zhang, Y.; Hernandez, M.; Choukroun, J. Platelet-Rich Fibrin and Soft Tissue Wound Healing: A Systematic Review. Tissue Eng. Part B Rev. 2017, 23, 83–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backly, R.E.; Ulivi, V.; Tonachini, L.; Cancedda, R.; Descalzi, F.; Mastrogiacomo, M. Platelet Lysate Induces In Vitro Wound Healing of Human Keratinocytes Associated with a Strong Proinflammatory Response. Tissue Eng. Part A 2011, 17, 1787–1800. [Google Scholar] [CrossRef]
- Burnouf, T.; Goubran, H.A.; Chen, T.-M.; Ou, K.-L.; El-Ekiaby, M.; Radosevic, M. Blood-Derived Biomaterials and Platelet Growth Factors in Regenerative Medicine. Blood Rev. 2013, 27, 77–89. [Google Scholar] [CrossRef]
- Saluja, H.; Dehane, V.; Mahindra, U. Platelet-Rich Fibrin: A Second Generation Platelet Concentrate and a New Friend of Oral and Maxillofacial Surgeons. Ann. Maxillofac. Sur. 2011, 1, 53. [Google Scholar] [CrossRef]
- Chiara Barsotti, M.; Losi, P.; Briganti, E.; Sanguinetti, E.; Magera, A.; Al Kayal, T.; Feriani, R.; Di Stefano, R.; Soldani, G. Effect of Platelet Lysate on Human Cells Involved in Different Phases of Wound Healing. PLoS ONE 2013, 8, e84753. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.L.W.; Trinder, S.; Labelle, M.; Rodriguez-Justo, M.; Hughes, S.; Holmes, A.M.; Scotton, C.J.; Porter, J.C. Platelet-derived Transforming Growth Factor-β1 Promotes Keratinocyte Proliferation in Cutaneous Wound Healing. J. Tissue Eng. Regen. Med. 2020, 14, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Barbul, A. Understanding the Role of Immune Regulation in Wound Healing. Am. J. Surg. 2004, 187, S11–S16. [Google Scholar] [CrossRef]
- Xian, L.J.; Roy Chowdhury, S.; Bin Saim, A.; Bt Hj Idrus, R. Concentration-Dependent Effect of Platelet-Rich Plasma on Keratinocyte and Fibroblast Wound Healing. Cytotherapy 2015, 17, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Tohidnezhad, M.; Lammel, J.; Lippross, S.; Behrendt, P.; Klüter, T.; Pufe, T.; Jahr, H.; Cremer, J.; Rademacher, F.; et al. Platelet-Released Growth Factors Induce Differentiation of Primary Keratinocytes. Mediat. Inflamm. 2017, 2017, 5671615. [Google Scholar] [CrossRef]
- Park, H.-B.; Yang, J.-H.; Chung, K.-H. Characterization of the Cytokine Profile of Platelet Rich Plasma (PRP) and PRP-Induced Cell Proliferation and Migration: Upregulation of Matrix Metalloproteinase-1 and -9 in HaCaT Cells. Korean J. Hematol. 2011, 46, 265. [Google Scholar] [CrossRef] [Green Version]
- Bayer, A.; Lammel, J.; Rademacher, F.; Groß, J.; Siggelkow, M.; Lippross, S.; Klüter, T.; Varoga, D.; Tohidnezhad, M.; Pufe, T.; et al. Platelet-Released Growth Factors Induce the Antimicrobial Peptide Human Beta-Defensin-2 in Primary Keratinocytes. Exp. Dermatol. 2016, 25, 460–465. [Google Scholar] [CrossRef]
- Ranzato, E.; Patrone, M.; Mazzucco, L.; Burlando, B. Platelet Lysate Stimulates Wound Repair of HaCaT Keratinocytes. Br. J. Dermatol. 2008, 159, 537–545. [Google Scholar] [CrossRef]
- Ranzato, E.; Martinotti, S.; Volante, A.; Mazzucco, L.; Burlando, B. Platelet Lysate Modulates MMP-2 and MMP-9 Expression, Matrix Deposition and Cell-to-Matrix Adhesion in Keratinocytes and Fibroblasts: Platelet Lysate and Matrix Metalloproteinases Modulation. Exp. Dermatol. 2011, 20, 308–313. [Google Scholar] [CrossRef]
- Cho, E.B.; Park, G.S.; Park, S.S.; Jang, Y.J.; Kim, K.H.; Kim, K.J.; Park, E.J. Effect of Platelet-Rich Plasma on Proliferation and Migration in Human Dermal Fibroblasts. J. Cosmet. Dermatol. 2019, 18, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.-C.; Liu, X.N.; Zhuan, Z.; Yang, C.-J.; Kim, Y.T.; Lee, G.W.; Choi, K.H.; Kim, K.-O. Leukocyte-Poor Platelet-Rich Plasma-Derived Growth Factors Enhance Human Fibroblast Proliferation In Vitro. Clin. Orthop. Surg. 2018, 10, 240. [Google Scholar] [CrossRef]
- Sovkova, V.; Vocetkova, K.; Hedvičáková, V.; Hefka Blahnová, V.; Buzgo, M.; Amler, E.; Filová, E. Cellular Response to Individual Components of the Platelet Concentrate. Int. J. Mol. Sci. 2021, 22, 4539. [Google Scholar] [CrossRef]
- Roy, S.; Driggs, J.; Elgharably, H.; Biswas, S.; Findley, M.; Khanna, S.; Gnyawali, U.; Bergdall, V.K.; Sen, C.K. Platelet-Rich Fibrin Matrix Improves Wound Angiogenesis via Inducing Endothelial Cell Proliferation: PRFM in Wound Angiogenesis. Wound Repair Regen. 2011, 19, 753–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naldini, A.; Morena, E.; Fimiani, M.; Campoccia, G.; Fossombroni, V.; Carraro, F. The Effects of Autologous Platelet Gel on Inflammatory Cytokine Response in Human Peripheral Blood Mononuclear Cells. Platelets 2008, 19, 268–274. [Google Scholar] [CrossRef]
- Scopelliti, F.; Cattani, C.; Dimartino, V.; Costanzo, G.; Mirisola, C.; Cavani, C. Platelet Lysate Converts M (IFNγ+LPS) Macrophages in CD206+ TGF-β+ Arginase+ M2-like Macrophages That Affect Fibroblast Activity and T Lymphocyte Migration. J. Tissue Eng. Regen. Med. 2021, 15, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Renn, T.-Y.; Kao, Y.-H.; Wang, C.-C.; Burnouf, T. Anti-Inflammatory Effects of Platelet Biomaterials in a Macrophage Cellular Model. Vox Sang. 2015, 109, 138–147. [Google Scholar] [CrossRef]
- Losi, P.; Barsotti, M.C.; Foffa, I.; Buscemi, M.; De Almeida, C.V.; Fabbri, M.; Gabbriellini, S.; Nocchi, F.; Ursino, S.; Urciuoli, P.; et al. In Vitro Human Cord Blood Platelet Lysate Characterisation with Potential Application in Wound Healing. Int. Wound J. 2020, 17, 65–72. [Google Scholar] [CrossRef]
- Nasirzade, J.; Kargarpour, Z.; Hasannia, S.; Strauss, F.J.; Gruber, R. Platelet-rich Fibrin Elicits an Anti-inflammatory Response in Macrophages in Vitro. J. Periodontol. 2020, 91, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Bieback, K. Platelet Lysate as Replacement for Fetal Bovine Serum in Mesenchymal Stromal Cell Cultures. Transfus. Med. Hemother. 2013, 40, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Kakudo, N.; Morimoto, N.; Ma, Y.; Kusumoto, K. Differences between the Proliferative Effects of Human Platelet Lysate and Fetal Bovine Serum on Human Adipose-Derived Stem Cells. Cells 2019, 8, 1218. [Google Scholar] [CrossRef] [Green Version]
- Švajger, U. Human Platelet Lysate Is a Successful Alternative Serum Supplement for Propagation of Monocyte-Derived Dendritic Cells. Cytotherapy 2017, 19, 486–499. [Google Scholar] [CrossRef]
- Date, I.; Koya, T.; Sakamoto, T.; Togi, M.; Kawaguchi, H.; Watanabe, A.; Kato, T.; Shimodaira, S. Interferon-α-Induced Dendritic Cells Generated with Human Platelet Lysate Exhibit Elevated Antigen Presenting Ability to Cytotoxic T Lymphocytes. Vaccines 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Papait, A.; Cancedda, R.; Mastrogiacomo, M.; Poggi, A. Allogeneic Platelet-Rich Plasma Affects Monocyte Differentiation to Dendritic Cells Causing an Anti-Inflammatory Microenvironment, Putatively Fostering Wound Healing: PRP Affects Monocyte Differentiation. J. Tissue Eng. Regen. Med. 2018, 12, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Tešić, N.; Pekle Simonič, I.; Roškar, K.; Rožman, P.; Švajger, U. Dendritic Cells Generated in the Presence of Platelet Lysate Have a Reduced Type 1 Polarization Capacity. Immunol. Investig. 2020, 49, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Scopelliti, F.; Cattani, C.; Dimartino, V.; Scarponi, C.; Madonna, S.; Albanesi, C.; Costanzo, G.; Mirisola, C.; Cavani, A. Platelet Lysate Promotes the Expansion of T Regulatory Cells That Favours in Vitro Wound Healing by Increasing Keratinocyte Migration and Fibroblast Production of Extracellular Matrix Components. Eur. J. Dermatol. 2020, 30, 3–11. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scopelliti, F.; Cattani, C.; Dimartino, V.; Mirisola, C.; Cavani, A. Platelet Derivatives and the Immunomodulation of Wound Healing. Int. J. Mol. Sci. 2022, 23, 8370. https://doi.org/10.3390/ijms23158370
Scopelliti F, Cattani C, Dimartino V, Mirisola C, Cavani A. Platelet Derivatives and the Immunomodulation of Wound Healing. International Journal of Molecular Sciences. 2022; 23(15):8370. https://doi.org/10.3390/ijms23158370
Chicago/Turabian StyleScopelliti, Fernanda, Caterina Cattani, Valentina Dimartino, Concetta Mirisola, and Andrea Cavani. 2022. "Platelet Derivatives and the Immunomodulation of Wound Healing" International Journal of Molecular Sciences 23, no. 15: 8370. https://doi.org/10.3390/ijms23158370
APA StyleScopelliti, F., Cattani, C., Dimartino, V., Mirisola, C., & Cavani, A. (2022). Platelet Derivatives and the Immunomodulation of Wound Healing. International Journal of Molecular Sciences, 23(15), 8370. https://doi.org/10.3390/ijms23158370