
Identification of Novel Inhibitors Targeting SGK1 via Ensemble-Based Virtual Screening Method, Biological Evaluation and Molecular Dynamics Simulation

Abstract
:1. Introduction
2. Results and Discussion
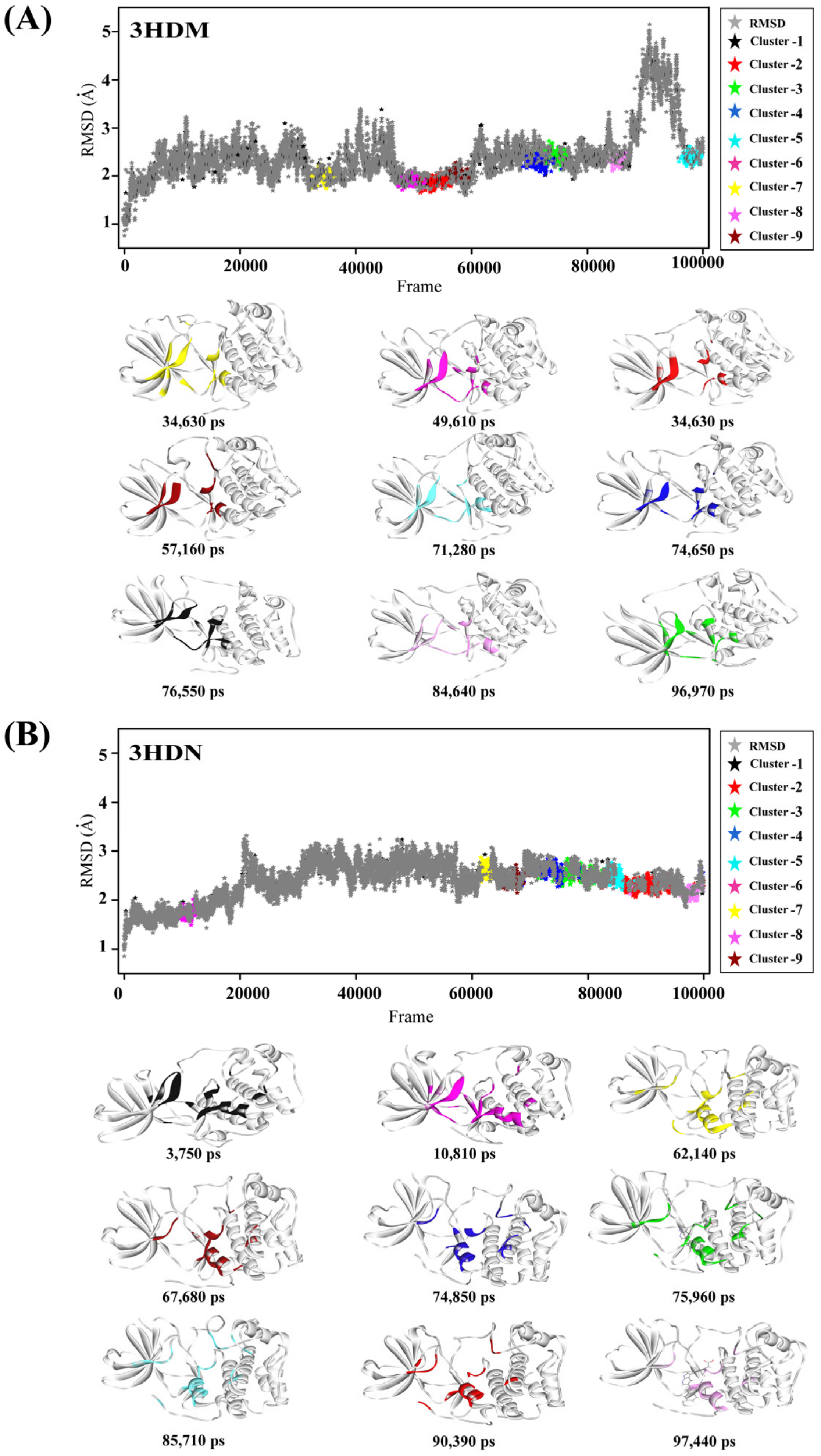
2.1. Representative Protein Conformer Selection Using Molecular Dynamics Simulation
2.2. Common Feature Pharmacophore Modeling and Validation
2.3. Structure-Based Pharmacophore Generation and Validation
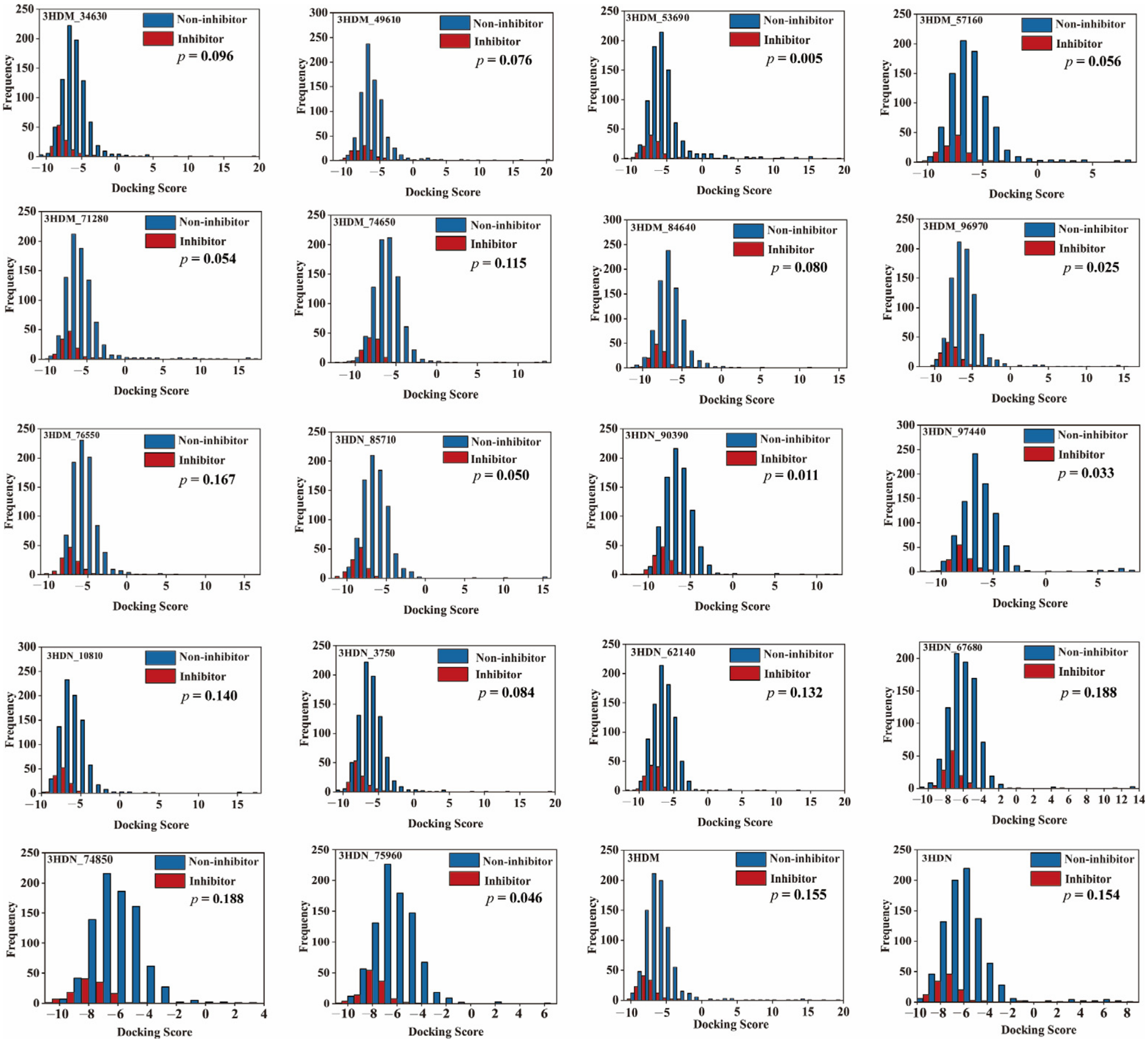
2.4. Molecular Docking Method Selection
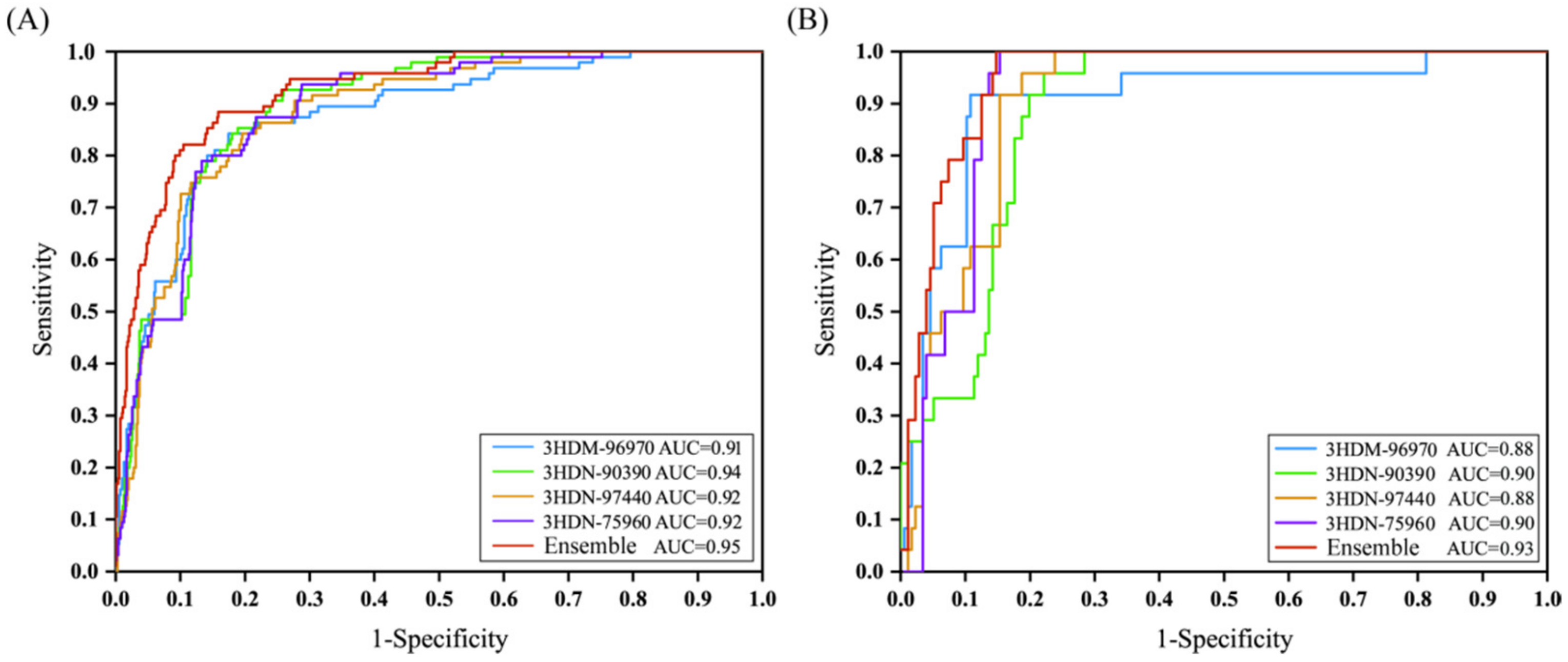
2.5. Performance of Bayesian Models Based on Molecular Docking Score
2.6. Virtual Screening
2.7. Kinase Inhibition Results
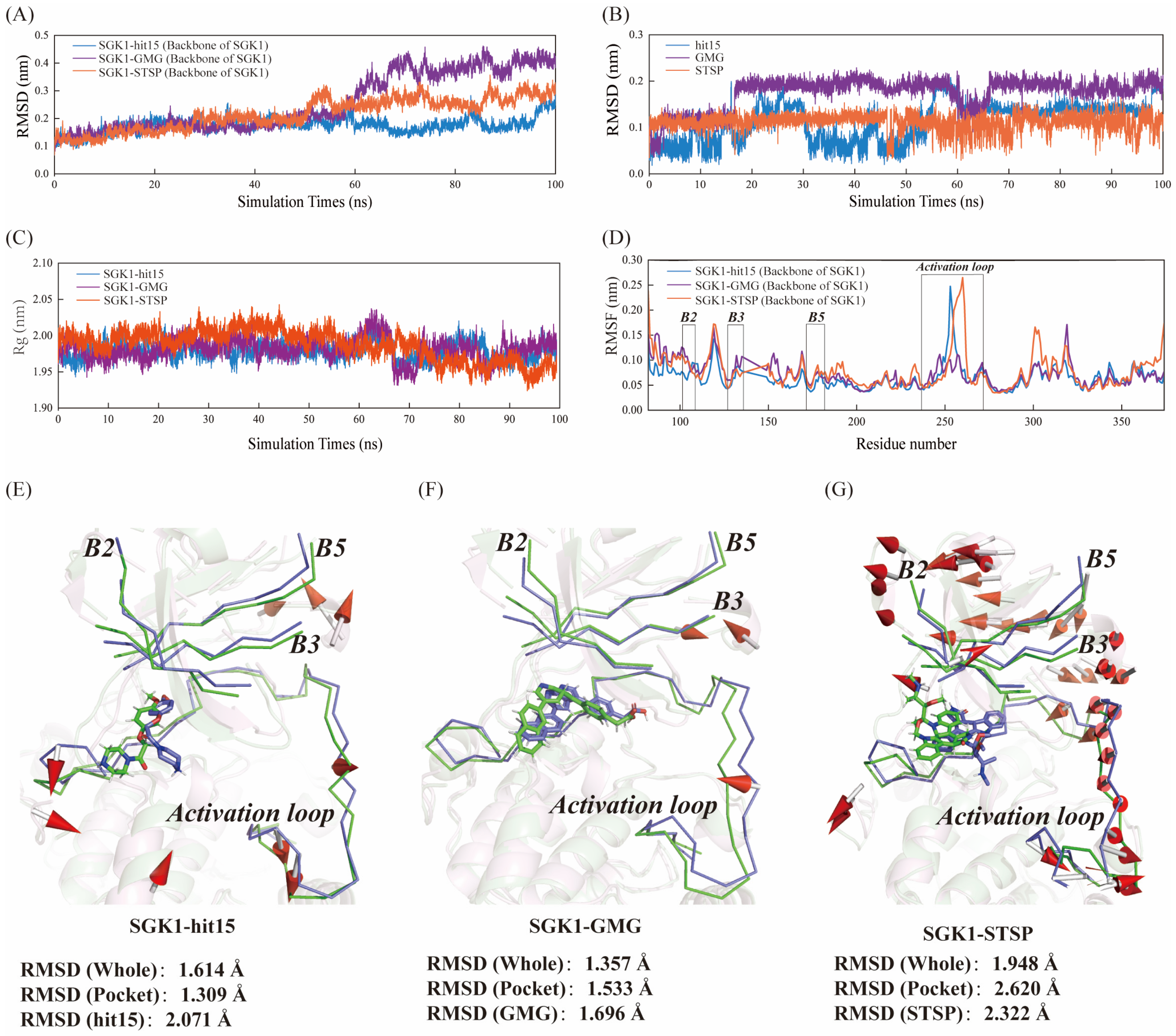
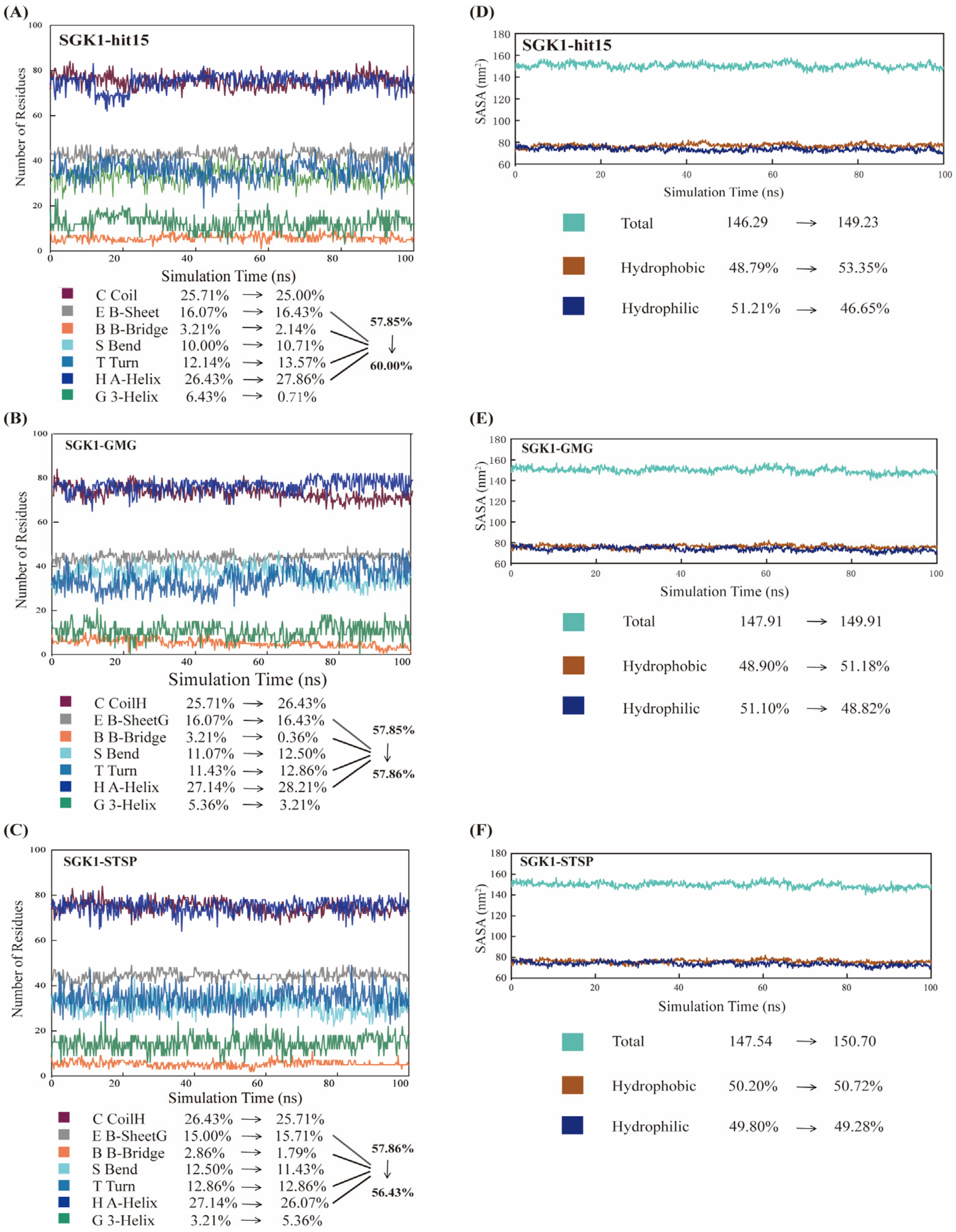
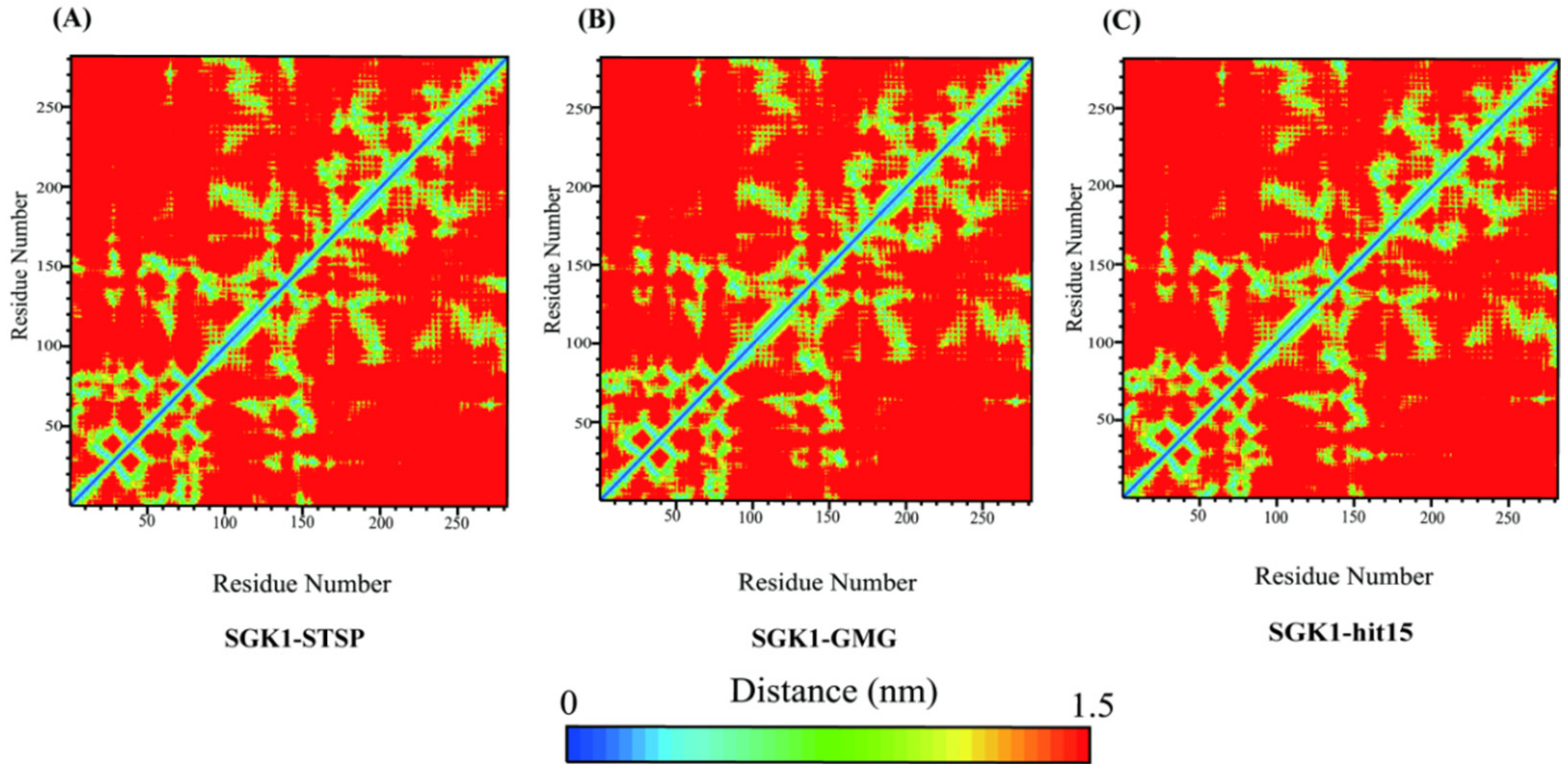
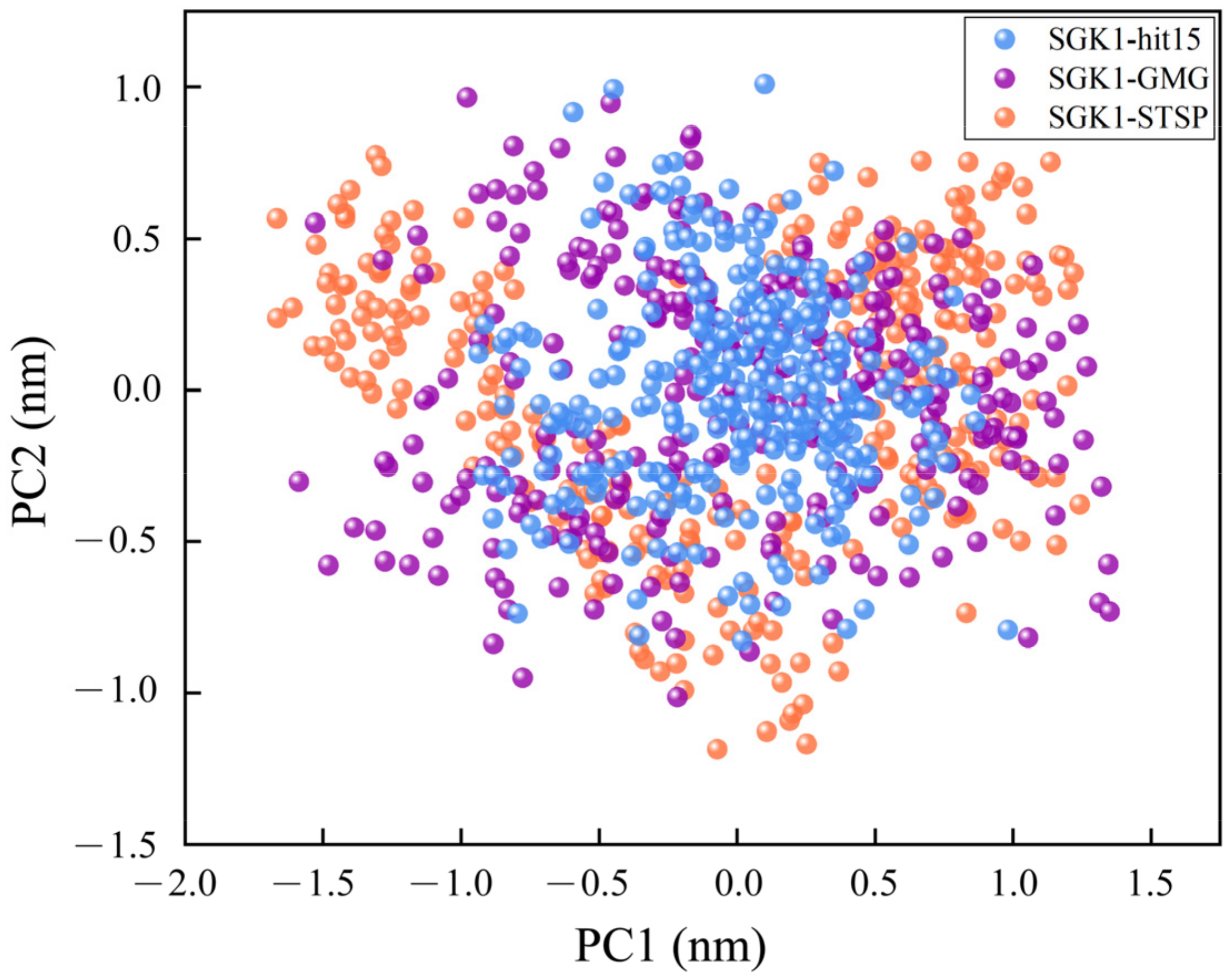
2.8. Molecular Dynamics Simulation and Analysis
2.8.1. Structural Deviations and Compactness Analysis
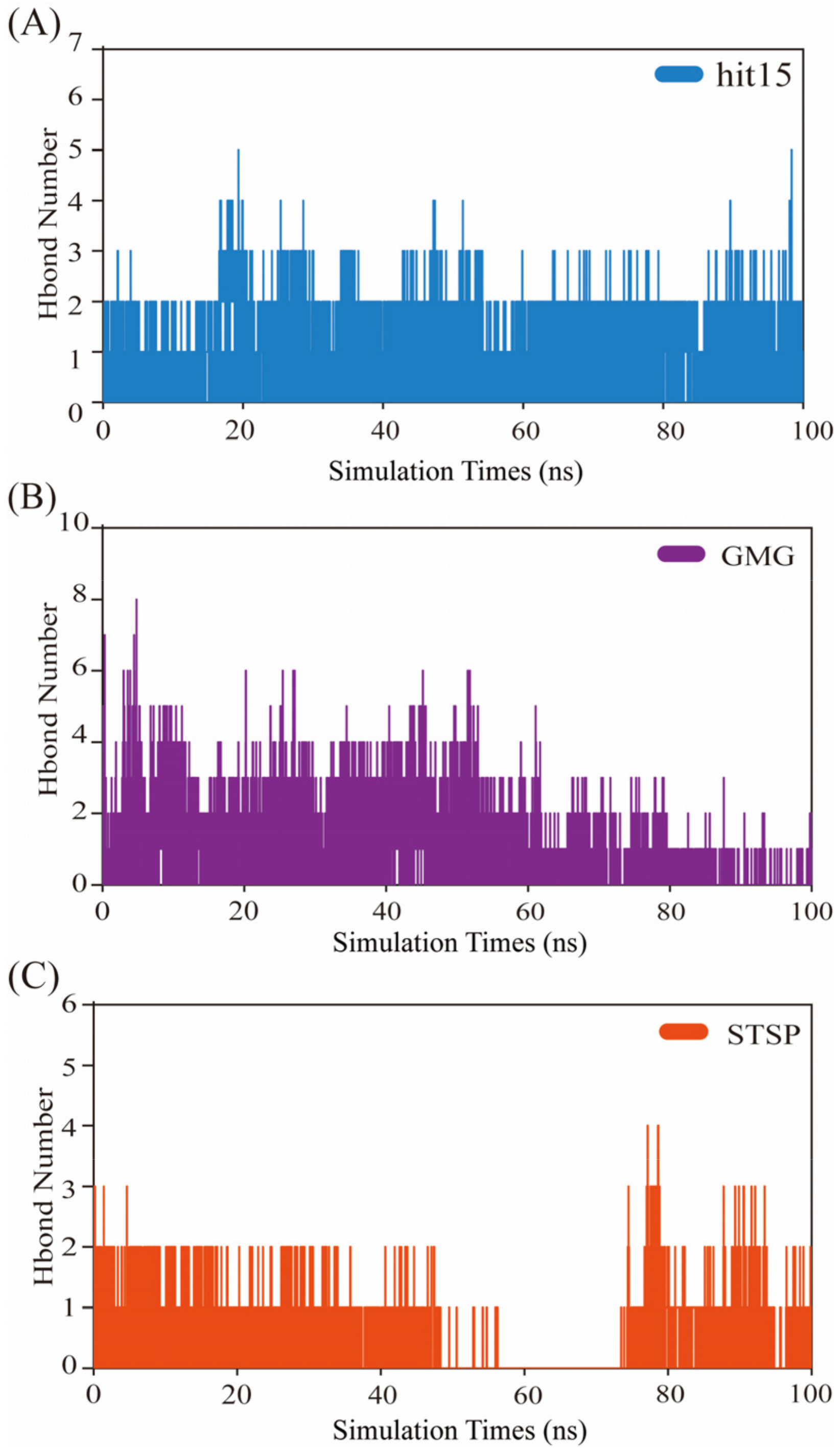
2.8.2. Hydrogen Bond Analysis
2.8.3. The Interaction Mechanism between SGK1 and hit15 Simulated by Molecular Dynamics
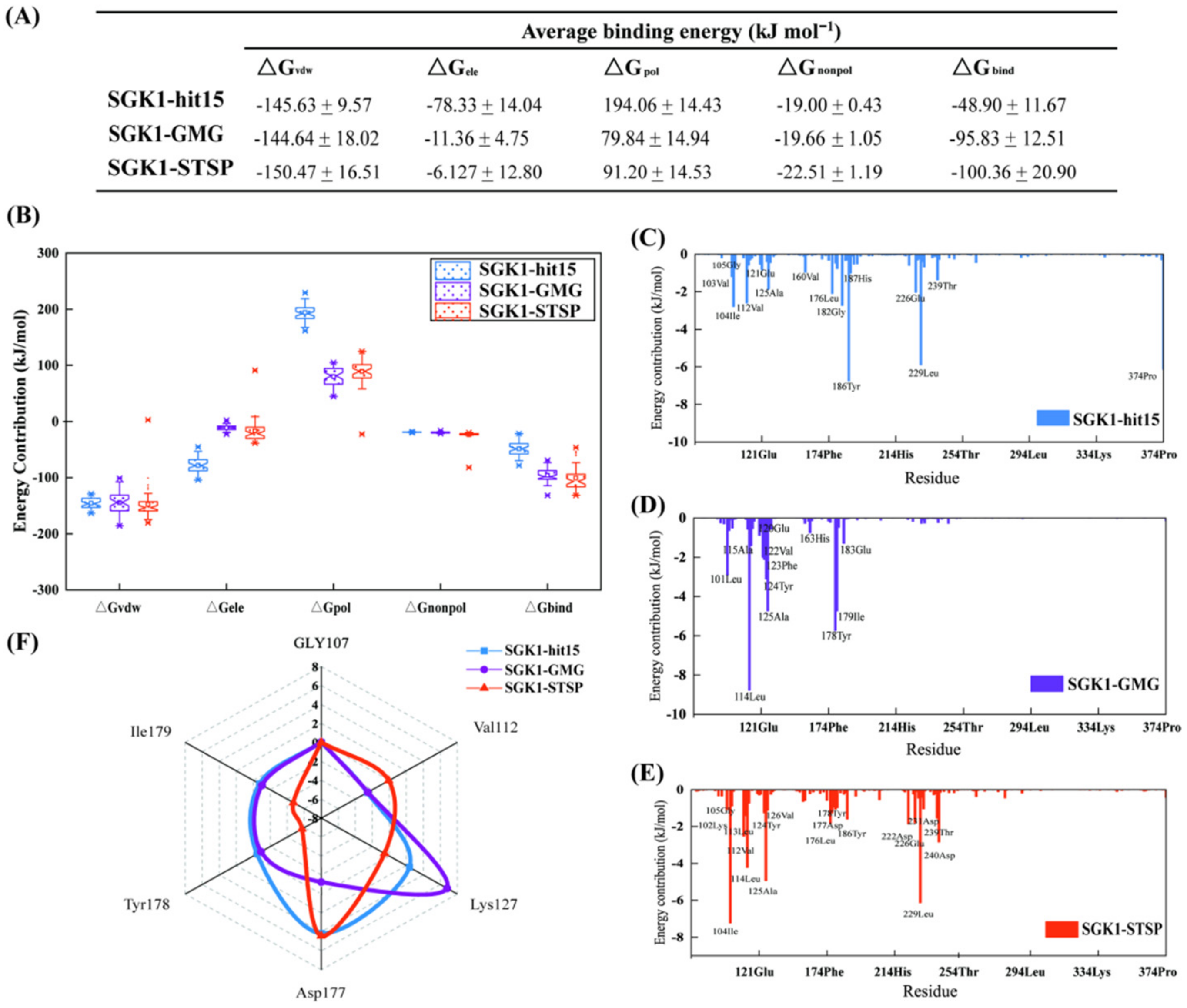
2.8.4. Binding Free-Energy Calculation and Decomposition
3. Materials and Methods
3.1. Crystal Structures Preparation
3.2. Dataset Collection
3.3. Pharmacophore Modeling
3.3.1. Common Feature Pharmacophore Modeling
3.3.2. Receptor–Ligand Pharmacophore Modeling
3.3.3. Validation of Pharmacophore Models
3.4. Molecular Docking
3.5. Bayesian Model
3.6. PAINS Filter, Drug-Likeness Assessment, and ADME/T Properties Prediction
3.7. Kinase Activity Assay
3.8. Molecular Dynamic Simulation
3.9. Binding Free-Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Basnet, R.; Gong, G.Q.; Li, C.; Wang, M.W. Serum and glucocorticoid inducible protein kinases (SGKs): A potential target for cancer intervention. Acta Pharm. Sin. B 2018, 8, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Vanhaelen, Q.; Mamoshina, P.; Aliper, A.M.; Artemov, A.; Lezhnina, K.; Ozerov, I.; Labat, I.; Zhavoronkov, A. Design of efficient computational workflows for in silico drug repurposing. Drug Discov. Today 2017, 22, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, G.; Lewington, S.; Sherliker, P. Body-mass index and cause-specific mortality in 900,000 adults: Collaborative analyses of 57 prospective studies. Lancet 2009, 373, 1083–1096. [Google Scholar] [PubMed] [Green Version]
- Lang, F.; Böhmer, C.; Palmada, M.; Seebohm, G.; Strutz-Seebohm, N.; Vallon, V. (Patho)physiological significance of the serum-and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 2006, 86, 1151–1178. [Google Scholar] [CrossRef]
- Kobayashi, T.; Cohen, P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J. 1999, 339, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Wormser, D.; Kaptoge, S.; Angelantonio, E.D.; Wood, A. Separate and combined associations of body-mass index and abdominal adiposity with cardiovascular disease: Collaborative analysis of 58 prospective studies. Lancet 2011, 377, 1085–1095. [Google Scholar] [PubMed] [Green Version]
- Lang, F.; Voelkl, J. Therapeutic potential of serum and glucocorticoid inducible kinase inhibition. Expert Opin. Investig. Drugs 2013, 22, 701–714. [Google Scholar] [CrossRef]
- Webster, M.K.; Goya, L.; Ge, Y.; Maiyar, A.C.; Firestone, G.L. Characterization of SGK, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol. Cell Biol. 1993, 13, 2031–2040. [Google Scholar]
- Leong, M.L.; Maiyar, A.C.; Kim, B.; O’Keeffe, B.A.; Firestone, G.L. Characterization of SGK, a novel member of the serine/threonine protein kin Expression of the serum- and glucocorticoid-inducible protein kinase, SGK is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J. Biol. Chem. 2003, 278, 5871–5882. [Google Scholar] [CrossRef] [Green Version]
- Sherk, A.B.; Frigo, D.E.; Schnackenberg, C.G.; Bray, J.D.; Laping, N.J.; Trizna, W.; Hammond, M.; Patterson, J.R.; Thompson, S.K.; Kazmin, D.; et al. Development of a smallmolecul; eserum-and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008, 68, 7475–7483. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, T.F.; Boinik, M.; Beier, N.; Scholz, W.; Fuchss, T.; Lang, F. EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell. Physiol. Biochem. 2011, 28, 137–146. [Google Scholar] [CrossRef]
- Gwak, E.H.; Yoo, H.Y.; Kim, S.H. Effects of Diabetes Mellitus on the Disposition of Tofacitinib, a Janus Kinase Inhibitor, in Rats. Biomol. Ther. 2020, 28, 361–369. [Google Scholar] [CrossRef]
- Halland, N.; Schmidt, F.; Weiss, T.; Saas, J.; Li, Z.; Czech, J.; Dreyer, M.; Hofmeister, A.; Mertsch, K.; Dietz, U.; et al. Discovery of N-[4-(1H-Pyrazolo[3,4-b]pyrazin-6-yl)-phenyl]-sulfonamides as Highly Active and Selective SGK1 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Kathrin, V.A.; Jonathan, B.; Nghi, N.H.; Leaver, D.J.; Cleary, B.L.; Rachel, L.H.; Nadeem, S.B.; John, T.T. Aryl Sulfonohydrazides. WO 2016198507 A1, 9 June 2016. [Google Scholar]
- Schneider, G. Virtual screening: An endless staircase? Nat. Rev. Drug Discov. 2010, 9, 273–276. [Google Scholar] [CrossRef]
- Santiago, D.N.; Pevzner, Y.; Durand, A.A.; Tran, M.; Scheerer, R.R.; Daniel, K.; Sung, S.-S.; Woodcock, H.L.; Guida, W.C.; Brooks, W.H. Virtual target screening: Validation using kinase inhibitors. J. Chem. Inform. Model. 2012, 52, 2192–2203. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Qi, H.Z.; Mao, J.; Zhang, H.R.; Luo, Q.Q.; Hu, M.L.; Shen, C.; Ding, L. Discovery of novel microtubule stabilizers targeting taxane binding site by applying molecular docking, molecular dynamics simulation, and anticancer activity testing. Bioorg. Chem. 2022, 122, 105–722. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Olanda, C.G.; Fokoue, H.H.; SantAnna, C.M.R. Virtual Screening Techniques in Drug Discovery: Review and Recent Applications. Curr. Top. Med. Chem. 2019, 19, 1751–1767. [Google Scholar]
- Kontoyianni, M. Docking and Virtual Screening in Drug Discovery. Methods Mol. Biol. 2017, 1647, 255–266. [Google Scholar]
- Wingert, B.M.; Camacho, C.J. Improving small molecule virtual screening strategies for the next generation of therapeutics. Cur. Opin. Chem. Biol. 2018, 44, 87–92. [Google Scholar] [CrossRef]
- Box, G.E.; Tiao, C.C. Bayesian Inference in Statistical Analysis; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Butina, D.; Segall, M.D.; Frankcombe, K. Predicting ADME properties in silico: Methods and models. Drug Discov. Today 2002, 7, S83–S88. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Gangwal, R.P.; Das, N.R.; Thanki, K.; Damre, M.V.; Dhoke, G.V.; Sharma, S.S.; Jain, S.; Sangamwar, A.T. Identification of p38α MAP kinase inhibitors by pharmacophore based virtual screening. J. Mol. Graph. Model. 2014, 49, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Inc, A.S. Discovery Studio 3.5; Accelrys Software Inc.: San Diego, CA, USA, 2010. [Google Scholar]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.O. Statistical Decision Theory and Bayesian Analysis; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Davis, M.I.; Sasaki, A.T.; Shen, M.; Emerling, B.M.; Thorne, N.; Michael, S.; Pragani, R.; Boxer, M.; Sumita, K.; Takeuchi, K.; et al. A homogeneous, high-throughput assay for phosphatidylinositol 5-phosphate 4-kinase with a novel, rapid substrate preparation. PLoS ONE 2013, 8, e54127. [Google Scholar] [CrossRef] [Green Version]
- Van, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Hypo 1 Results | Hypo 2 Results |
|---|---|---|
| Total molecules in database (D) | 1000 | 1000 |
| Total number of actives in database (A) | 119 | 119 |
| Total hits returned (Ht) | 135 | 118 |
| Active hits returned (Ha) | 97 | 92 |
| % Yield of actives [(Ha/Ht) × 100] | 71.85% | 77.97% |
| % Ratio of actives [(Ha/A) × 100] | 81.51% | 77.31% |
| Enrichment factor (E) [(Ha × D)/(Ht × A)] | 6.04 | 6.55 |
| False negatives [A–Ha] | 22 | 27 |
| False positives [Ht–Ha] | 38 | 26 |
| Goodness of hit score (GH) * | 0.71 | 0.76 |
| PDB ID | RMSD Values a | ||||
|---|---|---|---|---|---|
| Ligand | AutoDock | AutoDock Vina | GOLD | CODCKER | |
| 3HDM | GMG | 0.67 | 3.32 | 0.70 | 0.43 |
| 3HDN | MMG | 0.56 | 3.53 | 1.68 | 4.23 |
| Average RMSD | 0.62 | 3.42 | 1.19 | 2.33 | |
| Model Name | Roc Score | TP | FN | SE | FP | TN | SP | Q | MCC | |
|---|---|---|---|---|---|---|---|---|---|---|
| 3HDM_96970 | Training | 0.88 | 82 | 13 | 0.86 | 158 | 647 | 0.80 | 0.81 | 0.46 |
| Test | 0.91 | 22 | 2 | 0.92 | 29 | 147 | 0.84 | 0.85 | 0.56 | |
| 3HDN_90390 | Training | 0.90 | 83 | 12 | 0.87 | 155 | 650 | 0.81 | 0.81 | 0.48 |
| Test | 0.94 | 23 | 1 | 0.96 | 33 | 143 | 0.81 | 0.83 | 0.56 | |
| 3HDN_97440 | Training | 0.88 | 80 | 15 | 0.84 | 147 | 658 | 0.82 | 0.82 | 0.47 |
| Test | 0.92 | 22 | 2 | 0.92 | 29 | 147 | 0.84 | 0.85 | 0.56 | |
| 3HDN_75960 | Training | 0.90 | 83 | 12 | 0.87 | 157 | 649 | 0.81 | 0.81 | 0.47 |
| Test | 0.93 | 24 | 0 | 1.00 | 34 | 142 | 0.81 | 0.83 | 0.58 | |
| Model_ensemble | Training | 0.93 | 84 | 11 | 0.88 | 123 | 582 | 0.83 | 0.83 | 0.52 |
| Test | 0.95 | 24 | 0 | 1.00 | 28 | 148 | 0.84 | 0.86 | 0.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Shen, C.; Zhang, H.-R.; Qi, H.-Z.; Hu, M.-L.; Luo, Q.-Q. Identification of Novel Inhibitors Targeting SGK1 via Ensemble-Based Virtual Screening Method, Biological Evaluation and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2022, 23, 8635. https://doi.org/10.3390/ijms23158635
Zhang H, Shen C, Zhang H-R, Qi H-Z, Hu M-L, Luo Q-Q. Identification of Novel Inhibitors Targeting SGK1 via Ensemble-Based Virtual Screening Method, Biological Evaluation and Molecular Dynamics Simulation. International Journal of Molecular Sciences. 2022; 23(15):8635. https://doi.org/10.3390/ijms23158635
Chicago/Turabian StyleZhang, Hui, Chen Shen, Hong-Rui Zhang, Hua-Zhao Qi, Mei-Ling Hu, and Qing-Qing Luo. 2022. "Identification of Novel Inhibitors Targeting SGK1 via Ensemble-Based Virtual Screening Method, Biological Evaluation and Molecular Dynamics Simulation" International Journal of Molecular Sciences 23, no. 15: 8635. https://doi.org/10.3390/ijms23158635
APA StyleZhang, H., Shen, C., Zhang, H.-R., Qi, H.-Z., Hu, M.-L., & Luo, Q.-Q. (2022). Identification of Novel Inhibitors Targeting SGK1 via Ensemble-Based Virtual Screening Method, Biological Evaluation and Molecular Dynamics Simulation. International Journal of Molecular Sciences, 23(15), 8635. https://doi.org/10.3390/ijms23158635