
Characterization of the Basal and mTOR-Dependent Acute Pulmonary and Systemic Immune Response in a Murine Model of Combined Burn and Inhalation Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
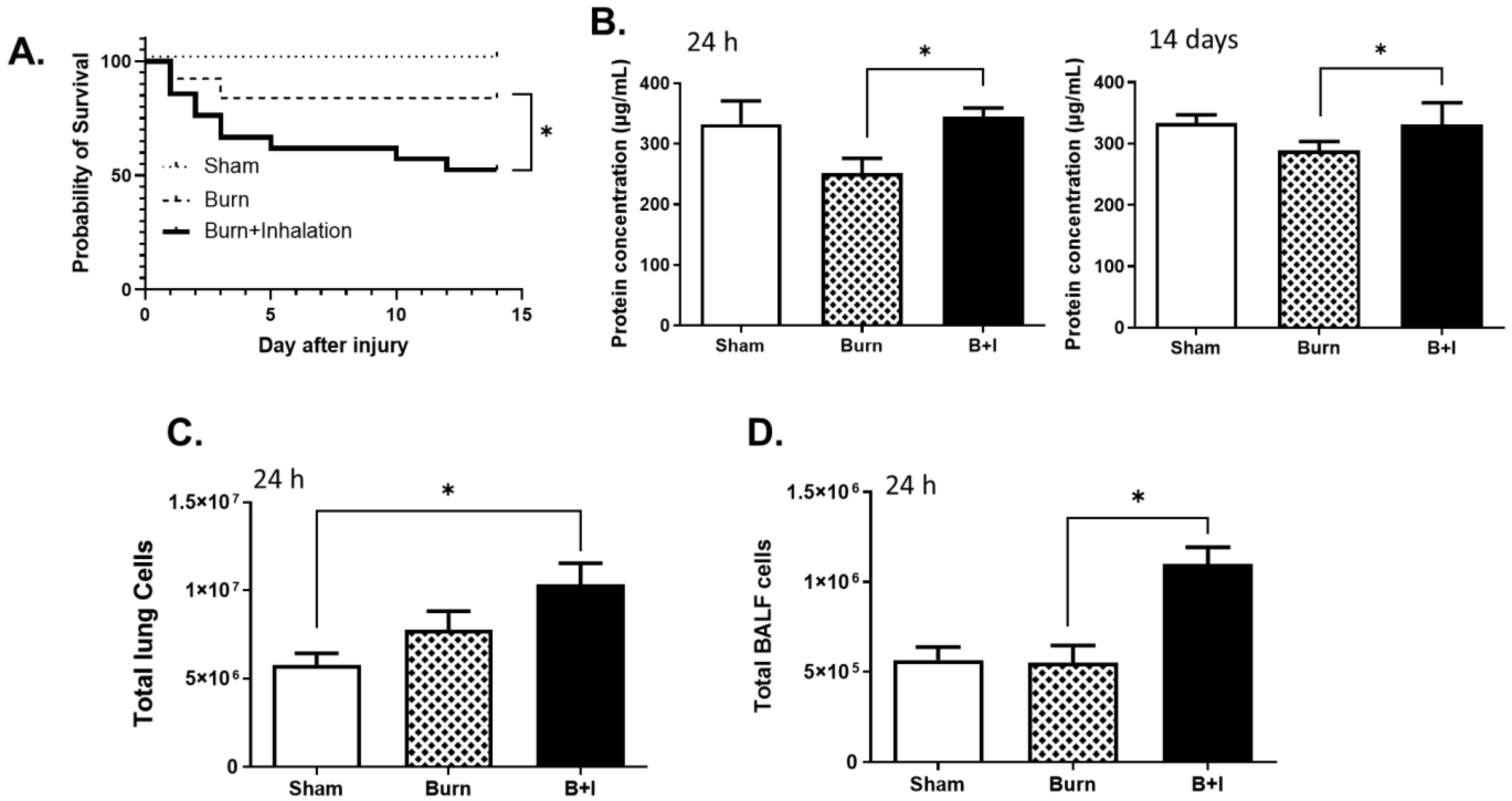
2.1. Combined Burn and Smoke Inhalation (B+I) Injury Leads to Higher Mortality than Burn Alone, with Elevated Levels of Protein and Cellular Infiltrate within the Airway Space and Lung Tissue
2.2. Immune Cells within the Lung Are Differentially Impacted by Burn and B+I Injury Early and Late after Injury
2.3. Lung and Soluble Immune Mediators in the Lung and Peripheral Blood Are Differentially Impacted by Burn and B+I Injury Early after Injury
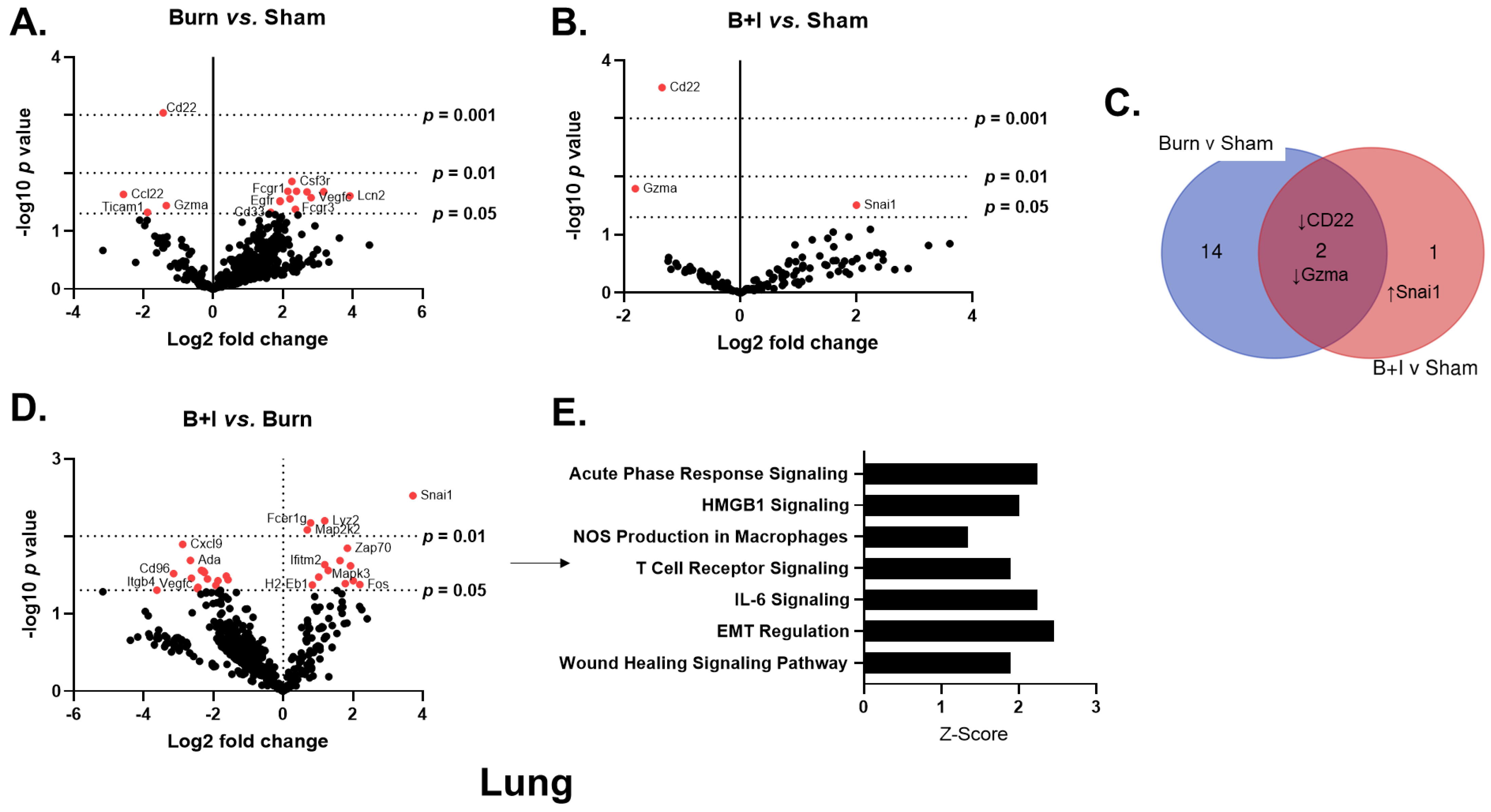
2.4. Immune Gene Transcriptome Analysis Revealed Pulmonary and Systemic Differential Immune Gene Expression in B+I Injury
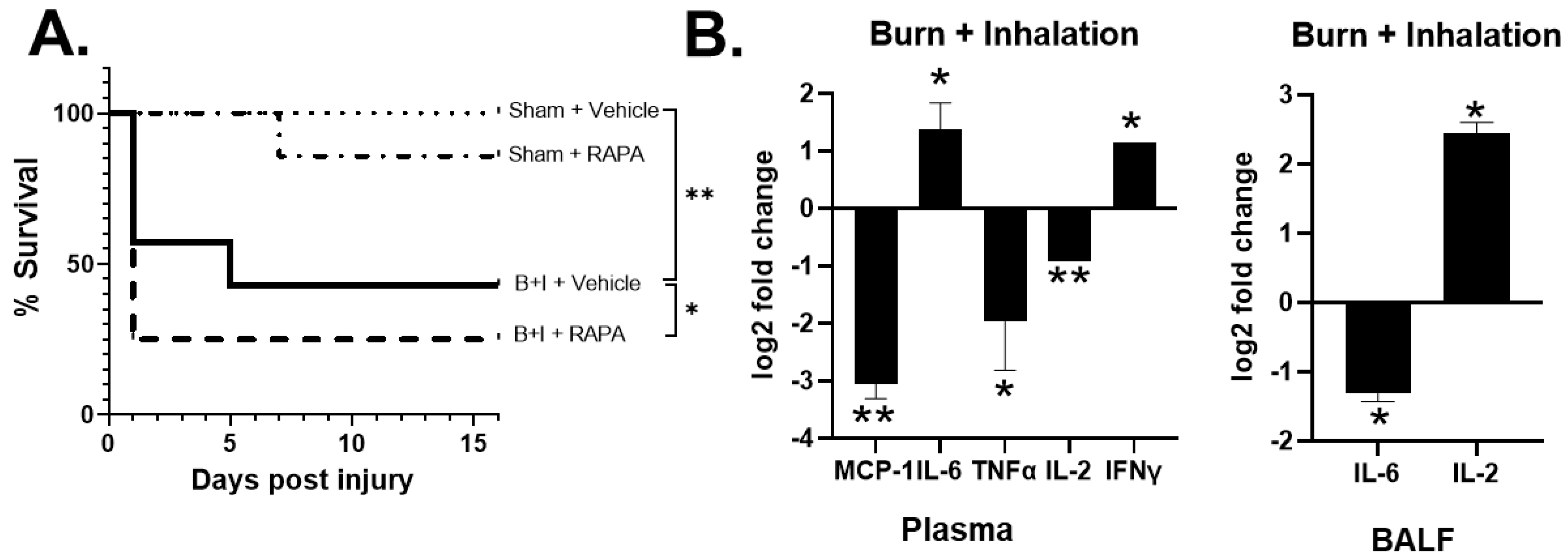
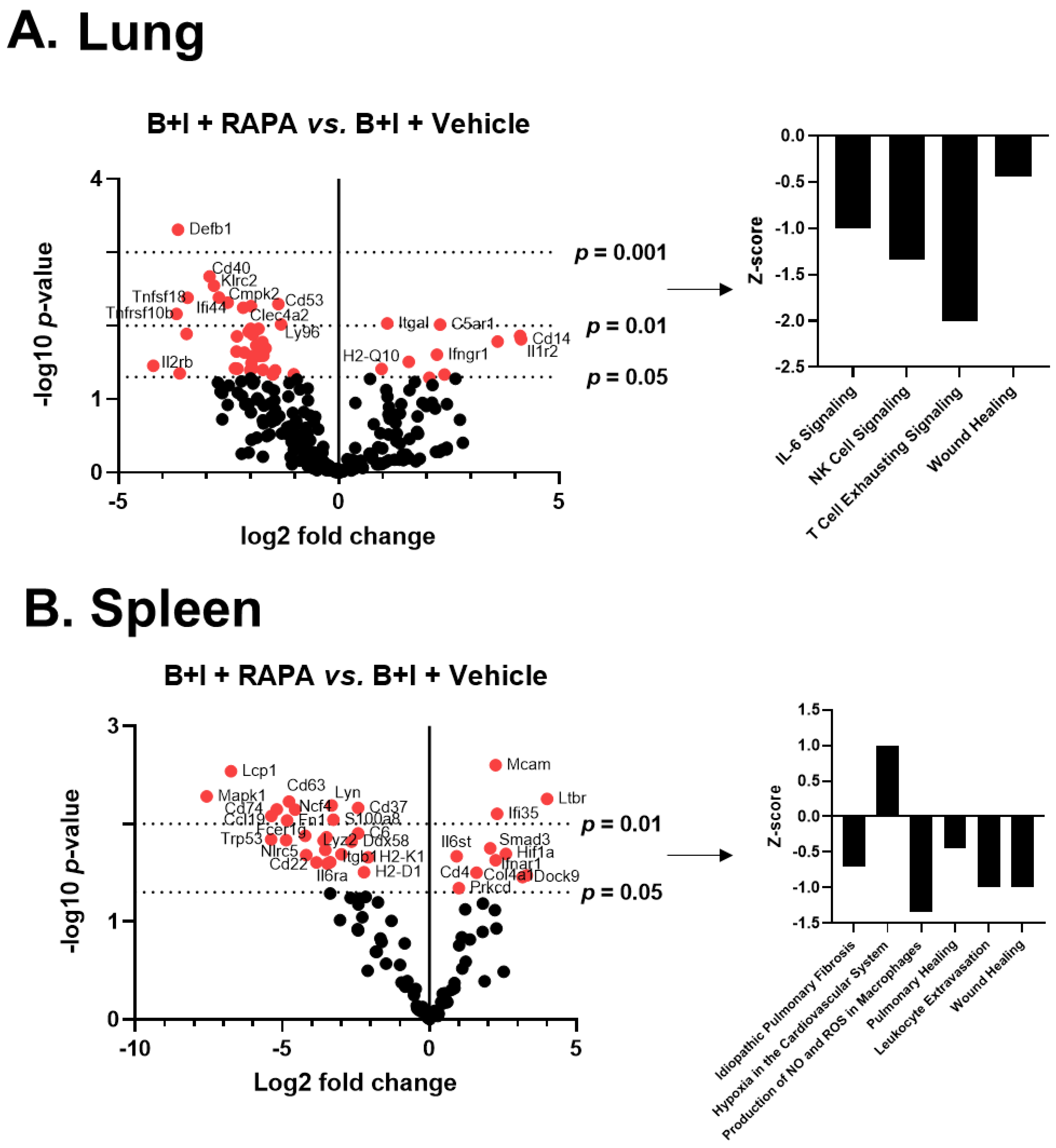
2.5. mTOR Regulates Systemic and Local Hyper-Responses to Burn and B+I Injury
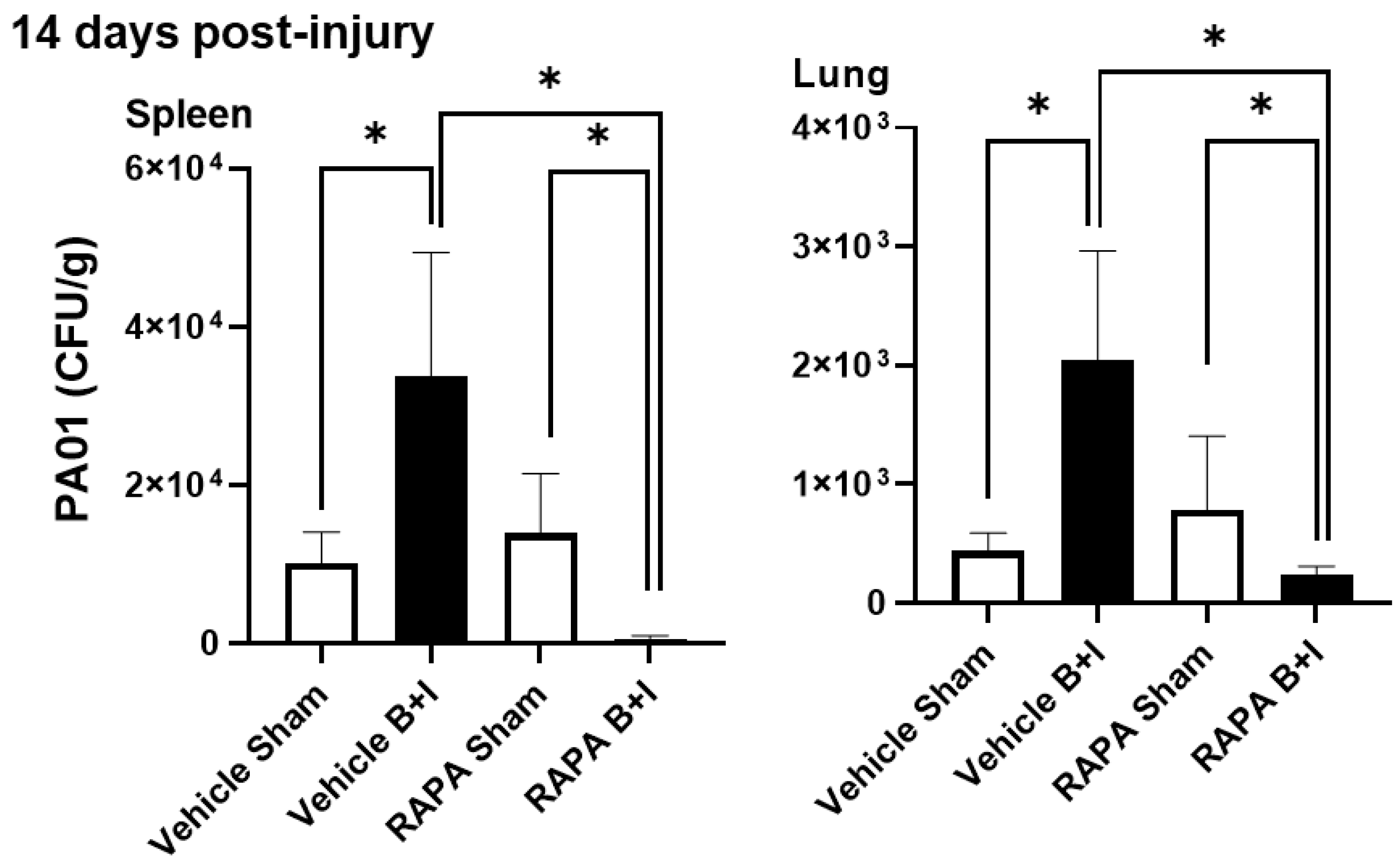
2.6. B+I Injury, Compared to Sham Injury, Increases Susceptibility to Bacterial Infections Early and Late after Injury; Susceptibility Early and Late after Injury Is Differentially Regulated by mTOR
3. Discussion
4. Materials and Methods
4.1. Murine Burn and Inhalation Injury Models
4.2. Peripheral Blood Plasma and Bronchoalveolar Lavage Collection
4.3. Lung and Spleen Tissue Isolation and Processing
4.4. Cytokine and Chemokine Assay
4.5. Flow Cytometry
4.6. Immune Gene Detection and Quantification
4.7. Rapamycin Blockade of mTOR
4.8. Infection with Pseudomonas aeruginosa
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Center for Injury Prevention and Control, CDC. Number of Injuries and Associated Costs 2022 WISQARS Cost Of Injury.
- Jeschke, M.G.; van Baar, M.E.; Choudhry, M.A.; Chung, K.K.; Gibran, N.S.; Logsetty, S. Burn injury. Nat. Rev. Dis Prim. 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.A.; Baun, M.M.; Meininger, J.C.; Wade, C.E. Comparison of mortality associated with sepsis in the burn, trauma, and general intensive care unit patient: A systematic review of the literature. Shock 2012, 37, 4–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snell, J.A.; Loh, N.H.; Mahambrey, T.; Shokrollahi, K. Clinical review: The critical care management of the burn patient. Crit. Care 2013, 17, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thombs, B.D.S.V.; Halonen, J.; Diallo, A.; Milner, S.M. The effects of preexisting medical comorbidities on mortality and length of hospital stay in acute burn injury: Evidence from a national sample of 31,338 adult patients. Ann. Surg. 2007, 245, 629–634. [Google Scholar] [CrossRef]
- Brandao, C.; Meireles, R.; Brito, I.; Ramos, S.; Cabral, L. The Role of Comorbidities on Outcome Prediction in Acute Burn Patients. Ann. Burn. Fire Disasters 2021, 34, 323–333. [Google Scholar]
- Jones, S.W.; Williams, F.N.; Cairns, B.A.; Cartotto, R. Inhalation Injury: Pathophysiology, Diagnosis, and Treatment. Clin. Plast. Surg. 2017, 44, 505–511. [Google Scholar] [CrossRef]
- Salibian, A.A.; Rosario, A.T.D.; Severo, L.A.M.; Nguyen, L.; Banyard, D.A.; Toranto, J.D.; Evans, G.R.D.; Widgerow, A.D. Current concepts on burn wound conversion-A review of recent advances in understanding the secondary progressions of burns. Burns 2016, 42, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Albright, J.M.; Davis, C.S.; Bird, M.D.; Ramirez, L.; Kim, H.; Burnham, E.L.; Gamelli, R.L.; Kovacs, E.J. The acute pulmonary inflammatory response to the graded severity of smoke inhalation injury. Crit. Care Med. 2012, 40, 1113–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, P.F.; Buehner, M.F.; Wood, L.A.; Boyer, N.L.; Driscoll, I.R.; Lundy, J.B.; Cancio, L.C.; Chung, K.K. Diagnosis and management of inhalation injury: An updated review. Crit. Care 2015, 19, 351. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, M.; Lepape, A.; Piriou, V.; Venet, F.; Friggeri, A. Innate danger signals in acute injury: From bench to bedside. Anaesth. Crit. Care Pain Med. 2016, 35, 283–292. [Google Scholar] [CrossRef]
- Rogobete, A.F.; Sandesc, D.; Papurica, M.; Stoicescu, E.R.; Popovici, S.E.; Bratu, L.M.; Vernic, C.; Sas, A.M.; Stan, A.T.; Bedreag, O.H. The influence of metabolic imbalances and oxidative stress on the outcome of critically ill polytrauma patients: A review. Burn. Trauma 2017, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, P. Burns as a model of SIRS. Front. Biosci. (Landmark Ed.). 2009, 14, 4962–4967. [Google Scholar] [CrossRef]
- Ward, N.S.; Casserly, B.; Ayala, A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin. Chest Med. 2008, 29, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.S.; Albright, J.M.; Carter, S.R.; Ramirez, L.; Kim, H.; Gamelli, R.L.; Kovacs, E.J. Early pulmonary immune hyporesponsiveness is associated with mortality after burn and smoke inhalation injury. J. Burn. Care Res. Off. Publ. Am. Burn. Assoc. 2012, 33, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.S.; Janus, S.E.; Mosier, M.J.; Carter, S.R.; Gibbs, J.T.; Ramirez, L.; Gamelli, R.L.; Kovacs, E.J. Inhalation injury severity and systemic immune perturbations in burned adults. Ann. Surg. 2013, 257, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perng, D.W.; Chang, T.M.; Wang, J.Y.; Lee, C.C.; Lu, S.H.; Shyue, S.K.; Lee, T.S.; Kou, Y.R. Inflammatory role of AMP-activated protein kinase signaling in an experimental model of toxic smoke inhalation injury. Crit. Care Med. 2013, 41, 120–132. [Google Scholar] [CrossRef]
- Clark, C.J.; Pollock, A.J.; Reid, W.H.; Campbell, D.; Gemmell, C. Role of pulmonary alveolar macrophage activation in acute lung injury after burns and smoke inhalation. Lancet 1988, 2, 872–874. [Google Scholar] [CrossRef]
- Quinn, D.A.; Moufarrej, R.; Volokhov, A.; Syrkina, O.; Hales, C.A. Combined smoke inhalation and scald burn in the rat. J. Burn. Care Rehabil. 2003, 24, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, H.; Yarmush, M.L.; Berthiaume, F. Dynamics of tissue neutrophil sequestration after cutaneous burns in rats. J. Surg. Res. 2000, 93, 88–96. [Google Scholar] [CrossRef]
- Sun, B.; Sun, H.; Liu, C.; Shen, J.; Chen, Z.; Chen, X. Role of CO-releasing molecules liberated CO in attenuating leukocytes sequestration and inflammatory responses in the lung of thermally injured mice. J. Surg. Res. 2007, 139, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H.; Mendiguren, I.; Fisher, D.; Fisher, A.B.; Thom, S.R. Role of neutrophils and nitric oxide in lung alveolar injury from smoke inhalation. Am. J. Respir. Crit. Care Med. 1994, 150, 337–341. [Google Scholar] [CrossRef]
- Zhu, F.; Qiu, X.; Wang, J.; Jin, Y.; Sun, Y.; Lv, T.; Xia, Z. A rat model of smoke inhalation injury. Inhal. Toxicol. 2012, 24, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Bjertnaes, L.J.; Schmalstieg, F.C.; McGuire, R.; Cox, R.A.; Hawkins, H.K.; Herndon, D.N.; Traber, L.D.; Traber, D.L. A novel animal model of sepsis after acute lung injury in sheep. Crit. Care Med. 2002, 30, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, D.M.; McIntyre, M.K.; Beely, B.; Jordan, B.; Walker, K.P., 3rd; Aden, J.K.; Batchinsky, A.; Chung, K.K.; Cancio, L.C.; Christy, R.J. A model of recovery from inhalation injury and cutaneous burn in ambulatory swine. Burn. J. Int. Soc. Burn. Inj. 2017, 43, 1295–1305. [Google Scholar] [CrossRef]
- Lange, M.; Hamahata, A.; Enkhbaatar, P.; Esechie, A.; Connelly, R.; Nakano, Y.; Jonkam, C.; Cox, R.A.; Traber, L.D.; Herndon, D.N.; et al. Assessment of vascular permeability in an ovine model of acute lung injury and pneumonia-induced Pseudomonas aeruginosa sepsis. Crit. Care Med. 2008, 36, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Pedraza-Chaverri, J.; Becerril, C.; Cisneros, J.; Gonzalez-Avila, G.; Rivera-Rosales, R.; Sommer, B.; Medina-Campos, O.N.; Montano, M. Oxidative stress and lung injury induced by short-term exposure to wood smoke in guinea pigs. Toxicol. Mech. Methods 2013, 23, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Grommes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. (Camb. Mass). 2011, 17, 293–307. [Google Scholar] [CrossRef]
- Martin, T.R. Neutrophils and lung injury: Getting it right. J. Clin. Investig. 2002, 110, 1603–1605. [Google Scholar] [CrossRef]
- Kartchner, L.B.; Gode, C.J.; Dunn, J.L.M.; Glenn, L.I.I.; Duncan, D.N.; Wolfgang, M.C.; Cairns, B.A.; Maile, R. One-hit wonder: Late after burn injury, granulocytes can clear one bacterial infection but cannot control a subsequent infection. Burns 2019, 45, 627–640. [Google Scholar] [CrossRef]
- Fuchs, P.C.; Demir, E.; Reuber, K.; Stromps, P.; Wolter, T.; Pallua, N. Intra-alveolar IL-6 levels following burn and inhalation injury. Burn. J. Int. Soc. Burn. Inj. 2009, 35, 840–844. [Google Scholar] [CrossRef]
- Xiao, W.; Mindrinos, M.N.; Seok, J.; Cuschieri, J.; Cuenca, A.G.; Gao, H.; Hayden, D.L.; Hennessy, L.; Moore, E.E.; Minei, J.P.; et al. A genomic storm in critically injured humans. J. Exp. Med. 2011, 208, 2581–2590. [Google Scholar] [CrossRef]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [Green Version]
- Tompkins, R.G. Genomics of injury: The Glue Grant experience. J. Trauma Acute Care Surg. 2015, 78, 671–686. [Google Scholar] [CrossRef] [Green Version]
- Lorne, E.; Zhao, X.; Zmijewski, J.W.; Liu, G.; Park, Y.J.; Tsuruta, Y.; Abraham, E. Participation of mammalian target of rapamycin complex 1 in Toll-like receptor 2- and 4-induced neutrophil activation and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2009, 41, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Liu, J.; Wu, Y.F.; Lou, J.; Mao, Y.Y.; Shen, H.H.; Chen, Z.H. mTOR and autophagy in regulation of acute lung injury: A review and perspective. Microbes Infect. 2014, 16, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes. Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dievart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remels, A.H.; Langen, R.C.; Gosker, H.R.; Russell, A.P.; Spaapen, F.; Voncken, J.W.; Schrauwen, P.; Schols, A.M. PPARgamma inhibits NF-kappaB-dependent transcriptional activation in skeletal muscle. Am. J. Physiology. Endocrinol. Metab. 2009, 297, E174–E183. [Google Scholar] [CrossRef]
- Dunn, J.L.M.; Kartchner, L.B.; Gast, K.; Sessions, M.; Hunter, R.A.; Thurlow, L.; Richardson, A.; Schoenfisch, M.; Cairns, B.A.; Maile, R. Mammalian target of rapamycin regulates a hyperresponsive state in pulmonary neutrophils late after burn injury. J. Leukoc. Biol. 2018, 103, 909–918. [Google Scholar] [CrossRef]
- Maile, R.; Barnes, C.M.; Nielsen, A.I.; Meyer, A.A.; Frelinger, J.A.; Cairns, B.A. Lymphopenia-induced homeostatic proliferation of CD8(+) T cells is a mechanism for effective allogeneic skin graft rejection following burn injury. J. Immunol. 2006, 176, 6717–6726. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.L.M.; Kartchner, L.B.; Stepp, W.H.; Glenn, L.I.; Malfitano, M.M.; Jones, S.; Doerschuk, C.M.; Maile, R.; Cairns, B.A. Blocking CXCL1-dependent neutrophil recruitment prevents immune damage and reduces pulmonary bacterial infection after inhalation injury. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2018, 314, L822–L834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, J.L.; Hunter, R.A.; Gast, K.; Maile, R.; Cairns, B.A.; Schoenfisch, M.H. Direct detection of blood nitric oxide reveals a burn-dependent decrease of nitric oxide in response to Pseudomonas aeruginosa infection. Burns 2016, 42, 1522–1527. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, M.M. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr. Protoc. Mol. Biol 2011, 4, 25B.10.1–25B.10.17. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Huang, S.; Houghton, P.J. Targeting mTOR signaling for cancer therapy. Curr. Opin. Pharmacol. 2003, 3, 371–377. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landh, E.; Moir, L.M.; Traini, D.; Young, P.M.; Ong, H.X. Properties of rapamycin solid lipid nanoparticles for lymphatic access through the lungs & part II: The effect of nanoparticle charge. Nanomedicine 2020, 15, 1947–1963. [Google Scholar] [CrossRef] [PubMed]
- Merle, D.A.; Provenzano, F.; Jarboui, M.A.; Kilger, E.; Clark, S.J.; Deleidi, M.; Armento, A.; Ueffing, M. mTOR Inhibition via Rapamycin Treatment Partially Reverts the Deficit in Energy Metabolism Caused by FH Loss in RPE Cells. Antioxid 2021, 10, 1994. [Google Scholar] [CrossRef]
- Noel, J.G.; Guo, X.; Wells-Byrum, D.; Schwemberger, S.; Caldwell, C.C.; Ogle, C.K. Effect of thermal injury on splenic myelopoiesis. Shock 2005, 23, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Noel, J.G.; Osterburg, A.; Wang, Q.; Guo, X.; Byrum, D.; Schwemberger, S.; Goetzman, H.; Caldwell, C.C.; Ogle, C.K. Thermal injury elevates the inflammatory monocyte subpopulation in multiple compartments. Shock 2007, 28, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Yang, Q.; Orman, M.A.; Ierapetritou, M.G.; Berthiaume, F.; Gale, S.C.; Androulakis, I.P. Impact of burn priming on immune and metabolic functions of whole Liver in a rat cecal ligation and puncture model. Int. J. Burn. Trauma 2013, 3, 55–65. [Google Scholar]
- Paterson, H.M.; Murphy, T.J.; Purcell, E.J.; Shelley, O.; Kriynovich, S.J.; Lien, E.; Mannick, J.A.; Lederer, J.A. Injury primes the innate immune system for enhanced Toll-like receptor reactivity. J. Immunol. 2003, 171, 1473–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, J.C.; Noel, J.G.; Nikolaidis, N.M.; Karns, R.; Aronow, B.J.; Ogle, C.K.; McCormack, F.X. G-CSF drives a posttraumatic immune program that protects the host from infection. J. Immunol. 2014, 192, 2405–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, G.; Wang, Q.; Schwemberger, S.; Hanson, C.; Giacalone, N.; Haar, L.; Ogle, C.K. Neutrophils, not monocyte/macrophages, are the major splenic source of postburn IL-10. Shock 2011, 36, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Huber, N.L.; Bailey, S.R.; Schuster, R.; Ogle, C.K.; Lentsch, A.B.; Pritts, T.A. Prior thermal injury accelerates endotoxin-induced inflammatory cytokine production and intestinal nuclear factor-kappaB activation in mice. J. Burn. Care Res. Off. Publ. Am. Burn. Assoc. 2012, 33, 279–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maile, R.; Jones, S.; Pan, Y.; Zhou, H.; Jaspers, I.; Peden, D.B.; Cairns, B.A.; Noah, T.L. Association between early airway damage-associated molecular patterns and subsequent bacterial infection in patients with inhalational and burn injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L855–L860. [Google Scholar] [CrossRef]
- Jones, S.W.; Zhou, H.; Ortiz-Pujols, S.M.; Maile, R.; Herbst, M.; Joyner, B.L., Jr.; Zhang, H.; Kesic, M.; Jaspers, I.; Short, K.A.; et al. Bronchoscopy-derived correlates of lung injury following inhalational injuries: A prospective observational study. PLoS ONE 2013, 8, e64250. [Google Scholar] [CrossRef] [Green Version]
- Eitas, T.K.; Stepp, W.; Sjeklocha, L.; Long, C.; Riley, C.; Callahan, J.; Sanchez, Y.; Gough, P.; Knowlin, L.; van Duin, D.; et al. Differential regulation of innate immune cytokine production through pharmacological activation of Nuclear Factor-Erythroid-2-Related Factor 2 (NRF2) in burn patient immune cells and monocytes. PLoS ONE 2017, 12, e0184164. [Google Scholar] [CrossRef]
- Hur, J.; Yang, H.T.; Chun, W.; Kim, J.H.; Shin, S.H.; Kang, H.J.; Kim, H.S. Inflammatory cytokines and their prognostic ability in cases of major burn injury. Ann. Lab. Med. 2015, 35, 105–110. [Google Scholar] [CrossRef]
- Hibi, M.; Murakami, M.; Saito, M.; Hirano, T.; Taga, T.; Kishimoto, T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 1990, 63, 1149–1157. [Google Scholar] [CrossRef]
- Andreev-Andrievskiy, A.A.; Zinovkin, R.A.; Mashkin, M.A.; Frolova, O.Y.; Kazaishvili, Y.G.; Scherbakova, V.S.; Rudoy, B.A.; Nesterenko, V.G. Gene Expression Pattern of Peyer’s Patch Lymphocytes Exposed to Kagocel Suggests Pattern-Recognition Receptors Mediate Its Action. Front. Pharmacol. 2021, 12, 679511. [Google Scholar] [CrossRef]
- Neely, C.J.; Kartchner, L.B.; Mendoza, A.E.; Linz, B.M.; Frelinger, J.A.; Wolfgang, M.C.; Maile, R.; Cairns, B.A. Flagellin treatment prevents increased susceptibility to systemic bacterial infection after injury by inhibiting anti-inflammatory IL-10+ IL-12- neutrophil polarization. PLoS ONE 2014, 9, e85623. [Google Scholar] [CrossRef] [PubMed]
- Linz, B.M.; Neely, C.J.; Kartchner, L.B.; Mendoza, A.E.; Khoury, A.L.; Truax, A.; Sempowski, G.; Eitas, T.; Brickey, J.; Ting, J.P.; et al. Innate Immune Cell Recovery Is Positively Regulated by NLRP12 during Emergency Hematopoiesis. J. Immunol. 2017, 198, 2426–2433. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, A.E.; Neely, C.J.; Charles, A.G.; Kartchner, L.B.; Brickey, W.J.; Khoury, A.L.; Sempowski, G.D.; Ting, J.P.; Cairns, B.A.; Maile, R. Radiation combined with thermal injury induces immature myeloid cells. Shock. (Augusta Ga.) 2012, 38, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Kebaier, C.; Chamberland, R.R.; Allen, I.C.; Gao, X.; Broglie, P.M.; Hall, J.D.; Jania, C.; Doerschuk, C.M.; Tilley, S.L.; Duncan, J.A. Staphylococcus aureus alpha-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J. Infect. Dis. 2012, 205, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, A.; Grubb, B.R.; Hudson, E.J.; Wilkinson, K.J.; Sheehan, J.K.; Mall, M.A.; O’Neal, W.K.; Boucher, R.C.; Randell, S.H. Airway and lung pathology due to mucosal surface dehydration in {beta}-epithelial Na+ channel-overexpressing mice: Role of TNF-{alpha} and IL-4R{alpha} signaling, influence of neonatal development, and limited efficacy of glucocorticoid treatment. J. Immunol. (Baltim. Md. 1950). 2009, 182, 4357–4367. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Gomez, J.C.; Chugh, P.E.; Lowell, C.A.; Dinauer, M.C.; Dittmer, D.P.; Doerschuk, C.M. Interferon-gamma production by neutrophils during bacterial pneumonia in mice. Am. J. Respir. Crit. Care Med. 2011, 183, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Coleman, L.G., Jr.; Maile, R.; Jones, S.W.; Cairns, B.A.; Crews, F.T. HMGB1/IL-1beta complexes in plasma microvesicles modulate immune responses to burn injury. PLoS ONE 2018, 13, e0195335. [Google Scholar] [CrossRef] [Green Version]
- Willis, M.L.; Mahung, C.; Wallet, S.M.; Barnett, A.; Cairns, B.A.; Coleman, L.G., Jr.; Maile, R. Plasma extracellular vesicles released after severe burn injury modulate macrophage phenotype and function. J. Leukoc. Biol. 2021, 111, 33–49. [Google Scholar] [CrossRef]
- Mahung, C.; Stepp, W.H.; Long, C.V.; Malfitano, M.M.; Saklayici, I.; Wallet, S.M.; Zhou, L.Y.; Zhou, H.; Cairns, B.A.; Maile, R. Early expression of IL-10, IL-12, ARG1 and NOS2 genes in peripheral blood mononuclear cells synergistically correlate with patient outcome after burn injury. J. Trauma Acute Care Surg. 2022. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hall, H.R.; Mahung, C.; Dunn, J.L.M.; Kartchner, L.M.; Seim, R.F.; Cairns, B.A.; Wallet, S.M.; Maile, R. Characterization of the Basal and mTOR-Dependent Acute Pulmonary and Systemic Immune Response in a Murine Model of Combined Burn and Inhalation Injury. Int. J. Mol. Sci. 2022, 23, 8779. https://doi.org/10.3390/ijms23158779
Hall HR, Mahung C, Dunn JLM, Kartchner LM, Seim RF, Cairns BA, Wallet SM, Maile R. Characterization of the Basal and mTOR-Dependent Acute Pulmonary and Systemic Immune Response in a Murine Model of Combined Burn and Inhalation Injury. International Journal of Molecular Sciences. 2022; 23(15):8779. https://doi.org/10.3390/ijms23158779
Chicago/Turabian StyleHall, Hannah R., Cressida Mahung, Julia L. M. Dunn, Laurel M. Kartchner, Roland F. Seim, Bruce A. Cairns, Shannon M. Wallet, and Robert Maile. 2022. "Characterization of the Basal and mTOR-Dependent Acute Pulmonary and Systemic Immune Response in a Murine Model of Combined Burn and Inhalation Injury" International Journal of Molecular Sciences 23, no. 15: 8779. https://doi.org/10.3390/ijms23158779