
Aberrant Splicing of INS Impairs Beta-Cell Differentiation and Proliferation by ER Stress in the Isogenic iPSC Model of Neonatal Diabetes

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation of iPSC-Based Isogenic System by CRISPR/Cas9 Gene Editing of the INS Gene
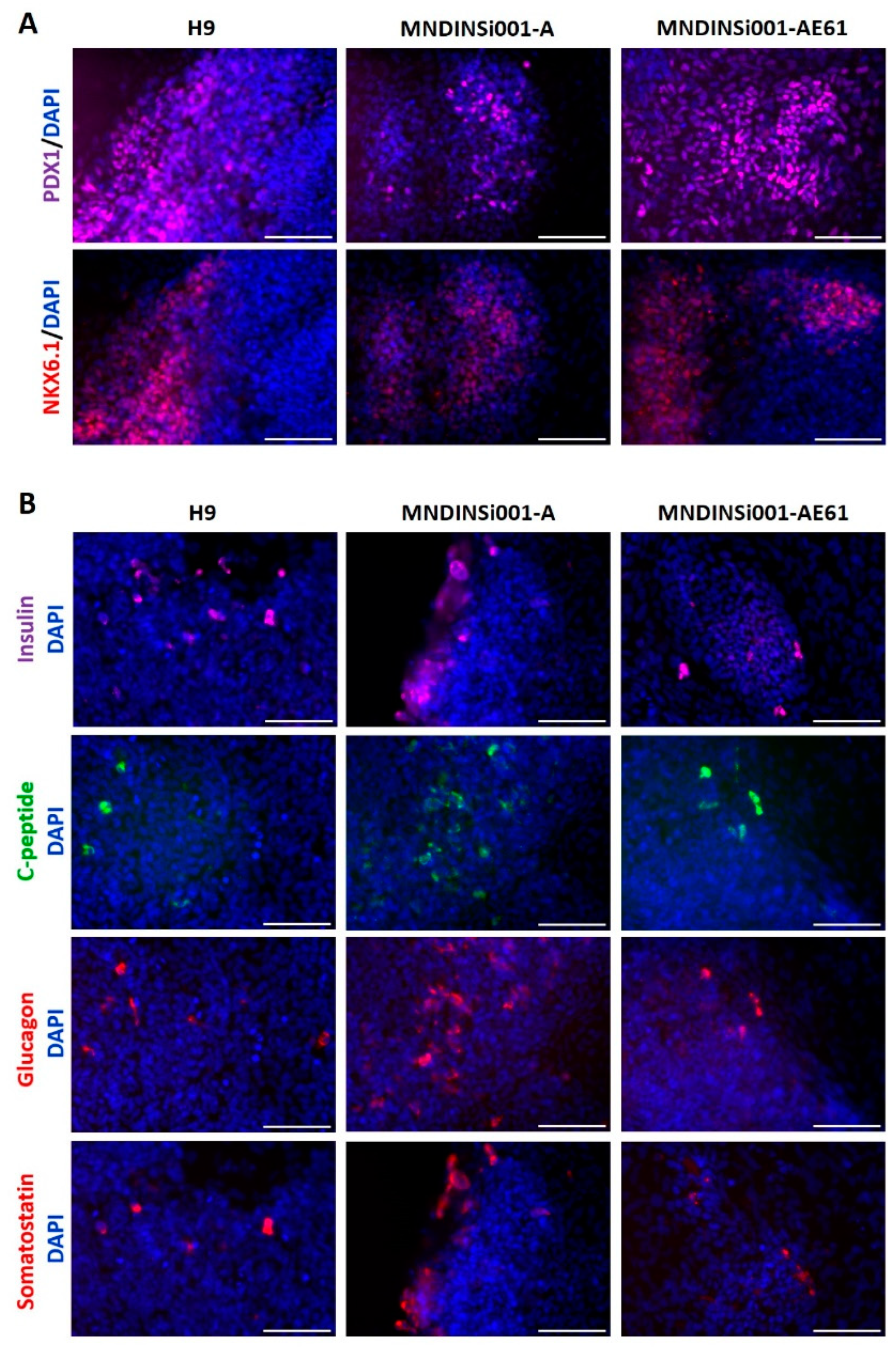
2.2. Differentiation of INS Mutant and Corrected iPSC Lines to Pancreatic Cells
2.3. INS c.188-31G>A Mutation Creates Insulin mRNA Isoform in Differentiated Human Beta-Like Cells without Insulin Production
2.4. Splice Variant Isoform Inhibits Proliferation of Mature Insulin Producing MIN6 Cells
2.5. ER Stress as a Possible Mechanism of Beta-Cell Dysfunction
3. Discussion
4. Materials and Methods
4.1. Ethics and Informed Consent
4.2. Cell Culture
4.3. In Vitro Differentiation of PSC Lines to Beta-Like Pancreatic Cells
4.4. sgRNA Design and Cloning
4.5. Cloning of the INS Gene and Plasmid Construction
4.6. Cells Transfection and iPSC Clone Selection
4.7. DNA, RNA Extraction, Sanger Sequencing
4.8. Immunocytochemistry
4.9. Cell Proliferation Measurement
4.10. Reporter Assays
4.11. ELISA
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- De Franco, E.; Flanagan, S.E.; AL Houghton, J.; Allen, H.L.; Mackay, D.J.; Temple, I.K.; Ellard, S.; Hattersley, A.T. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: An international cohort study. Lancet 2015, 386, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Cayabyab, F.; Nih, L.R.; Yoshihara, E. Advances in Pancreatic Islet Transplantation Sites for the Treatment of Diabetes. Front. Endocrinol. 2021, 12, 732431. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.-Y.; Watanabe, T.; Shirai, H.; Yamasaki, S.; Kinoshita, M.; Matsushita, K.; Hashiguchi, T.; Onoe, H.; Matsuyama, T.; Kuwahara, A.; et al. Medium- to Long-Term Survival and Functional Examination of Human iPSC-Derived Retinas in Rat and Primate Models of Retinal Degeneration. EBioMedicine 2019, 39, 562–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartfeld, S.; Clevers, H. Stem Cell-Derived Organoids and Their Application for Medical Research and Patient Treatment. J. Mol. Med. 2017, 95, 729–738. [Google Scholar] [CrossRef]
- Doi, D.; Magotani, H.; Kikuchi, T.; Ikeda, M.; Hiramatsu, S.; Yoshida, K.; Amano, N.; Nomura, M.; Umekage, M.; Morizane, A.; et al. Preclinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nat. Commun. 2020, 11, 3369. [Google Scholar] [CrossRef]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s Disease Pathology in Human Induced Plu-ripotent Stem Cell-Derived Neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef] [Green Version]
- Ilic, D.; Liovic, M. Industry updates from the field of stem cell research and regenerative medicine in February. Regen. Med. 2022, 17, 327–336. [Google Scholar] [CrossRef]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of Functional Human Pancreatic β Cells In Vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, K.G.; Augsornworawat, P.; Velazco-Cruz, L.; Kim, M.H.; Asada, R.; Hogrebe, N.J.; Morikawa, S.; Urano, F.; Millman, J.R. Gene-edited human stem cell–derived β cells from a patient with monogenic diabetes reverse preexisting diabetes in mice. Sci. Transl. Med. 2020, 12, eaax9106. [Google Scholar] [CrossRef]
- Ma, S.; Viola, R.; Sui, L.; Cherubini, V.; Barbetti, F.; Egli, D. β Cell Replacement after Gene Editing of a Neonatal Dia-betes-Causing Mutation at the Insulin Locus. Stem Cell Rep. 2018, 11, 1407–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balboa, D.; Saarimäki-Vire, J.; Borshagovski, D.; Survila, M.; Lindholm, P.; Galli, E.; Eurola, S.; Ustinov, J.; Grym, H.; Huopio, H.; et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. eLife 2018, 7, e38519. [Google Scholar] [CrossRef] [PubMed]
- Nair, G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.-L.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.G.; Millman, J.R. Applications of iPSC-Derived Beta Cells from Patients with Diabetes. Cell. Rep. Med. 2021, 2, 100238. [Google Scholar] [CrossRef]
- Rouhani, F.; Kumasaka, N.; de Brito, M.C.; Bradley, A.; Vallier, L.; Gaffney, D. Genetic Background Drives Transcrip-tional Variation in Human Induced Pluripotent Stem Cells. PLoS Genet. 2014, 10, e1004432. [Google Scholar] [CrossRef]
- Trombetta-Lima, M.; Sabogal-Guáqueta, A.M.; Dolga, A.M. Mitochondrial Dysfunction in Neurodegenerative Diseases: A Focus on iPSC-Derived Neuronal Models. Cell Calcium 2021, 94, 102362. [Google Scholar] [CrossRef]
- Miki, T.; Vazquez, L.; Yanuaria, L.; Lopez, O.; Garcia, I.M.; Ohashi, K.; Rodriguez, N.S. Induced Pluripotent Stem Cell Derivation and Ex Vivo Gene Correction Using a Mucopolysaccharidosis Type 1 Disease Mouse Model. Stem Cells Int. 2019, 2019, 6978303. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Shin, J.I.; Yang, J.W.; Lee, K.H.; Cha, D.H.; Hong, J.B.; Park, Y.; Choi, E.; Tizaoui, K.; Koyanagi, A.; et al. Genome Editing Using CRISPR-Cas9 and Autoimmune Diseases: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 1337. [Google Scholar] [CrossRef]
- Klementieva, N.; Goliusova, D.; Krupinova, J.; Yanvarev, V.; Panova, A.; Mokrysheva, N.; Kiselev, S.L. A Novel Iso-genic Human Cell-Based System for MEN1 Syndrome Generated by CRISPR/Cas9 Genome Editing. Int. J. Mol. Sci. 2021, 22, 12054. [Google Scholar] [CrossRef]
- Garin, I.; De Nanclares, G.P.; Gastaldo, E.; Harries, L.; Rubio-Cabezas, O.; Castaño, L. Permanent Neonatal Diabetes Caused by Creation of an Ectopic Splice Site within the INS Gene. PLoS ONE 2012, 7, e29205. [Google Scholar] [CrossRef] [Green Version]
- Tikhonovich, Y.V.; Petryaykina, E.E.; Timofeev, A.V.; Zubkova, N.A.; Kolodkina, A.A.; Sorkina, E.L.; Vasiliev, E.V.; Petrov, V.M.; Andrianova, E.A.; Zilberman, L.I.; et al. Clinical, hormonal and molecular-genetic characteristics of monogenic diabetes mellitus associated with the mutations in the INS gene. Diabetes Mellit. 2022, 24, 414–421. [Google Scholar] [CrossRef]
- Panova, A.V.; Klementieva, N.V.; Sycheva, A.V.; Goliusova, D.V.; Khokhlov, N.V.; Zubkova, N.A.; Tiulpakov, A.N.; Kiselev, S.L. Generation of an induced pluripotent stem cell line MNDINSi001-A from a patient with neonatal diabetes caused by a heterozygous INS mutation. Stem Cell Res. 2020, 47, 101929. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Kwart, D.; Paquet, D.; Teo, S.; Tessier-Lavigne, D.K.D.P.S.T.M. Precise and efficient scarless genome editing in stem cells using CORRECT. Nat. Protoc. 2017, 12, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J.A.; Cardenas-Diaz, F.L.; Gadue, P.; French, D.L. Highly Efficient CRISPR-Cas9-Mediated Genome Editing in Human Pluripotent Stem Cells. Curr. Protoc. Stem Cell Biol. 2019, 48, e64. [Google Scholar] [CrossRef] [Green Version]
- Colombo, C.; Porzio, O.; Liu, M.; Massa, O.; Vasta, M.; Salardi, S.; Beccaria, L.; Monciotti, C.; Toni, S.; Pedersen, O.; et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J. Clin. Investig. 2008, 118, 2148–2156. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shen, J.; Arenzana, N.; Tirasophon, W.; Kaufman, R.J.; Prywes, R. Activation of ATF6 and an ATF6 DNA Binding Site by the Endoplasmic Reticulum Stress Response. J. Biol. Chem. 2000, 275, 27013–27020. [Google Scholar] [CrossRef]
- Takayanagi, S.; Fukuda, R.; Takeuchi, Y.; Tsukada, S.; Yoshida, K. Gene regulatory network of unfolded protein response genes in endoplasmic reticulum stress. Cell Stress Chaperon 2013, 18, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Støy, J.; Steiner, D.F.; Park, S.-Y.; Ye, H.; Philipson, L.H.; Bell, G.I. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev. Endocr. Metab. Disord. 2010, 11, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Ye, H.; Steiner, D.F.; Bell, G.I. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem. Biophys. Res. Commun. 2010, 391, 1449–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmody, D.; Park, S.-Y.; Ye, H.; Perrone, M.E.; Alkorta-Aranburu, G.; Highland, H.M.; Hanis, C.L.; Philipson, L.H.; I Bell, G.; Greeley, S.A.W. Continued lessons from theINSgene: An intronic mutation causing diabetes through a novel mechanism. J. Med Genet. 2015, 52, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruin, J.E.; Erener, S.; Vela, J.; Hu, X.; Johnson, J.D.; Kurata, H.T.; Lynn, F.C.; Piret, J.M.; Asadi, A.; Rezania, A.; et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 2014, 12, 194–208. [Google Scholar] [CrossRef] [Green Version]
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone–expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; De Franco, E.; Arvan, P.; Cnop, M. Pathological β-Cell Endoplasmic Reticulum Stress in Type 2 Diabetes: Current Evidence. Front. Endocrinol. 2021, 12, 650158. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Riahi, Y.; Israeli, T.; Yeroslaviz, R.; Chimenez, S.; Avrahami, D.; Stolovich-Rain, M.; Alter, I.; Sebag, M.; Polin, N.; Bernal-Mizrachi, E.; et al. Inhibition of mTORC1 by ER stress impairs neonatal β-cell expansion and predisposes to diabetes in the Akita mouse. eLife 2018, 7, e38472. [Google Scholar] [CrossRef]
- Herbach, N.; Rathkolb, B.; Kemter, E.; Pichl, L.; Klaften, M.; de Angelis, M.H.; Halban, P.A.; Wolf, E.; Aigner, B.; Wanke, R. Dominant-Negative Effects of a Novel Mutated Ins2 Allele Causes Early-Onset Diabetes and Severe β-Cell Loss in Munich Ins2C95S Mutant Mice. Diabetes 2007, 56, 1268–1276. [Google Scholar] [CrossRef] [Green Version]
- McGrath, P.S.; Watson, C.L.; Ingram, C.; Helmrath, M.A.; Wells, J.M. The Basic Helix-Loop-Helix Transcription Factor NEUROG3 Is Required for Development of the Human Endocrine Pancreas. Diabetes 2015, 64, 2497–2505. [Google Scholar] [CrossRef] [Green Version]
- Novosyadlyy, R.; Kurshan, N.; Lann, D.; Vijayakumar, A.; Yakar, S.; Leroith, D. Insulin-like growth factor-I protects cells from ER stress-induced apoptosis via enhancement of the adaptive capacity of endoplasmic reticulum. Cell Death Differ. 2008, 15, 1304–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inageda, K. Insulin modulates induction of glucose-regulated protein 78 during endoplasmic reticulum stress via augmentation of ATF4 expression in human neuroblastoma cells. FEBS Lett. 2010, 584, 3649–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panova, A.V.; Klementieva, N.V.; Sycheva, A.V.; Korobko, E.V.; Sosnovtseva, A.O.; Krasnova, T.S.; Karpova, M.R.; Rubtsov, P.M.; Tikhonovich, Y.V.; Tiulpakov, A.N.; et al. Aberrant Splicing of INS Impairs Beta-Cell Differentiation and Proliferation by ER Stress in the Isogenic iPSC Model of Neonatal Diabetes. Int. J. Mol. Sci. 2022, 23, 8824. https://doi.org/10.3390/ijms23158824
Panova AV, Klementieva NV, Sycheva AV, Korobko EV, Sosnovtseva AO, Krasnova TS, Karpova MR, Rubtsov PM, Tikhonovich YV, Tiulpakov AN, et al. Aberrant Splicing of INS Impairs Beta-Cell Differentiation and Proliferation by ER Stress in the Isogenic iPSC Model of Neonatal Diabetes. International Journal of Molecular Sciences. 2022; 23(15):8824. https://doi.org/10.3390/ijms23158824
Chicago/Turabian StylePanova, Alexandra V., Natalia V. Klementieva, Anna V. Sycheva, Elena V. Korobko, Anastasia O. Sosnovtseva, Tatiana S. Krasnova, Maria R. Karpova, Petr M. Rubtsov, Yulia V. Tikhonovich, Anatoly N. Tiulpakov, and et al. 2022. "Aberrant Splicing of INS Impairs Beta-Cell Differentiation and Proliferation by ER Stress in the Isogenic iPSC Model of Neonatal Diabetes" International Journal of Molecular Sciences 23, no. 15: 8824. https://doi.org/10.3390/ijms23158824
APA StylePanova, A. V., Klementieva, N. V., Sycheva, A. V., Korobko, E. V., Sosnovtseva, A. O., Krasnova, T. S., Karpova, M. R., Rubtsov, P. M., Tikhonovich, Y. V., Tiulpakov, A. N., & Kiselev, S. L. (2022). Aberrant Splicing of INS Impairs Beta-Cell Differentiation and Proliferation by ER Stress in the Isogenic iPSC Model of Neonatal Diabetes. International Journal of Molecular Sciences, 23(15), 8824. https://doi.org/10.3390/ijms23158824