
Stellettin B-Induced Oral Cancer Cell Death via Endoplasmic Reticulum Stress–Mitochondrial Apoptotic and Autophagic Signaling Pathway

 , ,
, ,
Abstract
:1. Introduction
2. Results
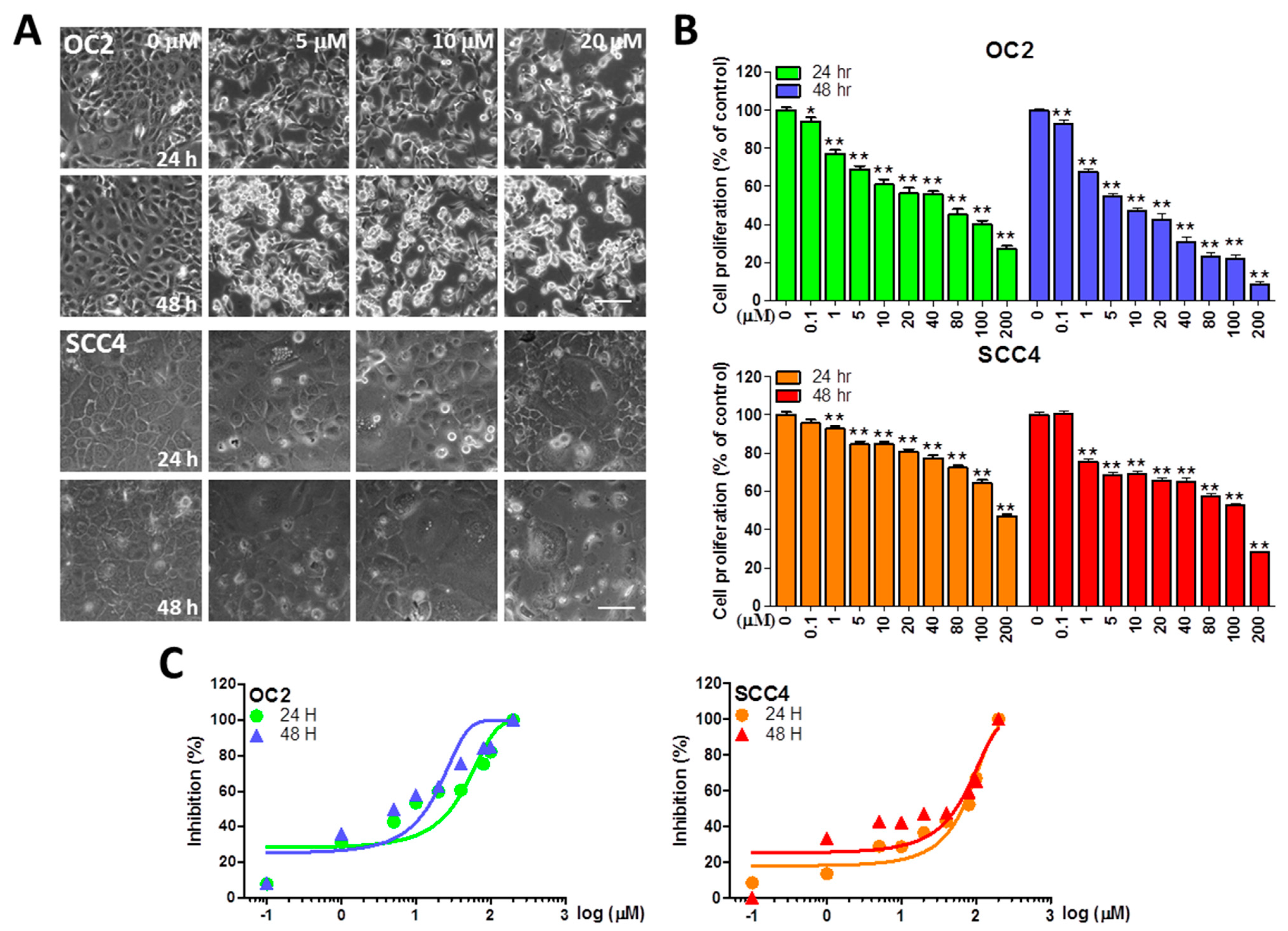
2.1. Stellettin B Inhibits the Viability of OSCC Cells
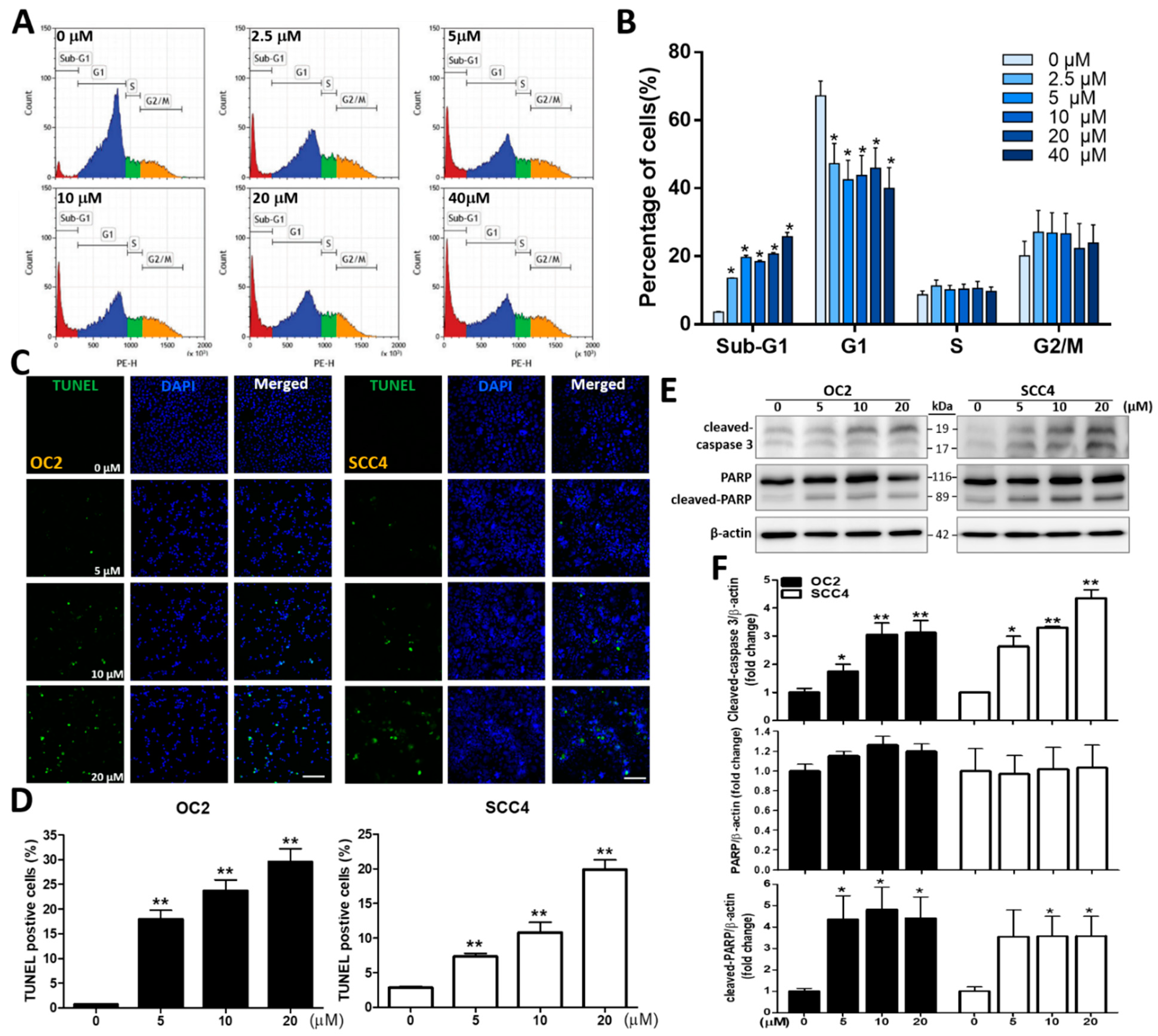
2.2. Stellettin B Induces Cell Cycle Arrest and Apoptosis
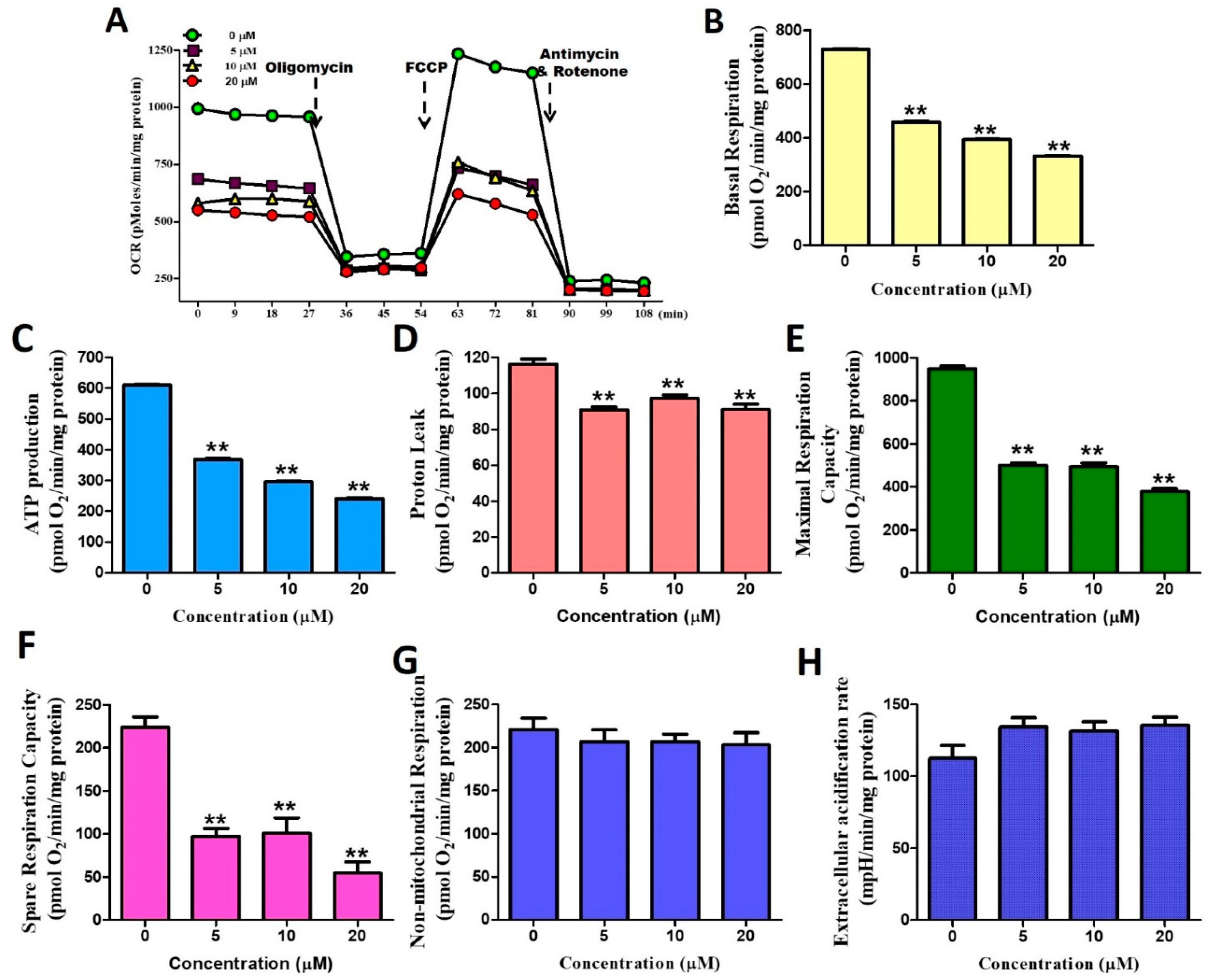
2.3. Stellettin B Down-Regulates Oxygen Consumption of Mitochondria
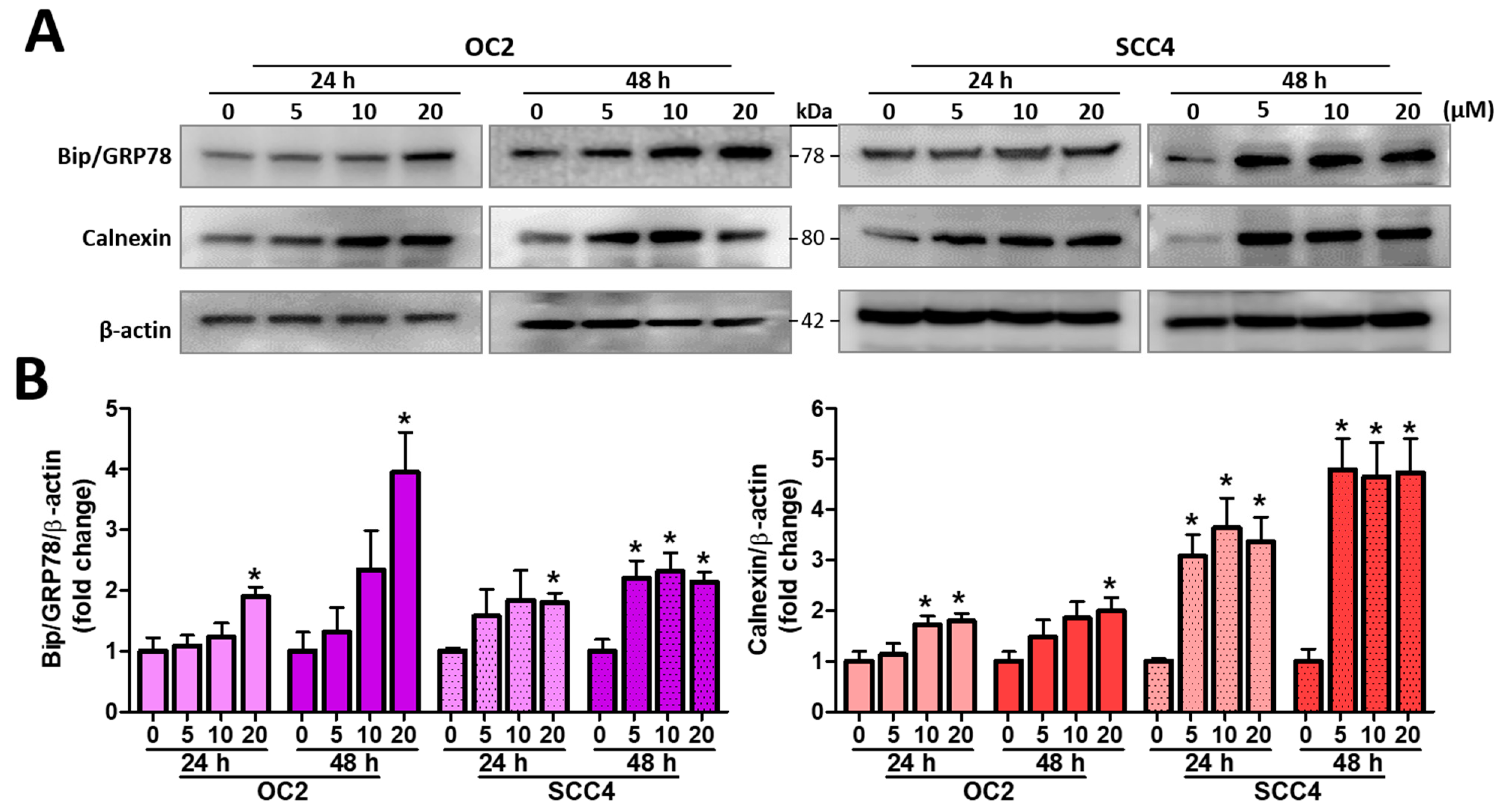
2.4. Stellettin B Induces ER Stress
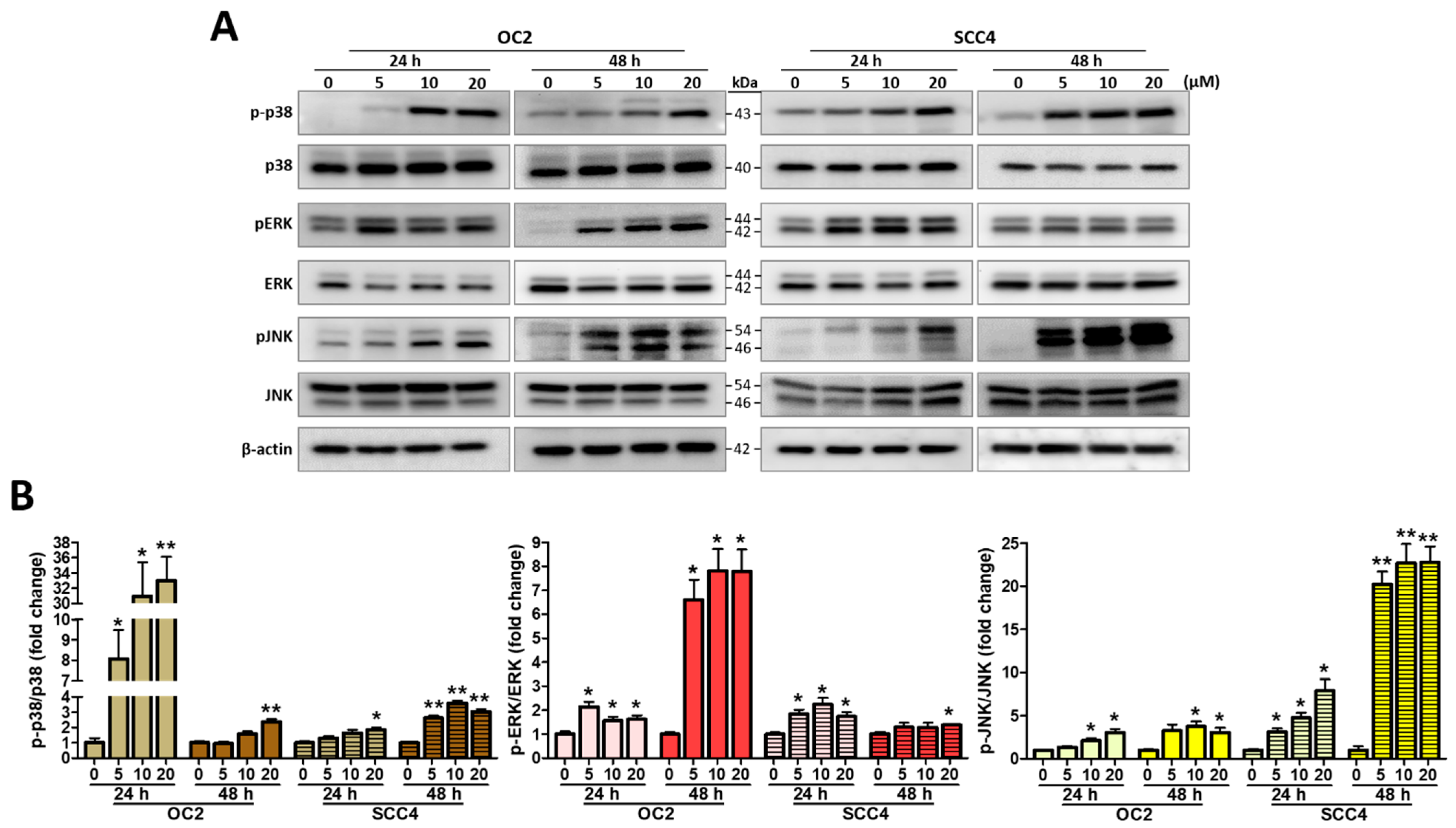
2.5. Stellettin B Induces MAPK Activation
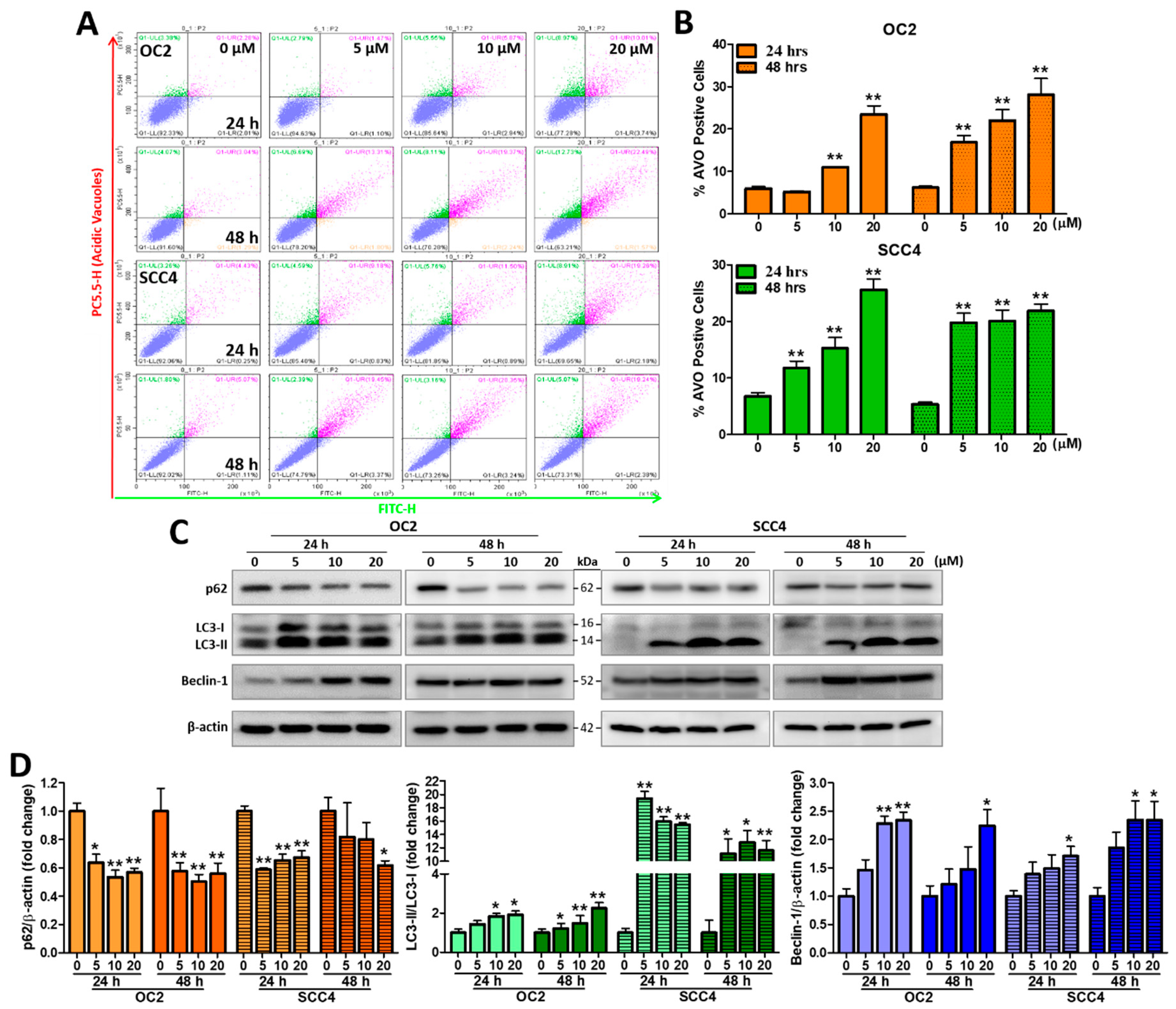
2.6. Stellettin B Induces Autophagy-Related Acidic Vesicular Organelles and Autophagic Protein Expression
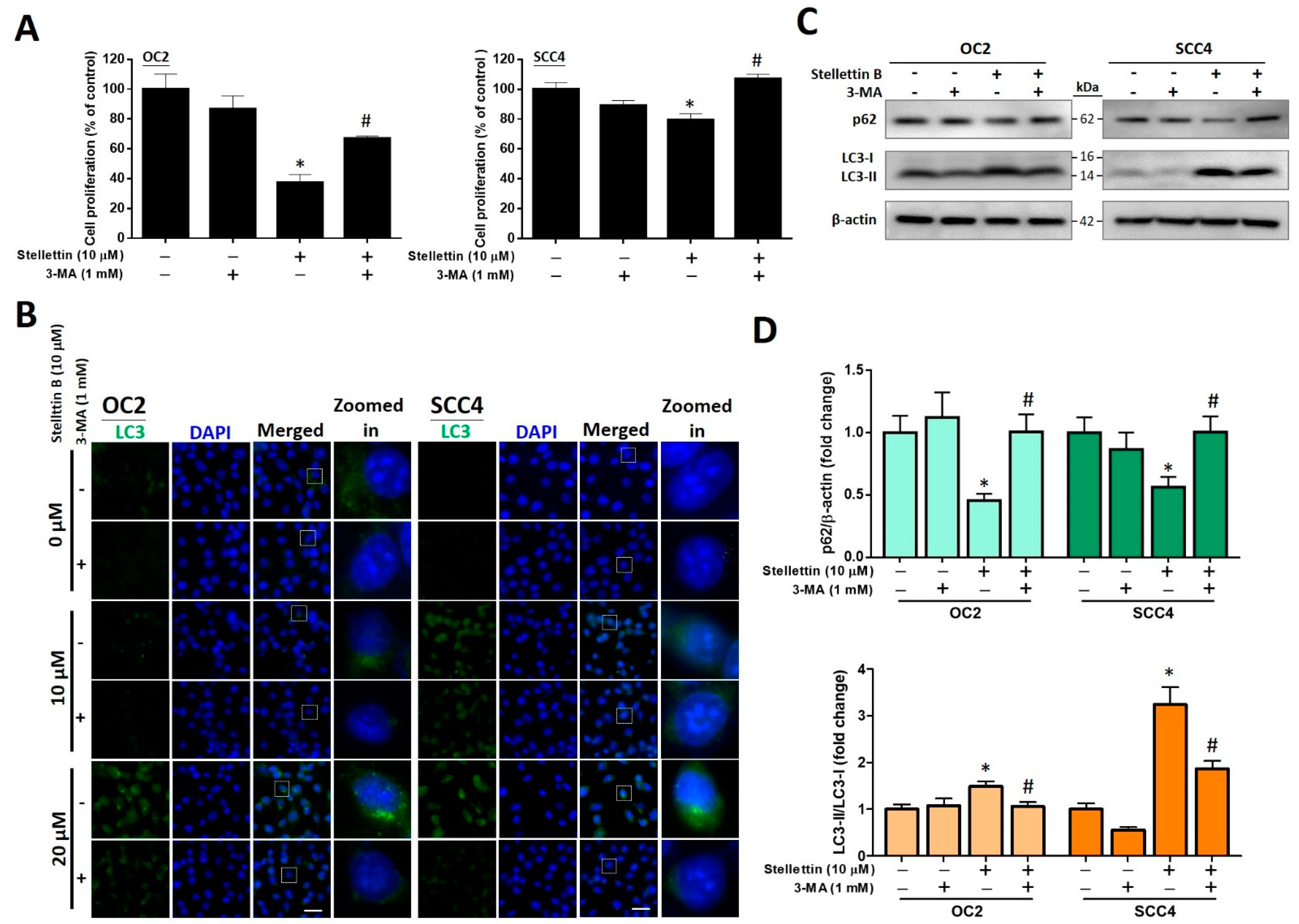
2.7. The Autophagy Inhibitor 3-MA Attenuates Stellettin B-Induced Cell Viability and Autophagic Proteins
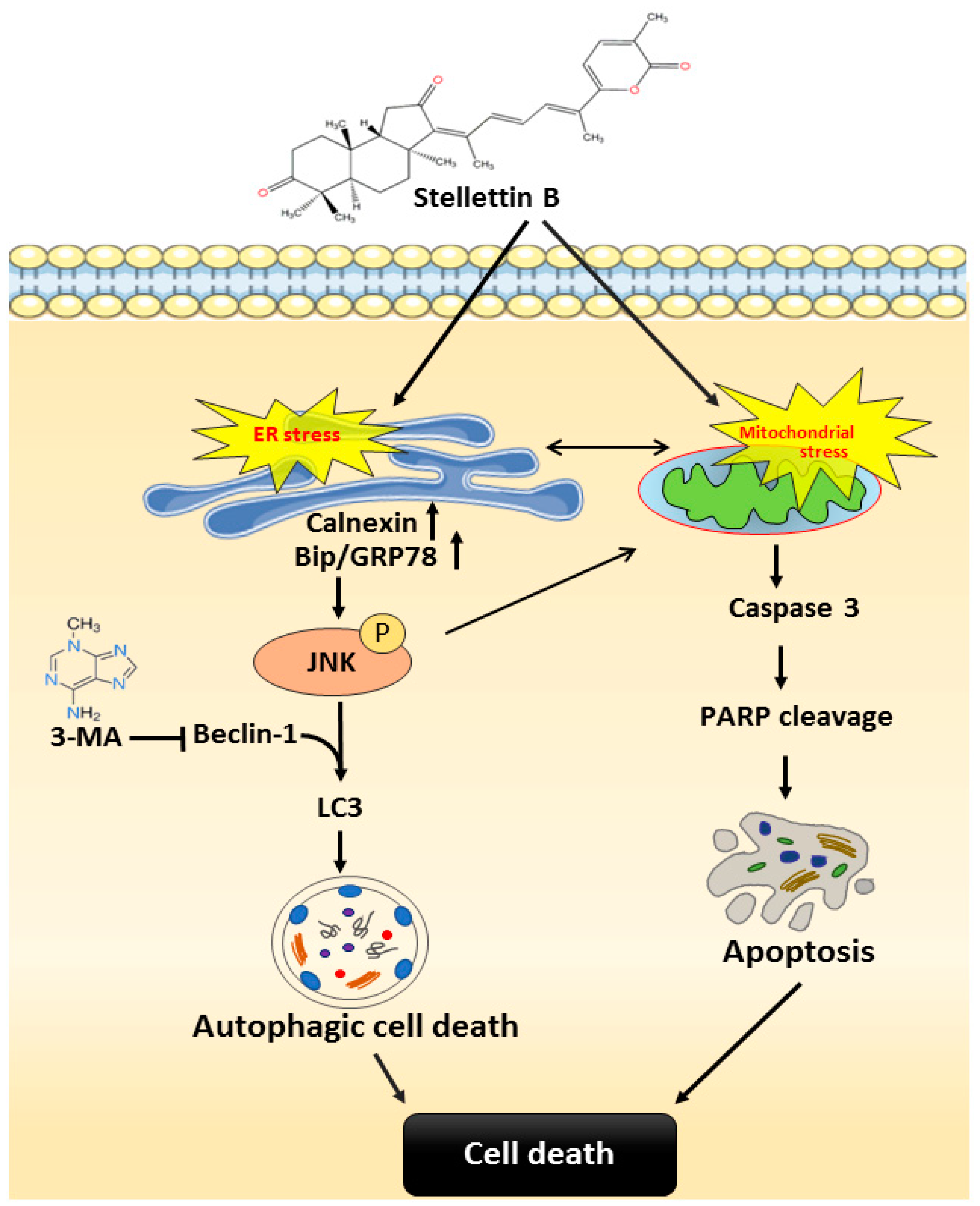
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Crystal Violet Assay
4.4. Cell Viability Assay: MTT Method
4.5. Cell Cycle Analysis
4.6. TUNEL Staining
4.7. Western Blot Analysis
4.8. Immunofluorescence Staining
4.9. Quantification of AVOs
4.10. Mitochondrial Respiration Measurement
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation & Definition
| 3-MA | 3-Methyladenine |
| AKT | Protein kinase B |
| AO | Acridine Orange |
| Atg5 | Autophagy related 5 |
| AVOs | Acidic Vesicular Organelles |
| BiP/GRP78 | Binding immunoglobulin Protein/Glucose Regulatory Protein 78 |
| BSA | Bovine Serum Albumin |
| DAPI | 4,6-Diamidino-2-Phenylindole |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl Sulfoxide |
| ECAR | Extra Cellular Acidification Rate |
| ER | Endoplasmic Reticulum |
| ERK | Extracellular signal-regulated kinases |
| FCCP | Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone |
| HO-1 | Heme-oxygenase 1 |
| HRP | Horseradish peroxidase |
| IC50 | 50% Inhibitory concentration |
| IgG | Immunoglobulin G |
| JNK | c-Jun N-terminal kinase |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| MAPK | Mitogen-activated protein kinase |
| mTOR | Mammalian target of rapamycin |
| MTT | 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NMMBA | National Museum of Marine Biology and Aquarium |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| OCRs | Oxygen Consumption Rates |
| OSCC | Oral Squamous Cell Carcinoma |
| OXPHOS | Oxidative phosphorylation |
| PARP | Poly (ADP-ribose) polymerase |
| PBS | Phosphate buffered saline |
| PI3Ks | Phosphoinositide 3-kinases |
| PVDF | Polyvinylidene difluoride |
| RPMI | Roswell Park Memorial Institute |
| RIPA | Radioimmunoprecipitation assay |
| p62/SQSTM1 | Ubiquitin-binding protein p62/Sequestosome-1 |
| ROS | Reactive Oxygen Species |
| SE | Standard error |
| TBST | Tris-buffered saline, 0.1% Tween 20 detergent |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick-end labeling |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Andreadis, C.; Vahtsevanos, K.; Sidiras, T.; Thomaidis, I.; Antoniadis, K.; Mouratidou, D. 5-fluorouracil and cisplatin in the treatment of advanced oral cancer. Oral Oncol. 2003, 39, 380–385. [Google Scholar] [CrossRef]
- Chin, D.; Boyle, G.M.; Porceddu, S.; Theile, D.R.; Parsons, P.G.; Coman, W.B. Head and neck cancer: Past, present and future. Expert Rev. Anticancer Ther. 2006, 6, 1111–1118. [Google Scholar] [CrossRef]
- Ko, C.Y.; Shih, P.C.; Huang, P.W.; Lee, Y.H.; Chen, Y.F.; Tai, M.H.; Liu, C.H.; Wen, Z.H.; Kuo, H.M. Sinularin, an anti-cancer agent causing mitochondria-modulated apoptosis and cytoskeleton disruption in human hepatocellular carcinoma. Int. J. Mol. Sci. 2021, 22, 3946. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.H.; Ma, Y.L.; Shih, P.C.; Chen, C.T.; Cheng, S.Y.; Pan, C.Y.; Jean, Y.H.; Chu, Y.M.; Lin, S.C.; Lai, Y.C.; et al. The antimicrobial peptide tilapia piscidin 3 induces mitochondria-modulated intrinsic apoptosis of osteosarcoma cells. Biochem. Pharmacol. 2020, 178, 114064–114075. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Shih, P.C.; Feng, C.W.; Wu, C.C.; Tsui, K.H.; Lin, Y.H.; Kuo, H.M.; Wen, Z.H. Pardaxin activates excessive mitophagy and mitochondria-mediated apoptosis in human ovarian cancer by inducing reactive oxygen species. Antioxidants 2021, 10, 1883. [Google Scholar] [CrossRef]
- Lin, Y.D.; Chen, S.; Yue, P.; Zou, W.; Benbrook, D.M.; Liu, S.; Le, T.C.; Berlin, K.D.; Khuri, F.R.; Sun, S.Y. Caat/enhancer binding protein homologous protein-dependent death receptor 5 induction is a major component of sheta2-induced apoptosis in lung cancer cells. Cancer Res. 2008, 68, 5335–5344. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Hubbard, S.R. How ire1 reacts to er stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under er stress. J. Cell. Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an atg7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell. Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.; Lopez, J.M. Understanding mapk signaling pathways in apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. P38 and jnk mapk pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.L.; McKee, T.C.; Cardellina, J.H., 2nd; Leid, M.; Boyd, M.R. Cytotoxic triterpenes from a marine sponge, stelletta sp. J. Nat. Prod. 1996, 59, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Chen, N.F.; Lin, P.Y.; Su, J.H.; Chen, B.H.; Kuo, H.M.; Sung, C.S.; Sung, P.J.; Wen, Z.H.; Chen, W.F. Anti-invasion and antiangiogenic effects of stellettin b through inhibition of the akt/girdin signaling pathway and vegf in glioblastoma cells. Cancers 2019, 11, 220. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.W.; Chen, N.F.; Wen, Z.H.; Yang, W.Y.; Kuo, H.M.; Sung, P.J.; Su, J.H.; Cheng, S.Y.; Chen, W.F. In vitro and in vivo neuroprotective effects of stellettin b through anti-apoptosis and the nrf2/ho-1 pathway. Mar. Drugs 2019, 17, 315. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.A.; Zhou, Q.; Guo, W.Z.; Qiu, Y.; Wang, R.; Jin, M.; Zhang, W.; Li, K.; Yamori, T.; Dan, S.; et al. In vitro antitumor activity of stellettin b, a triterpene from marine sponge jaspis stellifera, on human glioblastoma cancer sf295 cells. Mar. Drugs 2014, 12, 4200. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhang, Q.; Peng, X.; Zhou, C.; Zhong, Y.; Chen, X.; Qiu, Y.; Jin, M.; Gong, M.; Kong, D. Stellettin b induces g1 arrest, apoptosis and autophagy in human non-small cell lung cancer a549 cells via blocking pi3k/akt/mtor pathway. Sci. Rep. 2016, 6, 27071–27080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.K.; Ho, J.C.; Che, C.T. Apoptotic activity of isomalabaricane triterpenes on human promyelocytic leukemia hl60 cells. Cancer Lett. 2005, 230, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, Q.; Zhang, L.; Zhong, Y.; Fan, G.; Zhang, Z.; Wang, R.; Jin, M.; Qiu, Y.; Kong, D. Stellettin b induces apoptosis in human chronic myeloid leukemia cells via targeting pi3k and stat5. Oncotarget 2017, 8, 28906–28921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajstura, M.; Halicka, H.D.; Pryjma, J.; Darzynkiewicz, Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “sub-g1” peaks on DNA content histograms. Cytometry A 2007, 71, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the cell cycle: A complex landscape. Front. Oncol. 2017, 7, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Darling, N.J.; Cook, S.J. The role of mapk signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [Green Version]
- Cagnol, S.; Chambard, J.C. Erk and cell death: Mechanisms of erk-induced cell death—Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Stankiewicz, M.; Jonas, W.; Hadas, E.; Cabaj, W.; Douch, P.G. Supravital staining of eosinophils. Int. J. Parasitol. 1996, 26, 445–446. [Google Scholar] [CrossRef]
- Traganos, F.; Darzynkiewicz, Z. Lysosomal proton pump activity: Supravital cell staining with acridine orange differentiates leukocyte subpopulations. Methods Cell Biol. 1994, 41, 185–194. [Google Scholar]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- García-Heredia, J.M.; Carnero, A. Role of mitochondria in cancer stem cell resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Nay, B.; Yang, M.; Ni, Y.; Wang, H.; Yao, L.; Li, X. Marine sponges of the genus stelletta as promising drug sources: Chemical and biological aspects. Acta Pharm. Sin. B 2019, 9, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.K.; Ling, Y.H.; Cheung, F.W.; Che, C.T. Stellettin a induces endoplasmic reticulum stress in murine b16 melanoma cells. J. Nat. Prod. 2012, 75, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. Er stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Limonta, P.; Moretti, R.M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Montagnani Marelli, M. Role of endoplasmic reticulum stress in the anticancer activity of natural compounds. Int. J. Mol. Sci. 2019, 20, 961. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ni, M.; Lee, B.; Barron, E.; Hinton, D.R.; Lee, A.S. The unfolded protein response regulator grp78/bip is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell. Death Differ. 2008, 15, 1460–1471. [Google Scholar] [CrossRef]
- Li, C.; Johnson, D.E. Bortezomib induces autophagy in head and neck squamous cell carcinoma cells via jnk activation. Cancer Lett. 2012, 314, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Ansari, S.S.; Sharma, A.K.; Soni, H.; Ali, D.M.; Tews, B.; König, R.; Eibl, H.; Berger, M.R. Induction of er and mitochondrial stress by the alkylphosphocholine erufosine in oral squamous cell carcinoma cells. Cell. Death Dis. 2018, 9, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Russo, G.L. Autophagy inducers in cancer. Biochem. Pharmacol. 2018, 153, 51–61. [Google Scholar] [CrossRef]
- Hsieh, M.J.; Chien, S.Y.; Lin, J.T.; Yang, S.F.; Chen, M.K. Polyphyllin g induces apoptosis and autophagy cell death in human oral cancer cells. Phytomedicine 2016, 23, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lee, C.Y.; Lu, C.C.; Tsai, F.J.; Hsu, Y.M.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Yang, J.S.; Wang, C.C. Resveratrol-induced autophagy and apoptosis in cisplatin-resistant human oral cancer car cells: A key role of ampk and akt/mtor signaling. Int. J. Oncol. 2017, 50, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. Map kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Deng, Z.; Pan, H.; Gu, L.; Liu, O.; Tang, Z. Mitogen-activated protein kinase signaling pathway in oral cancer. Oncol. Lett. 2018, 15, 1379–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Kim, J.H.; Lee, G.H.; Kim, H.T.; Lim, J.M.; Chae, S.W.; Chae, H.J.; Kim, H.R. P38 mitogen-activated protein kinase is involved in endoplasmic reticulum stress-induced cell death and autophagy in human gingival fibroblasts. Biol Pharm. Bull. 2010, 33, 545–549. [Google Scholar] [CrossRef] [Green Version]
- Dhanasekaran, D.N.; Reddy, E.P. Jnk signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [Green Version]
- Gkouveris, I.; Nikitakis, N.G. Role of jnk signaling in oral cancer: A mini review. Tumour Biol. 2017, 39, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Konishi, A.; Nishida, Y.; Mizuta, T.; Nishina, H.; Yamamoto, A.; Tsujimoto, Y. Involvement of jnk in the regulation of autophagic cell death. Oncogene 2010, 29, 2070–2082. [Google Scholar] [CrossRef] [Green Version]
- Lou, M.; Zhang, L.N.; Ji, P.G.; Feng, F.Q.; Liu, J.H.; Yang, C.; Li, B.F.; Wang, L. Quercetin nanoparticles induced autophagy and apoptosis through akt/erk/caspase-3 signaling pathway in human neuroglioma cells: In vitro and in vivo. Biomed. Pharmacother 2016, 84, 1–9. [Google Scholar] [CrossRef]
- Shih, A.; Davis, F.B.; Lin, H.Y.; Davis, P.J. Resveratrol induces apoptosis in thyroid cancer cell lines via a mapk- and p53-dependent mechanism. J. Clin. Endocrinol. Metab. 2002, 87, 1223–1232. [Google Scholar] [CrossRef]
- Kim, H.J.; Chakravarti, N.; Oridate, N.; Choe, C.; Claret, F.X.; Lotan, R. N-(4-hydroxyphenyl)retinamide-induced apoptosis triggered by reactive oxygen species is mediated by activation of mapks in head and neck squamous carcinoma cells. Oncogene 2006, 25, 2785–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.C.; Hsieh, M.J.; Chen, C.J.; Lin, J.T.; Lo, Y.S.; Chuang, Y.C.; Chien, S.Y.; Chen, M.K. Polyphyllin g induce apoptosis and autophagy in human nasopharyngeal cancer cells by modulation of akt and mitogen-activated protein kinase pathways in vitro and in vivo. Oncotarget 2016, 7, 70276–70289. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A.; Dev, S. A new class of triterpenoids from ailanthus malabarica dc derivatives of malabaricane. Tetrahedron Lett. 1967, 8, 4837–4843. [Google Scholar] [CrossRef]
- Sobti, R.R.; Dev, S. A direct correlation of (+)-malabaricol with (+)-ambreinolide. Tetrahedron Lett. 1968, 9, 2215–2217. [Google Scholar] [CrossRef]
- Paton, W.F.; Paul, I.C.; Bajaj, A.G.; Dev, S. The structure of malabricol. Tetrahedron Lett. 1979, 20, 4153–4154. [Google Scholar] [CrossRef]
- Tasdemir, D.; Mangalindan, G.C.; Concepción, G.P.; Verbitski, S.M.; Rabindran, S.; Miranda, M.; Greenstein, M.; Hooper, J.N.A.; Harper, M.K.; Ireland, C.M. Bioactive isomalabaricane triterpenes from the marine sponge rhabdastrella globostellata. J. Nat. Prod. 2002, 65, 210–214. [Google Scholar] [CrossRef]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [Green Version]
- Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine sponge natural products with anticancer potential: An updated review. Mar. Drugs 2017, 15, 310. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.M.; Tseng, C.C.; Chen, N.F.; Tai, M.H.; Hung, H.C.; Feng, C.W.; Cheng, S.Y.; Huang, S.Y.; Jean, Y.H.; Wen, Z.H. Msp-4, an antimicrobial peptide, induces apoptosis via activation of extrinsic fas/fasl- and intrinsic mitochondria-mediated pathways in one osteosarcoma cell line. Mar. Drugs 2018, 16, 8. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Host | Catalog # | Supplier | Dilution Ratio |
|---|---|---|---|---|
| Cleaved caspase-3 | Rabbit | 9664S | Cell Signaling | 1:1000 |
| PARP | Rabbit | 9542S | Cell Signaling | 1:1000 |
| p62/SQSTM1 | Rabbit | 18420-1-ap | ProteinTech | 1:1000 |
| Beclin-1 | Rabbit | 11306-1-ap | ProteinTech | 1:1000 |
| LC3 | Rabbit | AP1802a | Abgent | 1:1000 |
| Bip/GRP78 | Rabbit | ab21685 | Abcam | 1:1000 |
| Calnexin | Rabbit | ab22595 | Abcam | 1:1000 |
| Phospho-p38 | Rabbit | 4511S | Cell Signaling | 1:1000 |
| p38 | Rabbit | 9212 | Cell Signaling | 1:1000 |
| Phospho-ERK | Rabbit | 4370S | Cell Signaling | 1:1000 |
| ERK | Rabbit | 4695 | Cell Signaling | 1:1000 |
| Phospho-JNK | Rabbit | 4668S | Cell Signaling | 1:1000 |
| JNK | Rabbit | 9252S | Cell Signaling | 1:1000 |
| β-actin (HRP conjugate) | Mouse | 12262S | Cell Signaling | 1:2000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, T.-J.; Jean, Y.-H.; Shih, P.-C.; Cheng, S.-Y.; Kuo, H.-M.; Lee, Y.-T.; Lai, Y.-C.; Tseng, C.-C.; Chen, W.-F.; Wen, Z.-H. Stellettin B-Induced Oral Cancer Cell Death via Endoplasmic Reticulum Stress–Mitochondrial Apoptotic and Autophagic Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 8813. https://doi.org/10.3390/ijms23158813
Kuo T-J, Jean Y-H, Shih P-C, Cheng S-Y, Kuo H-M, Lee Y-T, Lai Y-C, Tseng C-C, Chen W-F, Wen Z-H. Stellettin B-Induced Oral Cancer Cell Death via Endoplasmic Reticulum Stress–Mitochondrial Apoptotic and Autophagic Signaling Pathway. International Journal of Molecular Sciences. 2022; 23(15):8813. https://doi.org/10.3390/ijms23158813
Chicago/Turabian StyleKuo, Tsu-Jen, Yen-Hsuan Jean, Po-Chang Shih, Shu-Yu Cheng, Hsiao-Mei Kuo, Yi-Ting Lee, Yu-Cheng Lai, Chung-Chih Tseng, Wu-Fu Chen, and Zhi-Hong Wen. 2022. "Stellettin B-Induced Oral Cancer Cell Death via Endoplasmic Reticulum Stress–Mitochondrial Apoptotic and Autophagic Signaling Pathway" International Journal of Molecular Sciences 23, no. 15: 8813. https://doi.org/10.3390/ijms23158813
APA StyleKuo, T.-J., Jean, Y.-H., Shih, P.-C., Cheng, S.-Y., Kuo, H.-M., Lee, Y.-T., Lai, Y.-C., Tseng, C.-C., Chen, W.-F., & Wen, Z.-H. (2022). Stellettin B-Induced Oral Cancer Cell Death via Endoplasmic Reticulum Stress–Mitochondrial Apoptotic and Autophagic Signaling Pathway. International Journal of Molecular Sciences, 23(15), 8813. https://doi.org/10.3390/ijms23158813