
Increased Expression of Interferon-Induced Transmembrane 3 (IFITM3) in Stroke and Other Inflammatory Conditions in the Brain

 , and
, and
Abstract
:1. Introduction
2. Results
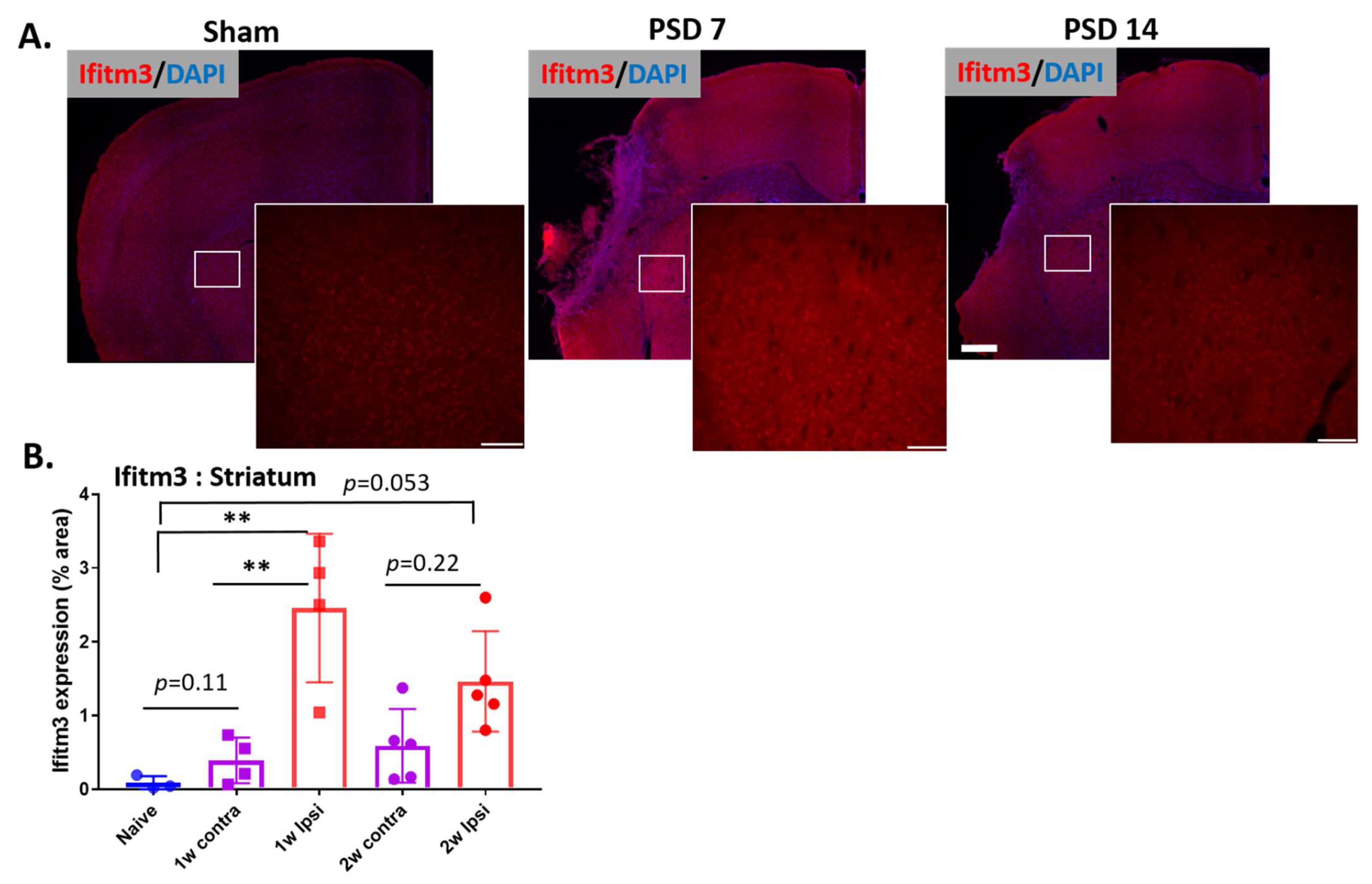
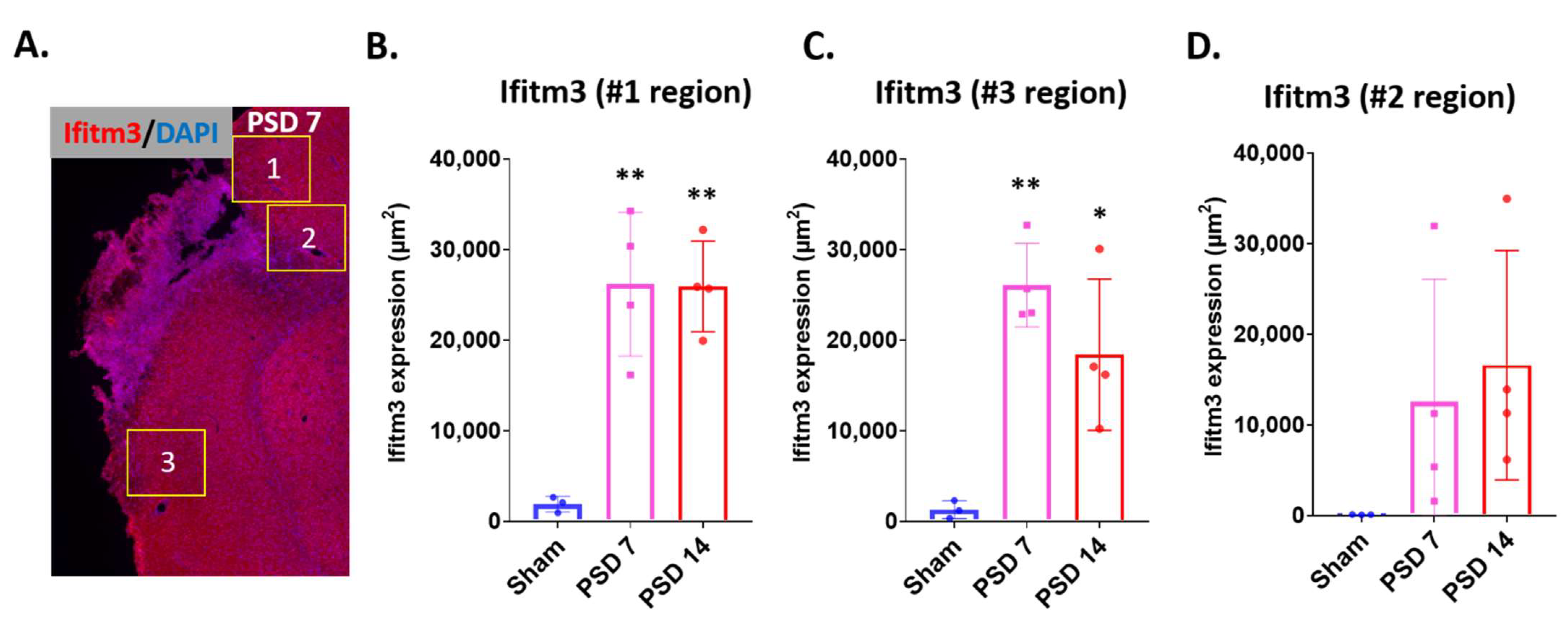
2.1. Ischemic Stroke Increases the Expression of IFITM3 Protein in the Striatum and Peri-Infarct Cortex in Aged Brains Following Stroke
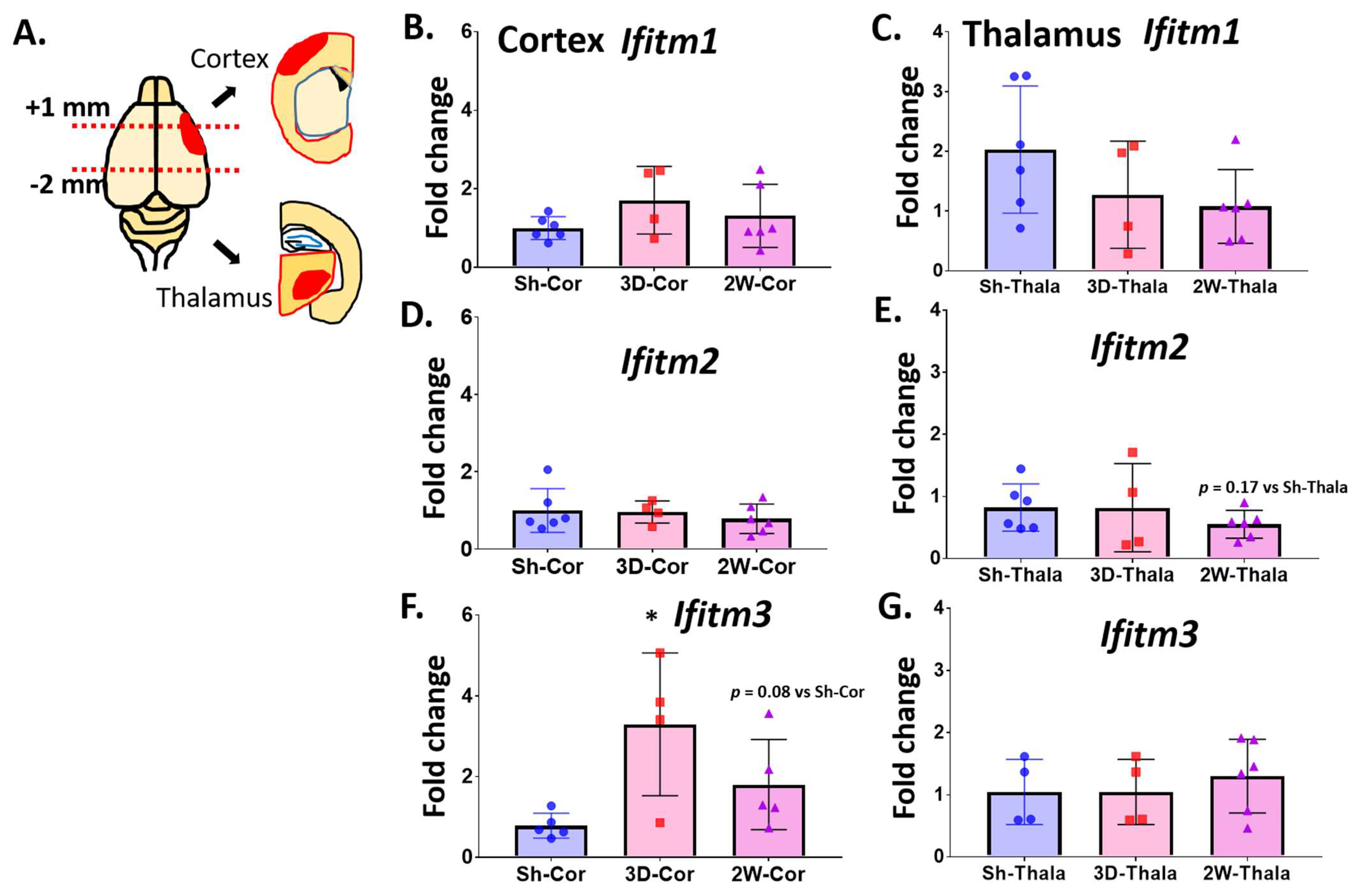
2.2. Ischemic Injury Causes the Induction of IFITM3 in Aged Brains Following Stroke
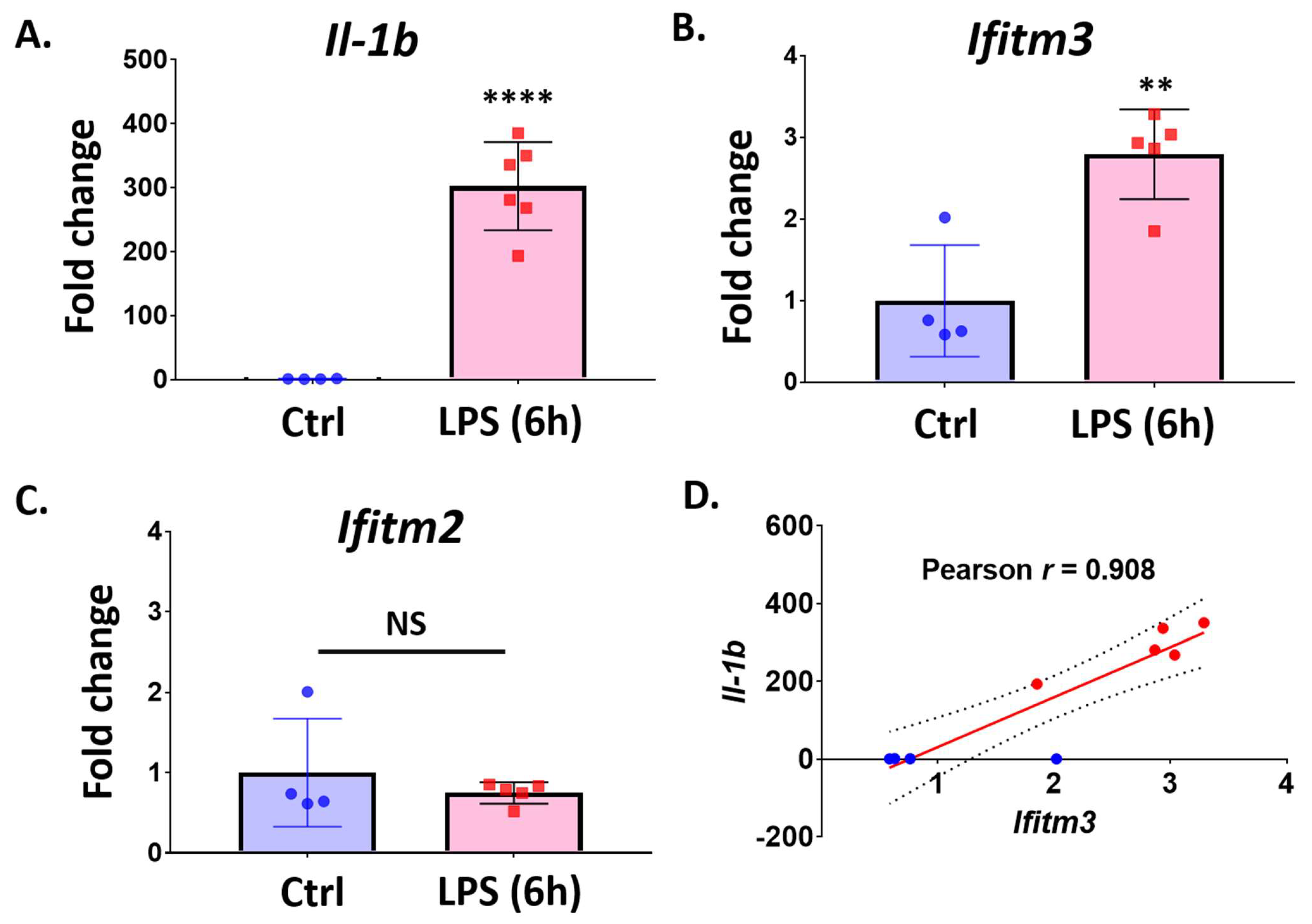
2.3. Lipopolysaccharide (LPS) Treatment Increases Ifitm3 mRNA in Primary Microglia
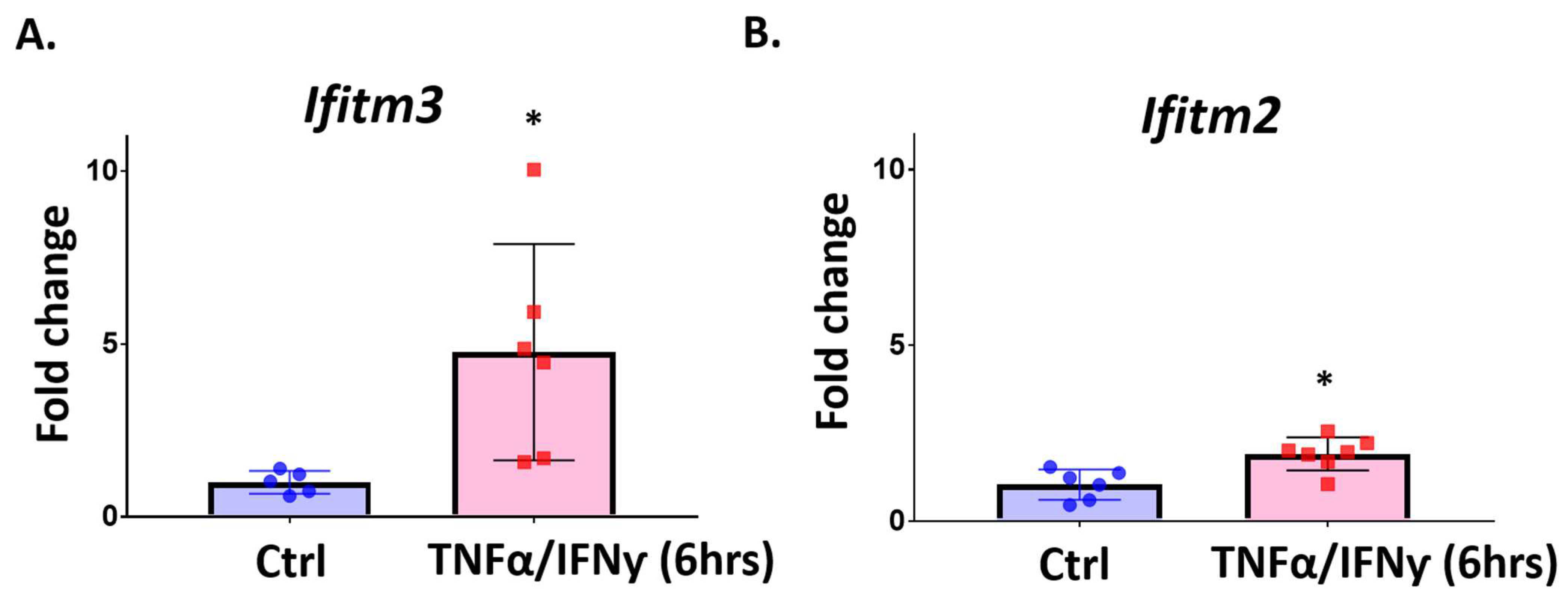
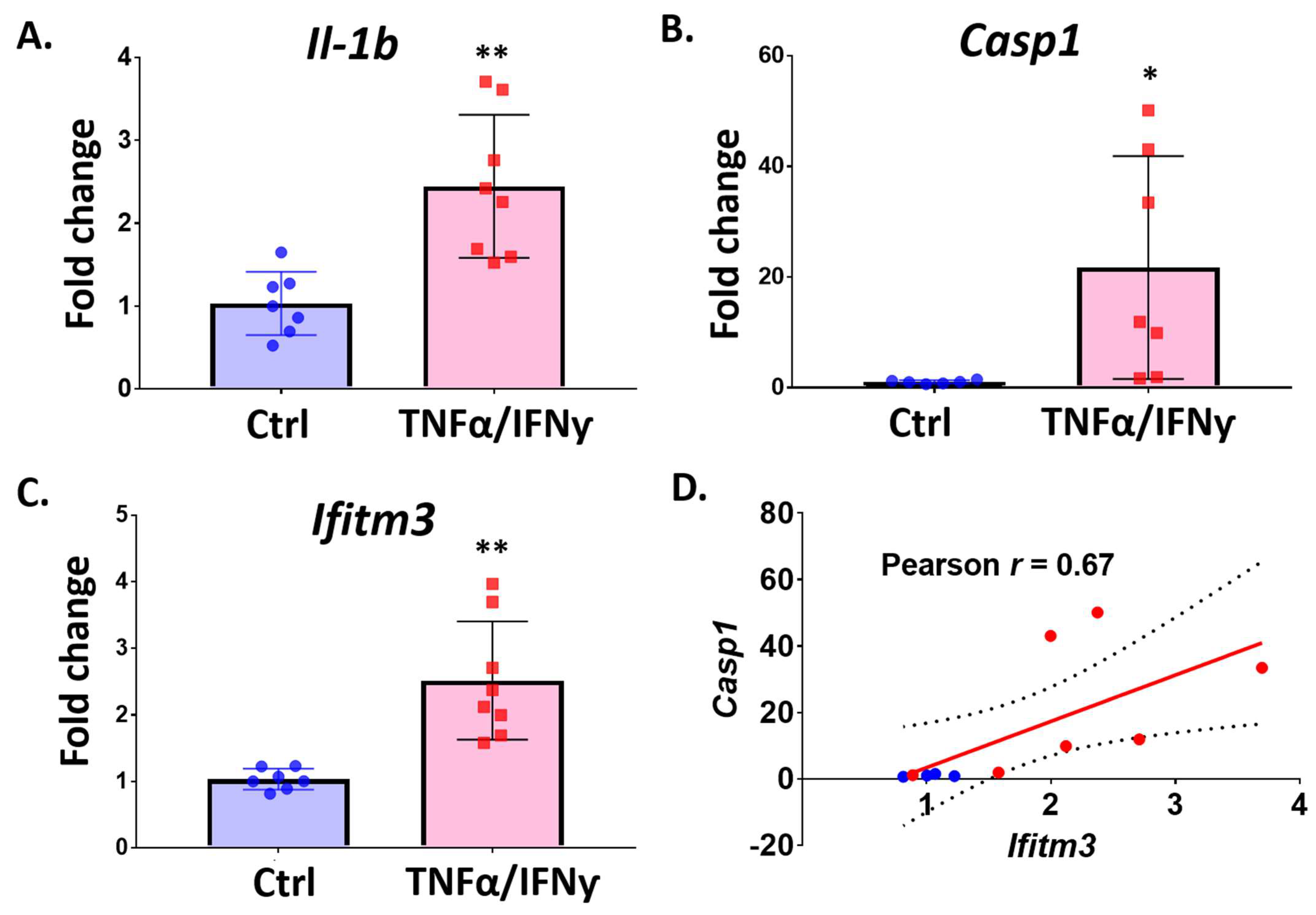
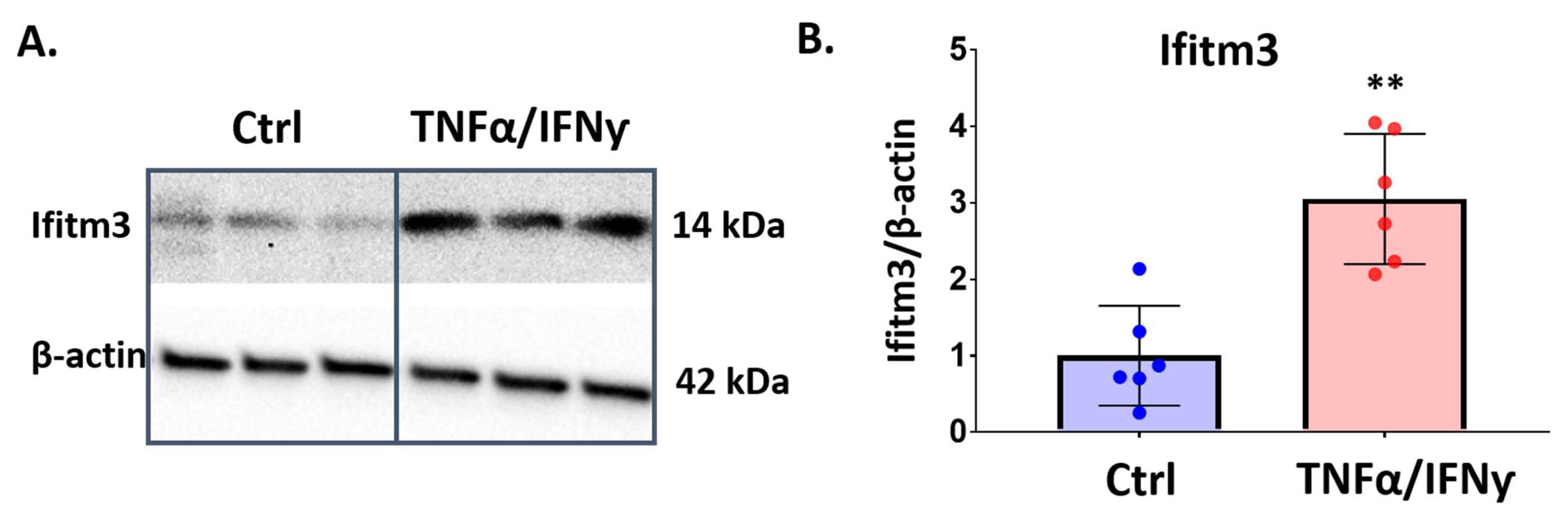
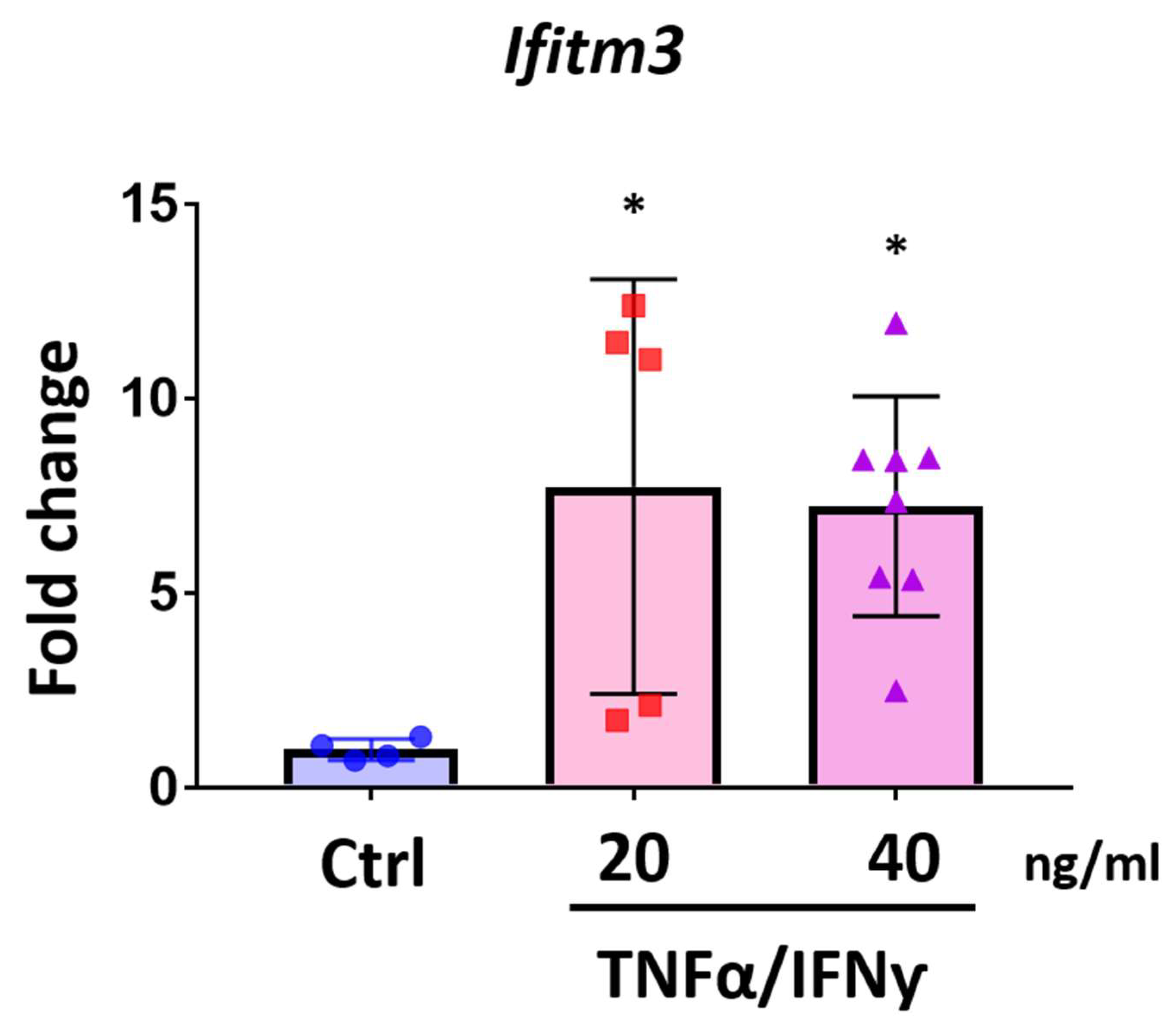
2.4. Treatment with Pro-Inflammatory Cytokines Induces Ifitm3 mRNA in Primary Microglia and Sim-A9 Cells
2.5. Amyloid-Beta Peptide Increases the Expression of Ifitm3
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Permanent Distal Middle Cerebral Artery Occlusion (PDMCAO) Model
4.3. Brain Sample Preparation and Immunostaining
4.4. Primary Microglia Culture
4.5. Sim-A9 Cell Culture
4.6. RNA Isolation and qRT-PCR
4.7. Western Blotting
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jakel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneriation. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the Central Nervous System. Cell Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.Q.; Ma, X.T.; Hu, Z.W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.J.; Tian, D.S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef]
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.T. Microglial aging in the healthy CNS: Phenotypes, drivers, and rejuvenation. Front. Cell Neurosci. 2013, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 606456. [Google Scholar] [CrossRef]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- de Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Nazmi, A.; Field, R.H.; Griffin, E.W.; Haugh, O.; Hennessy, E.; Cox, D.; Reis, R.; Tortorelli, L.; Murray, C.L.; Lopez-Rodriguez, A.B.; et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 2019, 67, 1254–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.P.; Henry, R.J.; Shirey, K.A.; Doran, S.J.; Makarevich, O.D.; Ritzel, R.M.; Meadows, V.A.; Vogel, S.N.; Faden, A.I.; Stoica, B.A.; et al. Interferon-beta Plays a Detrimental Role in Experimental Traumatic Brain Injury by Enhancing Neuroinflammation That Drives Chronic Neurodegeneration. J. Neurosci. 2020, 40, 2357–2370. [Google Scholar] [CrossRef] [PubMed]
- Roy, E.R.; Chiu, G.; Li, S.; Propson, N.E.; Kanchi, R.; Wang, B.; Coarfa, C.; Zheng, H.; Cao, W. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid beta plaques. Immunity 2022, 55, 879–894.e6. [Google Scholar] [CrossRef]
- Hur, J.Y.; Frost, G.R.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The innate immunity protein IFITM3 modulates gamma-secretase in Alzheimer’s disease. Nature 2020, 586, 735–740. [Google Scholar] [CrossRef]
- Kim, G.S.; Stephenson, J.M.; Al Mamun, A.; Wu, T.; Goss, M.G.; Min, J.W.; Li, J.; Liu, F.; Marrelli, S.P. Determining the effect of aging, recovery time, and post-stroke memantine treatment on delayed thalamic gliosis after cortical infarct. Sci. Rep. 2021, 11, 12613. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Winkler, C.A.; An, P.; Guo, J.T. IFITM Genes, Variants, and Their Roles in the Control and Pathogenesis of Viral Infections. Front. Microbiol. 2018, 9, 3228. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zeng, H.; Kumar, A.; Belser, J.A.; Maines, T.R.; Tumpey, T.M. Constitutively Expressed IFITM3 Protein in Human Endothelial Cells Poses an Early Infection Block to Human Influenza Viruses. J. Virol. 2016, 90, 11157–11167. [Google Scholar] [CrossRef] [Green Version]
- Spence, J.S.; He, R.; Hoffmann, H.H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J.; et al. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 2011, 7, e1002337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayana, S.K.; Helbig, K.J.; McCartney, E.M.; Eyre, N.S.; Bull, R.A.; Eltahla, A.; Lloyd, A.R.; Beard, M.R. The Interferon-induced Transmembrane Proteins, IFITM1, IFITM2, and IFITM3 Inhibit Hepatitis C Virus Entry. J. Biol. Chem. 2015, 290, 25946–25959. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.L.; Manly, S.P.; McMahon, M.; Kerr, I.M.; Stark, G.R. Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell 1984, 38, 745–755. [Google Scholar] [CrossRef]
- Lewin, A.R.; Reid, L.E.; McMahon, M.; Stark, G.R.; Kerr, I.M. Molecular analysis of a human interferon-inducible gene family. Eur. J. Biochem. 1991, 199, 417–423. [Google Scholar] [CrossRef]
- Yanez, D.C.; Ross, S.; Crompton, T. The IFITM protein family in adaptive immunity. Immunology 2020, 159, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef]
- Kummer, S.; Avinoam, O.; Krausslich, H.G. IFITM3 Clusters on Virus Containing Endosomes and Lysosomes Early in the Influenza A Infection of Human Airway Epithelial Cells. Viruses 2019, 11, 548. [Google Scholar] [CrossRef] [Green Version]
- Yanez, D.C.; Sahni, H.; Ross, S.; Solanki, A.; Lau, C.I.; Papaioannou, E.; Barbarulo, A.; Powell, R.; Lange, U.C.; Adams, D.J.; et al. IFITM proteins drive type 2 T helper cell differentiation and exacerbate allergic airway inflammation. Eur. J. Immunol. 2019, 49, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percie du Sert, N.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Hong, J.H.; Lahey, M.T.; Zhu, L.; Stephenson, J.M.; Marrelli, S.P. A low-cost mouse cage warming system provides improved intra-ischemic and post-ischemic body temperature control—Application for reducing variability in experimental stroke studies. J. Neurosci. Methods 2021, 360, 109228. [Google Scholar] [CrossRef] [PubMed]
- Nagamoto-Combs, K.; Kulas, J.; Combs, C.K. A novel cell line from spontaneously immortalized murine microglia. J. Neurosci. Methods 2014, 233, 187–198. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward | Reverse |
|---|---|---|
| Ifitm3 | TTCTGCTGCCTGGGCTTCATAG | ACCAAGGTGCTGATGTTCAGGC |
| Ifitm1 | GCCACCACAATCAACATGCCTG | ACCCACCATCTTCCTGTCCCTA |
| Ifitm2 | GTTCCAGAGTCAGTACCATGAG | GGCGTTGAAGAAGAGTGTATTG |
| Casp1 | TCTGTATTCACGCCCTGTTG | GATAAATTGCTTCCTCTTTGCCC |
| Il1b | CTGTGTCTTTCCCGTGGACC | CAGCTCATATGGGTCCGACA |
| Gapdh | GATGGCAACAATCTCCACTTTGC | GCCGCATCTTCTTGTGCAGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harmon, E.; Doan, A.; Bautista-Garrido, J.; Jung, J.E.; Marrelli, S.P.; Kim, G.S. Increased Expression of Interferon-Induced Transmembrane 3 (IFITM3) in Stroke and Other Inflammatory Conditions in the Brain. Int. J. Mol. Sci. 2022, 23, 8885. https://doi.org/10.3390/ijms23168885
Harmon E, Doan A, Bautista-Garrido J, Jung JE, Marrelli SP, Kim GS. Increased Expression of Interferon-Induced Transmembrane 3 (IFITM3) in Stroke and Other Inflammatory Conditions in the Brain. International Journal of Molecular Sciences. 2022; 23(16):8885. https://doi.org/10.3390/ijms23168885
Chicago/Turabian StyleHarmon, Elisabeth, Andrea Doan, Jesus Bautista-Garrido, Joo Eun Jung, Sean P. Marrelli, and Gab Seok Kim. 2022. "Increased Expression of Interferon-Induced Transmembrane 3 (IFITM3) in Stroke and Other Inflammatory Conditions in the Brain" International Journal of Molecular Sciences 23, no. 16: 8885. https://doi.org/10.3390/ijms23168885