
Characterization of Transposon-Derived Accessible Chromatin Regions in Rice (Oryza Sativa)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
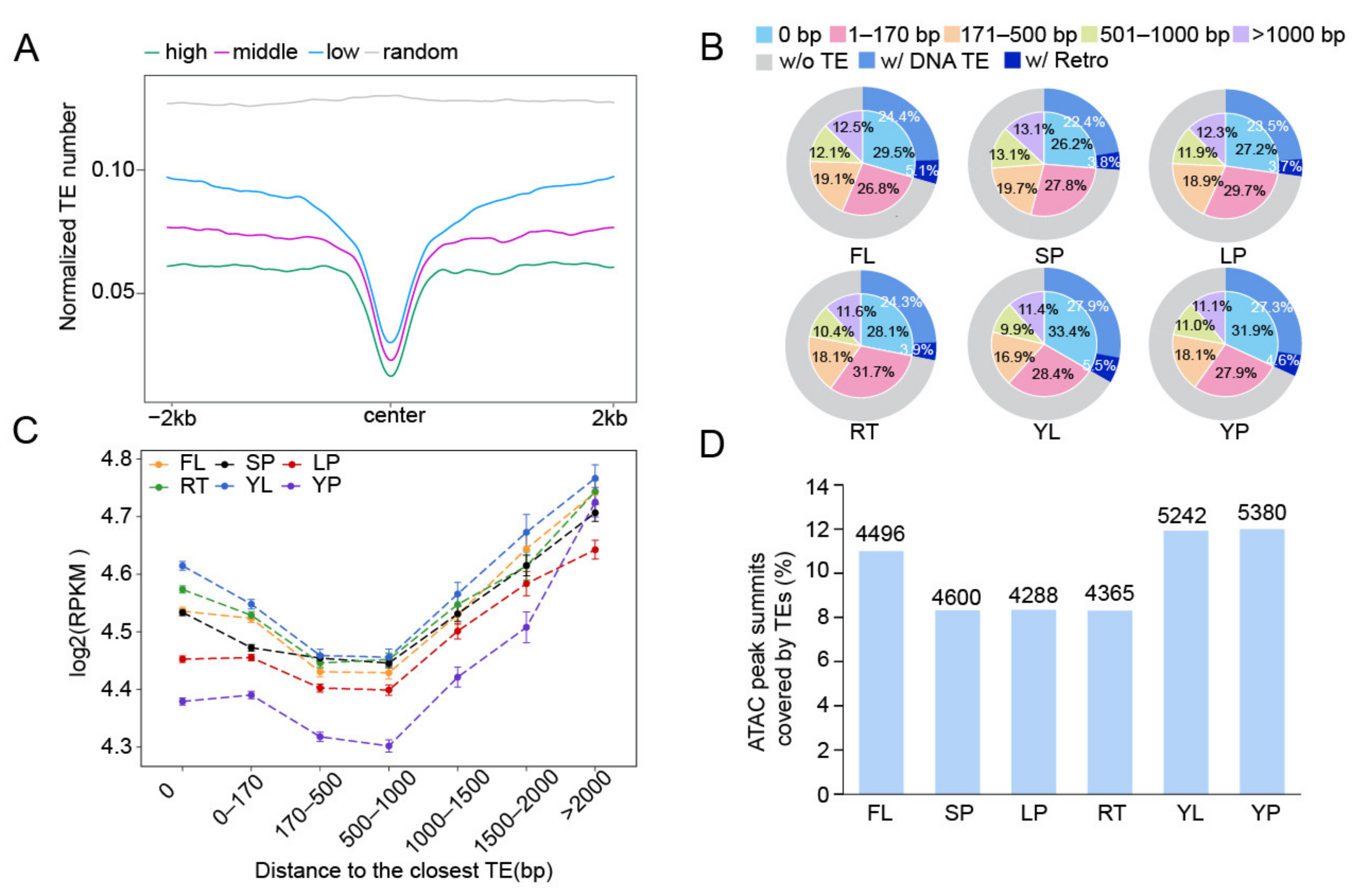
2.1. Identification of TE-Related Accessible Chromatin Regions in Rice
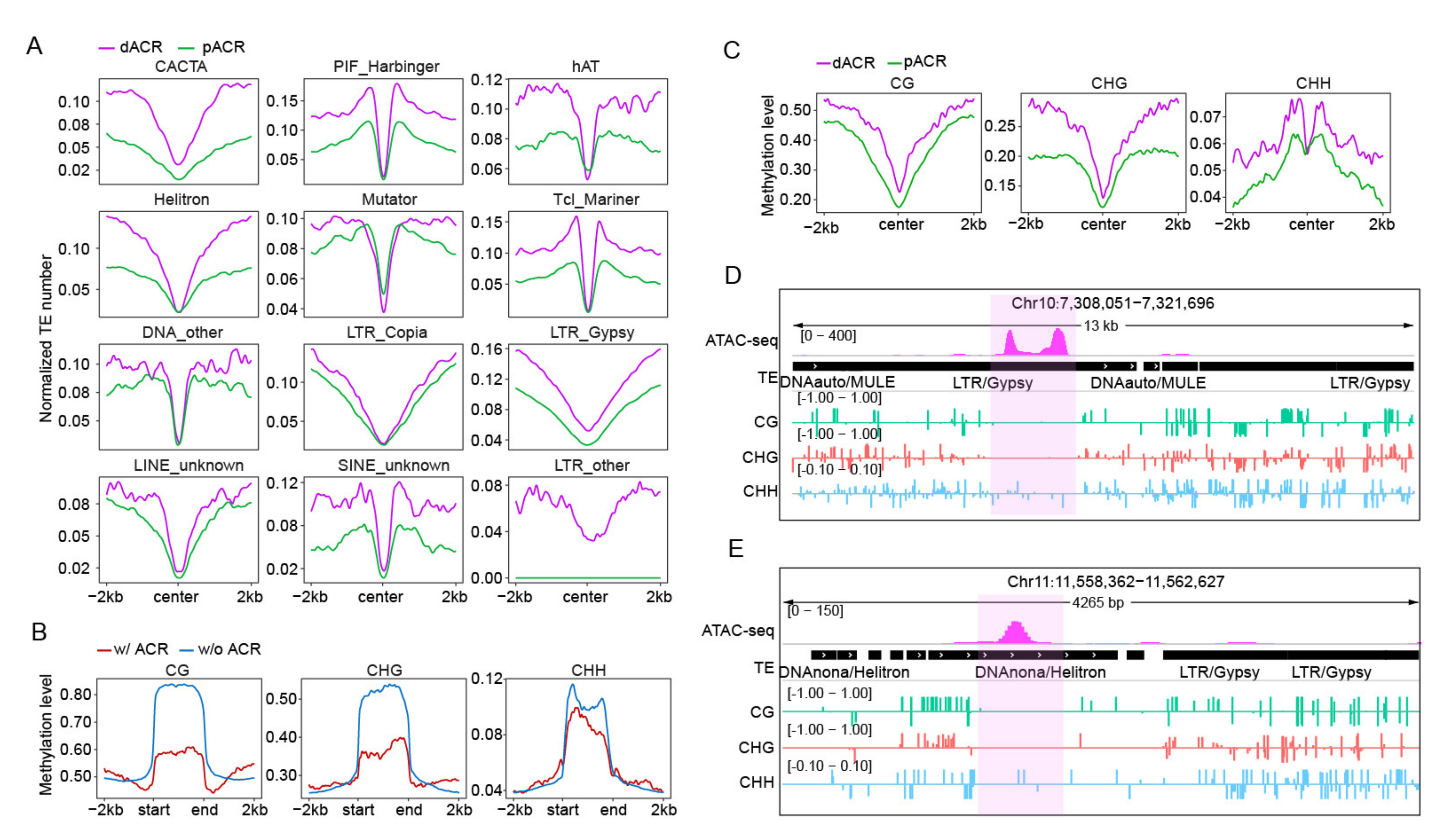
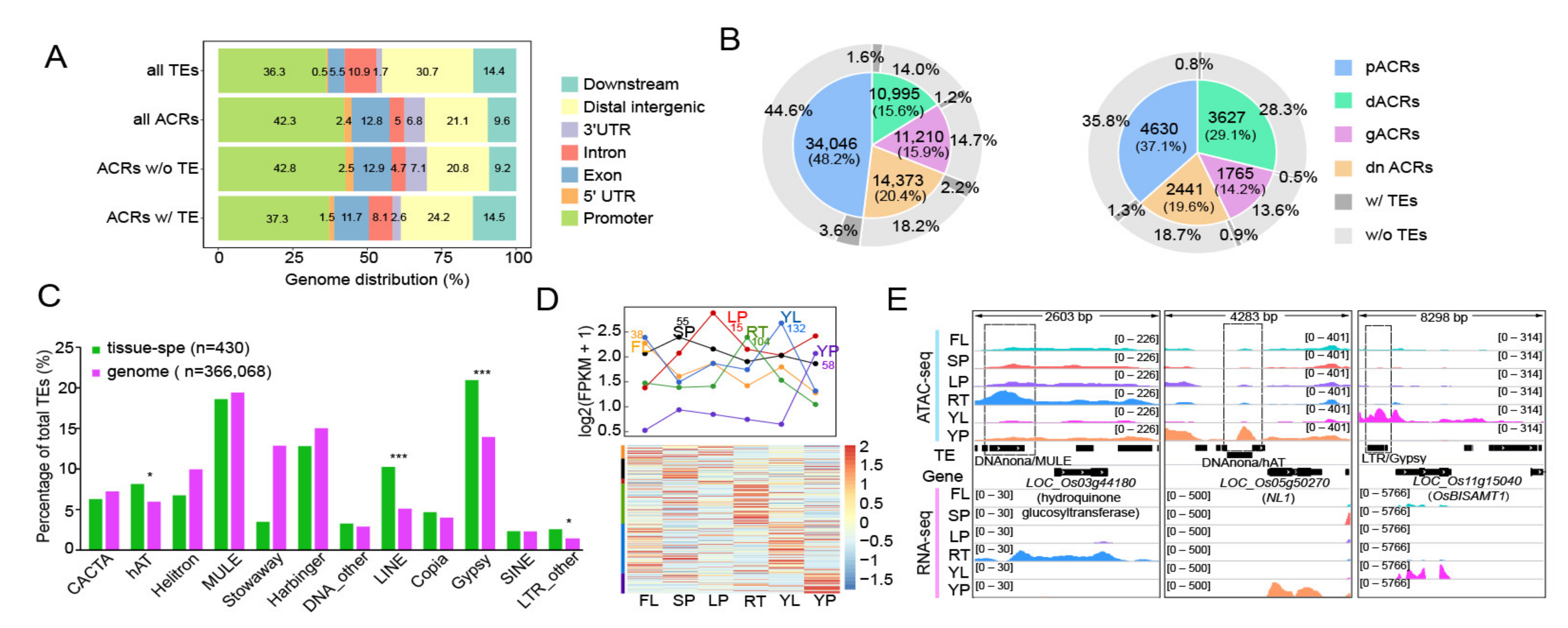
2.2. Profiling of TEs in Proximal and Distal ACRs
2.3. Involvement of TE-Derived ACRs in the Regulation of Tissue-Specific Gene Expression
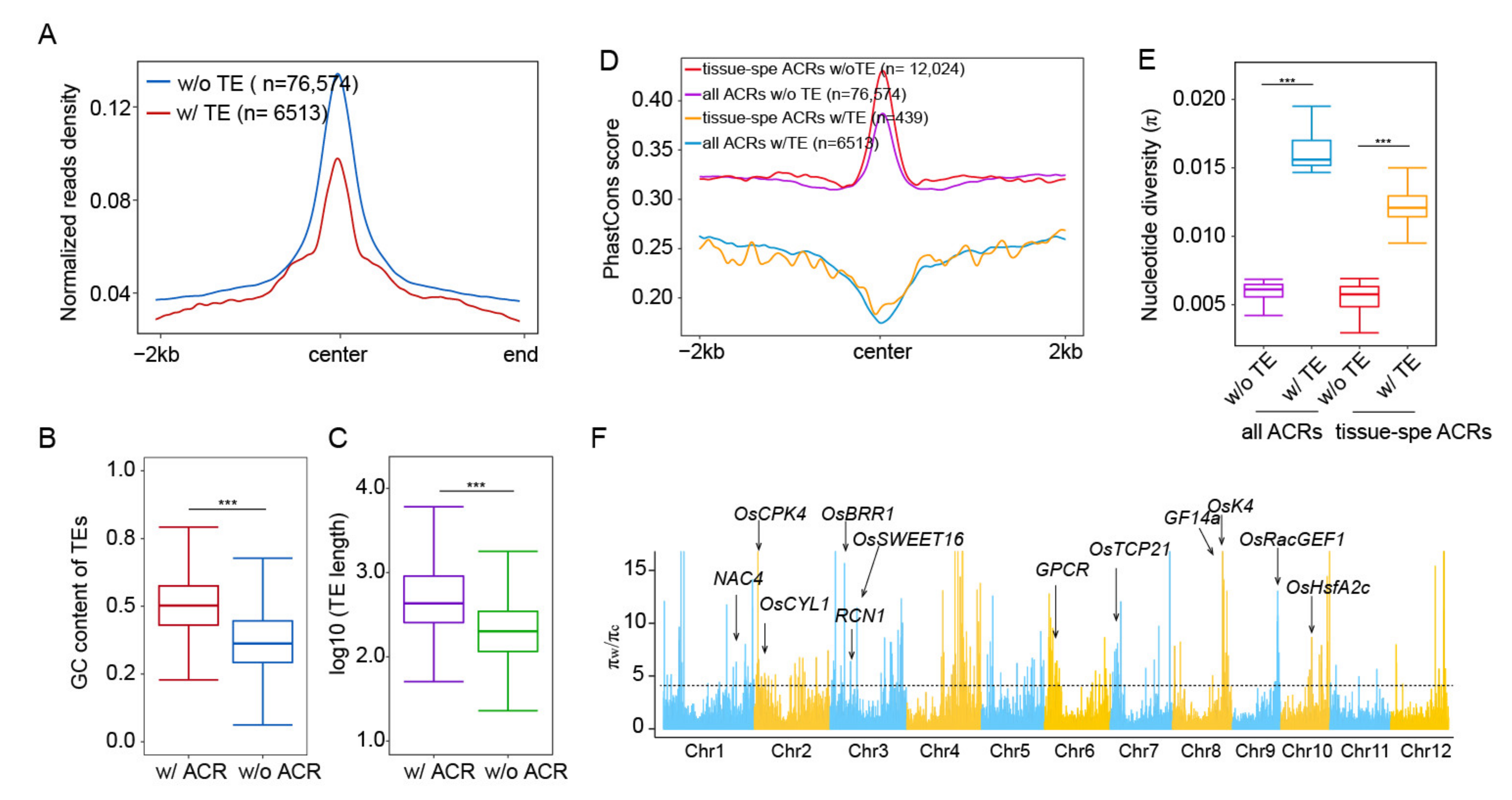
2.4. Impacts of TEs on Chromatin Accessibility and Its Conservation
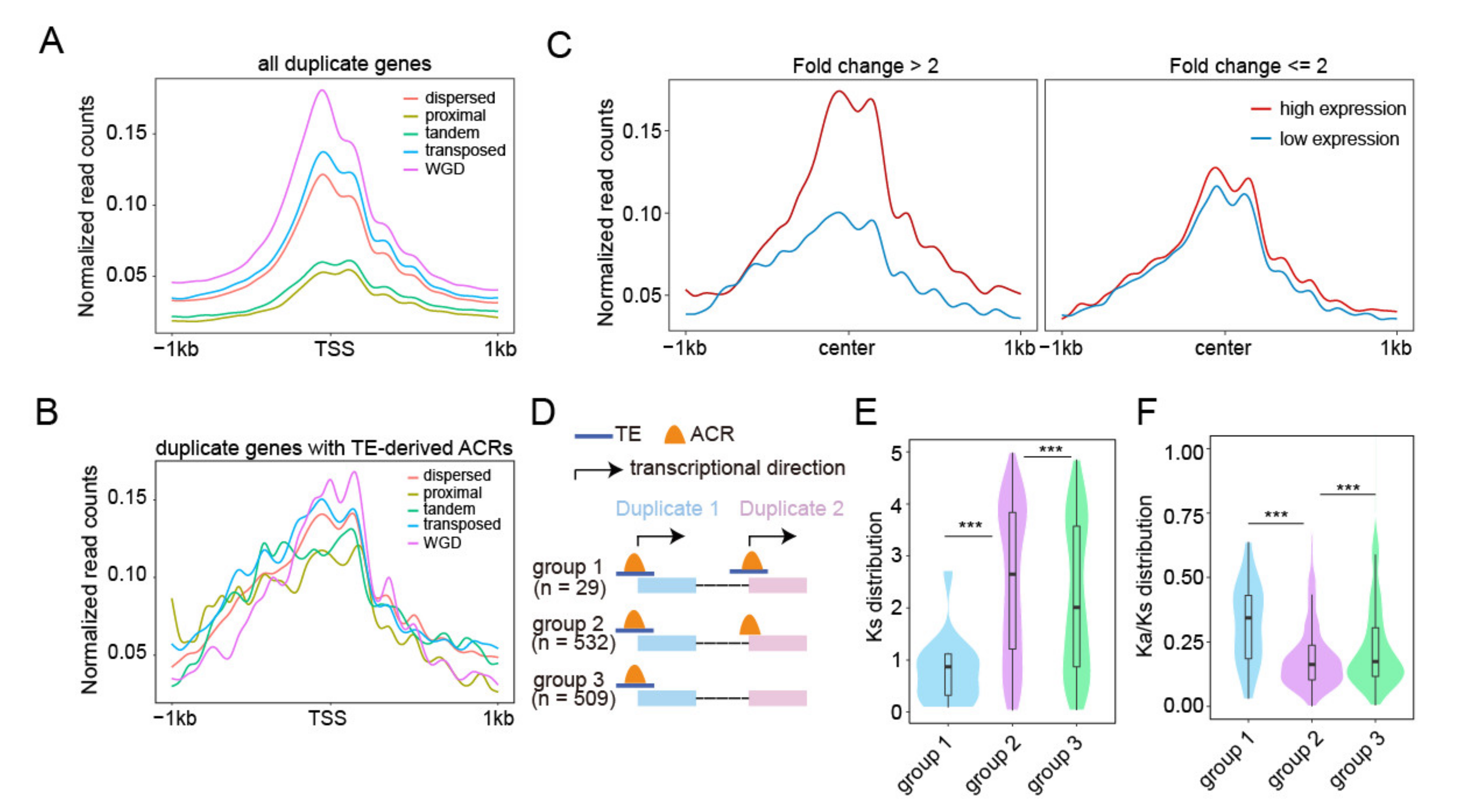
2.5. Relationship between TE-Derived ACR around TSS and Duplicated Genes
3. Discussion
4. Methods
4.1. Processing of Public RNA-Seq, ATAC-Seq and ChIP-Seq Data
4.2. BS-Seq Data Analyses
4.3. Association Analyses of ACRs with Other Genomic Loci
4.4. Identification of Tissue Specific ACRs
4.5. Estimation of the PhastCons Scores and π Values for ACRs
4.6. Identification of Rice Duplicated Genes and Calculation of Ka/Ks Values
4.7. Motif Enrichment Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McClintock, B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 337, 1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicker, T.; Gundlach, H.; Spannagl, M.; Uauy, C.; Borrill, P.; Ramirez-Gonzalez, R.H.; de Oliveira, R.; International Wheat Genome Sequencing Consortium; Mayer, K.F.X.; Paux, E.; et al. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 2018, 19, 103. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Sun, G.; He, S.; Gong, W.; Peng, Z.; Wang, R.; Lin, Z.; Du, X. Comparative effect of allopolyploidy on transposable element composition and gene expression between Gossypium hirsutum and its two diploid progenitors. J. Integr. Plant Biol. 2019, 61, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Naito, K.; Zhang, F.; Tsukiyama, T.; Saito, H.; Hancock, C.N.; Richardson, A.O.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 2009, 461, 1130–1134. [Google Scholar] [CrossRef]
- Buchmann, R.C.; Asad, S.; Wolf, J.N.; Mohannath, G.; Bisaro, D.M. Geminivirus AL2 and L2 proteins suppress transcriptional gene silencing and cause genome-wide reductions in cytosine methylation. J. Virol. 2009, 83, 5005–5013. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119. [Google Scholar] [CrossRef]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef] [Green Version]
- Agren, J.A.; Clark, A.G. Selfish genetic elements. PLoS Genet. 2018, 14, e1007700. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, V.V.; Jurka, J. Self-synthesizing DNA transposons in eukaryotes. Proc. Natl. Acad. Sci. USA 2006, 103, 4540–4545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedges, D.J.; Deininger, P.L. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. 2007, 616, 46–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucher, E.; Reinders, J.; Mirouze, M. Epigenetic control of transposon transcription and mobility in Arabidopsis. Curr. Opin. Plant Biol. 2012, 15, 503–510. [Google Scholar] [CrossRef]
- Cui, X.; Cao, X. Epigenetic regulation and functional exaptation of transposable elements in higher plants. Curr. Opin. Plant Biol. 2014, 21, 83–88. [Google Scholar] [CrossRef]
- Fultz, D.; Choudury, S.G.; Slotkin, R.K. Silencing of active transposable elements in plants. Curr. Opin. Plant Biol. 2015, 27, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Bousios, A.; Gaut, B.S. Mechanistic and evolutionary questions about epigenetic conflicts between transposable elements and their plant hosts. Curr. Opin. Plant Biol. 2016, 30, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, C.D.; Springer, N.M. Transposable element influences on gene expression in plants. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Sahebi, M.; Hanafi, M.M.; van Wijnen, A.J.; Rice, D.; Rafii, M.Y.; Azizi, P.; Osman, M.; Taheri, S.; Bakar, M.F.A.; Isa, M.N.M.; et al. Contribution of transposable elements in the plant’s genome. Gene 2018, 665, 155–166. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, T.; Wu, Y.; Jiang, J. Open chromatin in plant genomes. Cytogenet. Genome Res. 2014, 143, 18–27. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.C.J.; Lopez Gutierrez, R.; Gambetta, G.A.; Castellarin, S.D. Genome-wide analysis of cis-regulatory element structure and discovery of motif-driven gene co-expression networks in grapevine. DNA Res. 2017, 24, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wu, Y.; Schnable, J.C.; Zeng, Z.; Freeling, M.; Crawford, G.E.; Jiang, J. High-resolution mapping of open chromatin in the rice genome. Genome Res. 2012, 22, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Zhang, W.; Zhang, T.; Liu, B.; Jiang, J. Genome-wide prediction and validation of intergenic enhancers in Arabidopsis using open chromatin signatures. Plant Cell 2015, 27, 2415–2426. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Marand, A.P.; Jiang, J. PlantDHS: A database for DNase I hypersensitive sites in plants. Nucleic Acids Res. 2016, 44, D1148–D1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, R.; Zicola, J.; Weber, B.; Anderson, S.N.; Hodgman, C.; Gent, J.I.; Wesselink, J.J.; Springer, N.M.; Hoefsloot, H.C.J.; Turck, F.; et al. Genome-wide mapping of transcriptional enhancer candidates using DNA and chromatin features in maize. Genome Biol. 2017, 18, 137. [Google Scholar] [CrossRef]
- Sun, Y.; Dong, L.; Zhang, Y.; Lin, D.; Xu, W.; Ke, C.; Han, L.; Deng, L.; Li, G.; Jackson, D.; et al. 3D genome architecture coordinates trans and cis regulation of differentially expressed ear and tassel genes in maize. Genome Biol. 2020, 21, 143. [Google Scholar] [CrossRef]
- Zhao, L.; Xie, L.; Zhang, Q.; Ouyang, W.; Deng, L.; Guan, P.; Ma, M.; Li, Y.; Zhang, Y.; Xiao, Q.; et al. Integrative analysis of reference epigenomes in 20 rice varieties. Nat. Commun. 2020, 11, 2658. [Google Scholar] [CrossRef]
- Jacques, P.E.; Jeyakani, J.; Bourque, G. The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet. 2013, 9, e1003504. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Zhang, W.; Chen, L.; Wang, L.; Marand, A.P.; Wu, Y.; Jiang, J. Proliferation of regulatory DNA elements derived from transposable elements in the maize genome. Plant Physiol. 2018, 176, 2789–2803. [Google Scholar] [CrossRef] [Green Version]
- Miao, B.; Fu, S.; Lyu, C.; Gontarz, P.; Wang, T.; Zhang, B. Tissue-specific usage of transposable element-derived promoters in mouse development. Genome Biol. 2020, 21, 255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Z.; Zhang, Y.; Lin, K.; Peng, Y.; Ye, L.; Zhuang, Y.; Wang, M.; Xie, Y.; Guo, J.; et al. Evolutionary rewiring of the wheat transcriptional regulatory network by lineage-specific transposable elements. Genome Res. 2021, 31, 2276–2289. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, J.; Yang, L.; Qin, G.; Xia, C.; Xu, X.; Su, Y.; Liu, Y.; Ming, L.; Chen, L.L.; et al. An inferred functional impact map of genetic variants in rice. Mol. Plant 2021, 14, 1584–1599. [Google Scholar] [CrossRef]
- Ye, M.; Goudot, C.; Hoyler, T.; Lemoine, B.; Amigorena, S.; Zueva, E. Specific subfamilies of transposable elements contribute to different domains of T lymphocyte enhancers. Proc. Natl. Acad. Sci. USA 2020, 117, 7905–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisp, P.A.; Marand, A.P.; Noshay, J.M.; Zhou, P.; Lu, Z.; Schmitz, R.J.; Springer, N.M. Stable unmethylated DNA demarcates expressed genes and their cis-regulatory space in plant genomes. Proc. Natl. Acad. Sci. USA 2020, 117, 23991–24000. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.K.; Zhang, L.; Zhou, L.M.; Yung, W.S.; Li, M.W.; Lam, H.M. Genomic features of open chromatin regions (OCRs) in wild soybean and their effects on gene expressions. Genes 2021, 12, 640. [Google Scholar] [CrossRef] [PubMed]
- Schwope, R.; Magris, G.; Miculan, M.; Paparelli, E.; Celii, M.; Tocci, A.; Marroni, F.; Fornasiero, A.; de Paoli, E.; Morgante, M. Open chromatin in grapevine marks candidate CREs and with other chromatin features correlates with gene expression. Plant J. 2021, 107, 1631–1647. [Google Scholar] [CrossRef]
- Wang, L.; Ming, L.; Liao, K.; Xia, C.; Sun, S.; Chang, Y.; Wang, H.; Fu, D.; Xu, C.; Wang, Z.; et al. Bract suppression regulated by the miR156/529-SPLs-NL1-PLA1 module is required for the transition from vegetative to reproductive branching in rice. Mol. Plant 2021, 14, 1168–1184. [Google Scholar] [CrossRef]
- Xu, R.; Song, F.; Zheng, Z. OsBISAMT1, a gene encoding S-adenosyl-L-methionine: Salicylic acid carboxyl methyltransferase, is differentially expressed in rice defense responses. Mol. Biol. Rep. 2006, 33, 223–231. [Google Scholar] [CrossRef]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thastrom, A.; Field, Y.; Moore, I.K.; Wang, J.P.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef]
- Zhao, D.; Ferguson, A.A.; Jiang, N. What makes up plant genomes: The vanishing line between transposable elements and genes. Biochim. Biophys. Acta 2016, 1859, 366–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, Z.; Wu, J.; Cui, X.; Feng, D.; Wang, K.; Xu, M.; Zhou, L.; Han, X.; Gu, X.; et al. The Oryza sativa regulator HDR1 associates with the kinase OsK4 to control photoperiodic flowering. PLoS Genet. 2016, 12, e1005927. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Shen, X.; Wang, N.; Ding, X. Characterization of a novel cyclase-like gene family involved in controlling stress tolerance in rice. J. Plant Physiol. 2015, 181, 30–41. [Google Scholar] [CrossRef]
- Campo, S.; Baldrich, P.; Messeguer, J.; Lalanne, E.; Coca, M.; San Segundo, B. Overexpression of a calcium-dependent protein kinase confers salt and drought tolerance in rice by preventing membrane lipid peroxidation. Plant Physiol. 2014, 165, 688–704. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Zhang, Q.; Li, Y.; Lei, C.; Zhai, Y.; Sun, X.; Sun, D.; Sun, Y.; Lu, T. A putative leucine-rich repeat receptor kinase, OsBRR1, is involved in rice blast resistance. Planta 2009, 230, 377–385. [Google Scholar] [CrossRef]
- Matsuda, S.; Takano, S.; Sato, M.; Furukawa, K.; Nagasawa, H.; Yoshikawa, S.; Kasuga, J.; Tokuji, Y.; Yazaki, K.; Nakazono, M.; et al. Rice stomatal closure requires guard cell plasma membrane ATP-binding cassette transporter RCN1/OsABCG5. Mol. Plant 2016, 9, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Streubel, J.; Pesce, C.; Hutin, M.; Koebnik, R.; Boch, J.; Szurek, B. Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae. New Phytol. 2013, 200, 808–819. [Google Scholar] [CrossRef]
- Yadav, D.K.; Tuteja, N. Rice G-protein coupled receptor (GPCR): In silico analysis and transcription regulation under abiotic stress. Plant Signal Behav. 2011, 6, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Tan, L.; Sun, H.; Zhao, X.; Liu, F.; Cai, H.; Fu, Y.; Sun, X.; Gu, P.; Zhu, Z.; et al. Natural variations at TIG1 encoding a TCP transcription factor contribute to plant architecture domestication in rice. Mol. Plant 2019, 12, 1075–1089. [Google Scholar] [CrossRef]
- Chen, F.; Li, Q.; Sun, L.; He, Z. The rice 14-3-3 gene family and its involvement in responses to biotic and abiotic stress. DNA Res. 2006, 13, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akamatsu, A.; Wong, H.L.; Fujiwara, M.; Okuda, J.; Nishide, K.; Uno, K.; Imai, K.; Umemura, K.; Kawasaki, T.; Kawano, Y.; et al. An OsCEBiP/OsCERK1-OsRacGEF1-OsRac1 module is an essential early component of chitin-induced rice immunity. Cell Host Microbe 2013, 13, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, D.; Chakrabarti, S.; Sarkar, A.; Singh, A.; Grover, A. Heat shock factor gene family in rice: Genomic organization and transcript expression profiling in response to high temperature, low temperature and oxidative stresses. Plant Physiol. Biochem. 2009, 47, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Freeling, M. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef] [PubMed]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.H. Evolution of gene duplication in plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, X.; Lee, T.H.; Mansoor, S.; Paterson, A.H. Gene body methylation shows distinct patterns associated with different gene origins and duplication modes and has a heterogeneous relationship with gene expression in Oryza sativa (rice). New Phytol. 2013, 198, 274–283. [Google Scholar] [CrossRef]
- Xu, C.; Nadon, B.D.; Kim, K.D.; Jackson, S.A. Genetic and epigenetic divergence of duplicate genes in two legume species. Plant Cell Environ. 2018, 41, 2033–2044. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, T.; Qi, Y.; Song, J.; Han, Z.; Ma, C. Evolution of the RNA N (6)-methyladenosine methylome mediated by genomic duplication. Plant Physiol. 2020, 182, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Orlov, Y.L.; Li, X.; Zhou, Y.; Liu, Y.; Yuan, C.; Chen, M. In situ dissecting the evolution of gene duplication with different histone modification patterns based on high-throughput data analysis in Arabidopsis thaliana. PeerJ 2021, 9, e10426. [Google Scholar] [CrossRef]
- Shi, T.; Rahmani, R.S.; Gugger, P.F.; Wang, M.; Li, H.; Zhang, Y.; Li, Z.; Wang, Q.; van de Peer, Y.; Marchal, K.; et al. Distinct expression and methylation patterns for genes with different fates following a single whole-genome duplication in flowering plants. Mol. Biol. Evol. 2020, 37, 2394–2413. [Google Scholar] [CrossRef] [PubMed]
- Lannes, R.; Rizzon, C.; Lerat, E. Does the presence of transposable elements impact the epigenetic environment of human duplicated genes? Genes 2019, 10, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhao, H.; Yan, Y.; Wei, M.X.; Zheng, Y.C.; Yue, E.K.; Alam, M.S.; Smartt, K.O.; Duan, M.H.; Xu, J.H. Extensively current activity of transposable elements in natural rice accessions revealed by singleton insertions. Front. Plant Sci. 2021, 12, 745526. [Google Scholar] [CrossRef] [PubMed]
- Lisch, D.; Bennetzen, J.L. Transposable element origins of epigenetic gene regulation. Curr. Opin. Plant Biol. 2011, 14, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, K.; Lee, C.C. Reciprocal regulation of haem biosynthesis and the circadian clock in mammals. Nature 2004, 430, 467–471. [Google Scholar] [CrossRef]
- Noshay, J.M.; Marand, A.P.; Anderson, S.N.; Zhou, P.; Mejia Guerra, M.K.; Lu, Z.; O’Connor, C.H.; Crisp, P.A.; Hirsch, C.N.; Schmitz, R.J.; et al. Assessing the regulatory potential of transposable elements using chromatin accessibility profiles of maize transposons. Genetics 2021, 217, 1–13. [Google Scholar] [CrossRef]
- Moschetti, R.; Palazzo, A.; Lorusso, P.; Viggiano, L.; Marsano, R.M. “What You Need, Baby, I Got It”: Transposable elements as suppliers of cis-operating sequences in Drosophila. Biology 2020, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Kunarso, G.; Chia, N.Y.; Jeyakani, J.; Hwang, C.; Lu, X.; Chan, Y.S.; Ng, H.H.; Bourque, G. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 2010, 42, 631–634. [Google Scholar] [CrossRef]
- Thornburg, B.G.; Gotea, V.; Makalowski, W. Transposable elements as a significant source of transcription regulating signals. Gene 2006, 365, 104–110. [Google Scholar] [CrossRef]
- Nicolau, M.; Picault, N.; Moissiard, G. The evolutionary volte-face of transposable elements: From harmful jumping genes to major drivers of genetic innovation. Cells 2021, 10, 2952. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Xia, E.H.; Yao, Q.Y.; Liu, X.D.; Gao, L.Z. Autotetraploid rice methylome analysis reveals methylation variation of transposable elements and their effects on gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, E7022–E7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.Y.; Purugganan, M.D. Evolutionary epigenomics of retrotransposon-mediated methylation spreading in rice. Mol. Biol. Evol. 2018, 35, 365–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deniz, O.; Frost, J.M.; Branco, M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhou, Y.; Lin, N.; Lowdon, R.F.; Hong, C.; Nagarajan, R.P.; Cheng, J.B.; Li, D.; Stevens, M.; Lee, H.J.; et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013, 23, 1522–1540. [Google Scholar]
- Todd, C.D.; Deniz, O.; Taylor, D.; Branco, M.R. Functional evaluation of transposable elements as enhancers in mouse embryonic and trophoblast stem cells. elife 2019, 8, e44344. [Google Scholar] [CrossRef]
- Jin, P.; Qin, S.; Chen, X.; Song, Y.; Li-Ling, J.; Xu, X.; Ma, F. Evolutionary rate of human tissue-specific genes are related with transposable element insertions. Genetica 2012, 140, 513–523. [Google Scholar] [CrossRef]
- Chishima, T.; Iwakiri, J.; Hamada, M. Identification of transposable elements contributing to tissue-specific expression of long non-coding RNAs. Genes 2018, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Trizzino, M.; Kapusta, A.; Brown, C.D. Transposable elements generate regulatory novelty in a tissue-specific fashion. BMC Genomics. 2018, 19, 468. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhu, T.; Fang, W.; Yu, R.; He, Z.; Chen, D. Systematic annotation of conservation states provides insights into regulatory regions in rice. J. Genet. Genomics, 2022; in press. [Google Scholar] [CrossRef]
- Qian, W.; Liao, B.Y.; Chang, A.Y.; Zhang, J. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet. 2010, 26, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, Z.; Fu, T.; Hu, L.; Xu, C.; Gong, L.; Wendel, J.F.; Liu, B. Gene-body CG methylation and divergent expression of duplicate genes in rice. Sci. Rep. 2017, 7, 2675. [Google Scholar] [CrossRef] [PubMed]
- Mortada, H.; Vieira, C.; Lerat, E. Genes devoid of full-length transposable element insertions are involved in development and in the regulation of transcription in human and closely related species. J. Mol. Evol. 2010, 71, 180–191. [Google Scholar] [CrossRef]
- Nellaker, C.; Keane, T.M.; Yalcin, B.; Wong, K.; Agam, A.; Belgard, T.G.; Flint, J.; Adams, D.J.; Frankel, W.N.; Ponting, C.P. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012, 13, R45. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhu, P.; Pellegrini, M.; Zhang, M.Q.; Wang, X.; Ni, Z. CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data. Bioinformatics 2018, 34, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Schug, J.; Schuller, W.P.; Kappen, C.; Salbaum, J.M.; Bucan, M.; Stoeckert, C.J., Jr. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 2005, 6, R33. [Google Scholar] [CrossRef] [Green Version]
- Pang, H.; Chen, Q.; Li, Y.; Wang, Z.; Wu, L.; Yang, Q.; Zheng, X. Comparative analysis of the transcriptomes of two rice subspecies during domestication. Sci. Rep. 2021, 11, 3660. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tu, L.; Lin, M.; Lin, Z.; Wang, P.; Yang, Q.; Ye, Z.; Shen, C.; Li, J.; Zhang, L.; et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017, 49, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Defoort, J.; Tasdighian, S.; Maere, S.; van de Peer, Y.; de Smet, R. Gene duplicability of core genes is highly consistent across all angiosperms. Plant Cell 2016, 28, 326–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machanick, P.; Bailey, T.L. MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 2011, 27, 1696–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 2016, 165, 1280–1292. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, A.; Zhang, W. Characterization of Transposon-Derived Accessible Chromatin Regions in Rice (Oryza Sativa). Int. J. Mol. Sci. 2022, 23, 8947. https://doi.org/10.3390/ijms23168947
Zhang A, Zhang W. Characterization of Transposon-Derived Accessible Chromatin Regions in Rice (Oryza Sativa). International Journal of Molecular Sciences. 2022; 23(16):8947. https://doi.org/10.3390/ijms23168947
Chicago/Turabian StyleZhang, Aicen, and Wenli Zhang. 2022. "Characterization of Transposon-Derived Accessible Chromatin Regions in Rice (Oryza Sativa)" International Journal of Molecular Sciences 23, no. 16: 8947. https://doi.org/10.3390/ijms23168947
APA StyleZhang, A., & Zhang, W. (2022). Characterization of Transposon-Derived Accessible Chromatin Regions in Rice (Oryza Sativa). International Journal of Molecular Sciences, 23(16), 8947. https://doi.org/10.3390/ijms23168947