
Mouse Models for Immune Checkpoint Blockade Therapeutic Research in Oral Cancer


Abstract
:1. Introduction
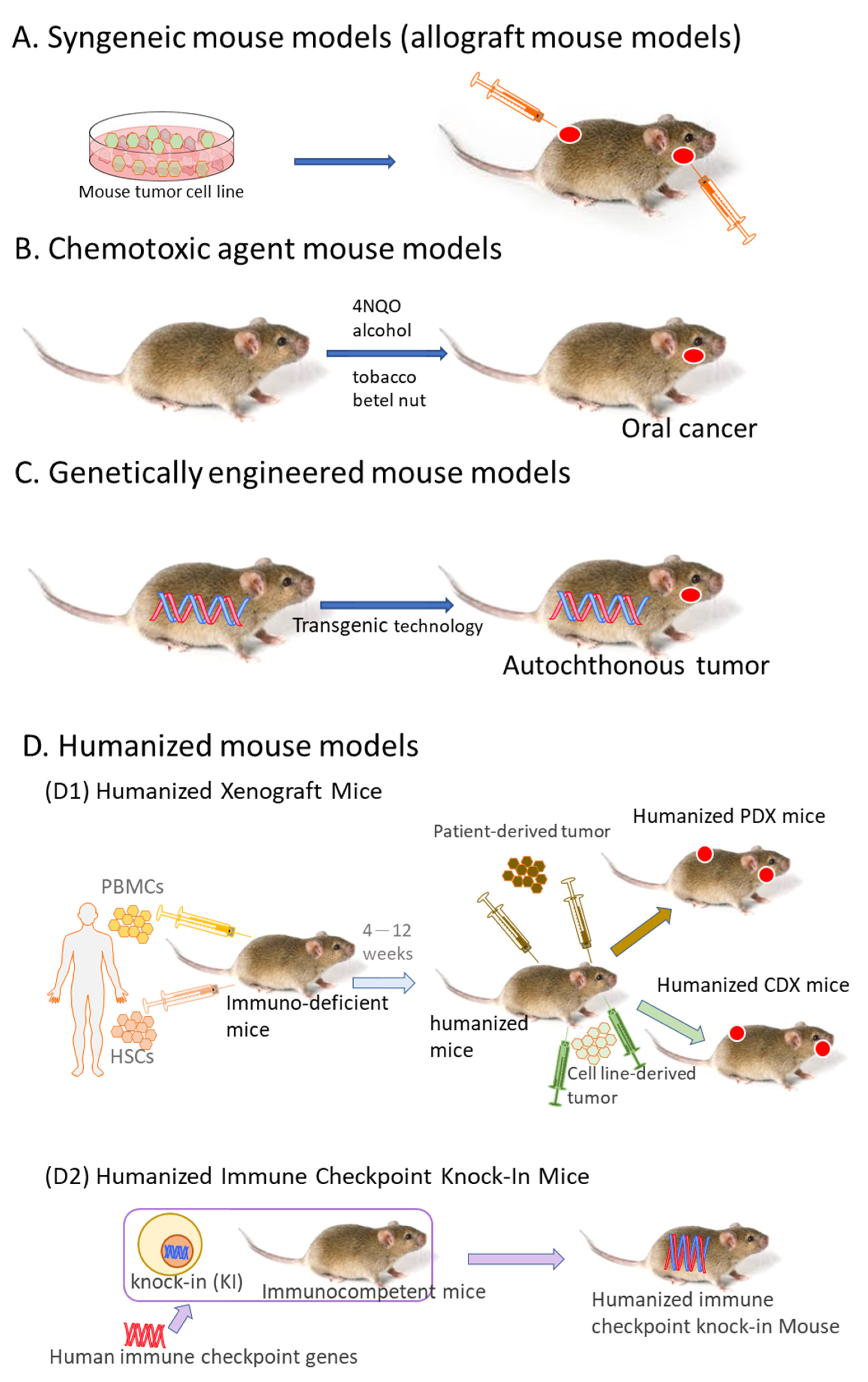
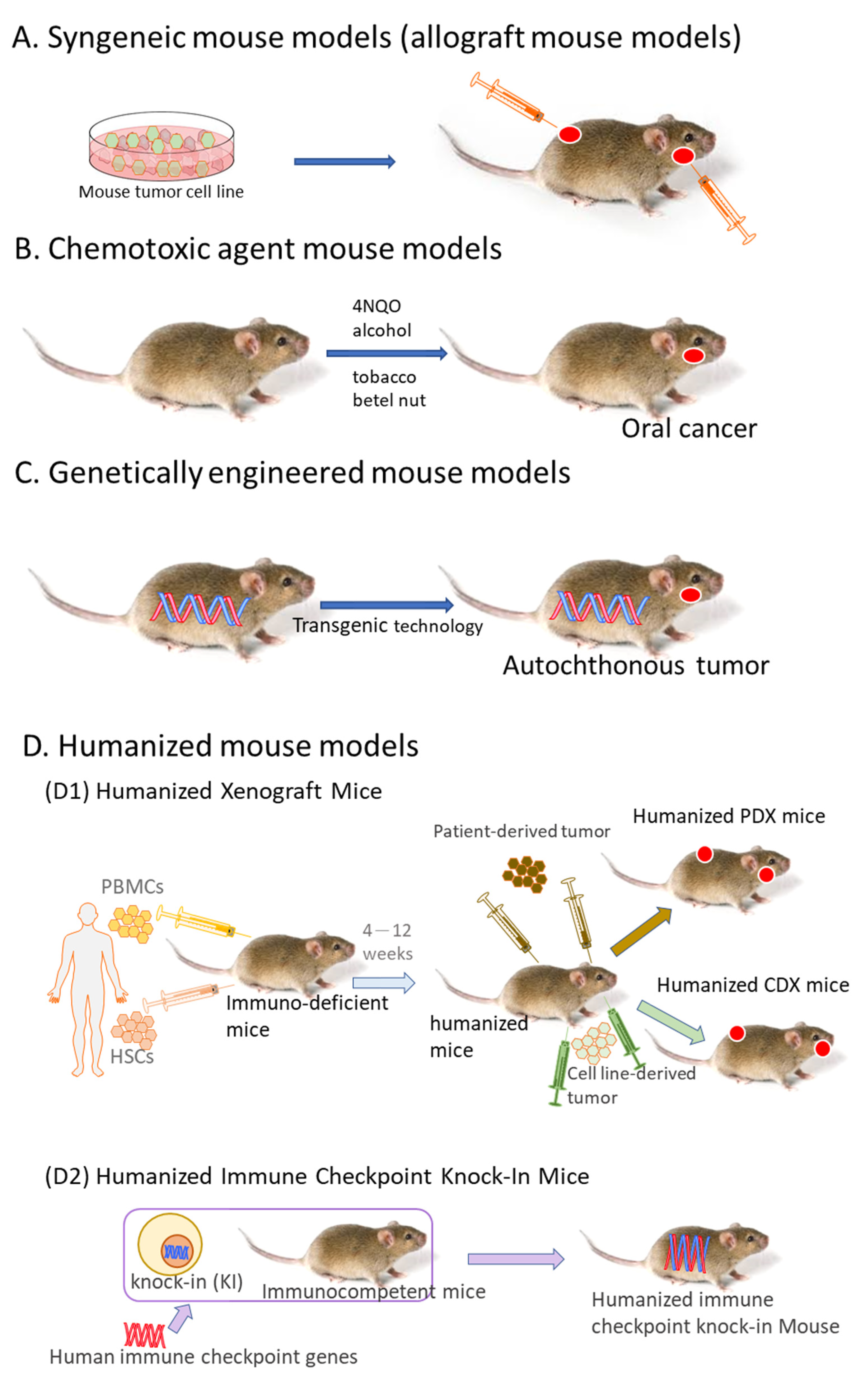
2. Syngeneic Mouse Models
3. Chemotoxic Agent Mouse Models
3.1. 4NQO-Induced OSCC Mouse Model
3.2. Tobacco-Related Chemical Carcinogens-Induced OSCC
3.3. 4NQO Combined with Other Chemical Carcinogens
4. Genetically Engineered Mouse Models
4.1. LSL-KrasG12D Mice
4.2. L2D1+/p53+/− and L2D1+/p53−/− Mice
4.3. Tgfbr1/Pten 2cKO (Tgfbr1flox/flox; Ptenflox/flox; K14-CreERtam) Mice
4.4. p53R172H; K5.CrePR1 and p53flox/flox; K5 CrePR1 Mice
5. Humanized Mouse Models
5.1. Humanized Xenograft Mice
5.2. Humanized Immune Checkpoint Knock-In Mice
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, J.; Zhang, X.; Lee, S.K.; Song, N.-Y.; Son, S.H.; Kim, K.R.; Shim, J.H.; Park, K.-K.; Chung, W.-Y. CCL28-induced RARβ expression inhibits oral squamous cell carcinoma bone invasion. J. Clin. Investig. 2019, 129, 5381–5399. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Tian, H.; Cheng, X.; Chen, Y.; Liang, S.; Zhang, Z.; Liao, Y.; Xu, P. Aberrant Kank1 expression regulates YAP to promote apoptosis and inhibit proliferation in OSCC. J. Cell. Physiol. 2020, 235, 1850–1865. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ali, K. Oral cancer-the fight must go on against all odds... Evid.-Based Dent. 2022, 23, 4–5. [Google Scholar] [CrossRef]
- Bandhary, S.K.; Shetty, V.; Saldanha, M.; Gatti, P.; Devegowda, D.; R, P.S.; Shetty, A.K. Detection of Human Papilloma Virus and Risk Factors among Patients with Head and Neck Squamous Cell Carcinoma Attending a Tertiary Referral Centre in South India. Asian Pac. J. Cancer Prev. 2018, 19, 1325–1330. [Google Scholar] [CrossRef]
- Auguste, A.; Deloumeaux, J.; Joachim, C.; Gaete, S.; Michineau, L.; Herrmann-Storck, C.; Duflo, S.; Luce, D. Joint effect of tobacco, alcohol, and oral HPV infection on head and neck cancer risk in the French West Indies. Cancer Med. 2020, 9, 6854–6863. [Google Scholar] [CrossRef]
- Ahmad, P.; Nawaz, R.; Qurban, M.; Shaikh, G.M.; Mohamed, R.N.; Nagarajappa, A.K.; Asif, J.A.; Alam, M.K. Risk factors associated with the mortality rate of oral squamous cell carcinoma patients: A 10-year retrospective study. Medicine 2021, 100, e27127. [Google Scholar] [CrossRef]
- Aghiorghiesei, O.; Zanoaga, O.; Nutu, A.; Braicu, C.; Campian, R.S.; Lucaciu, O.; Berindan Neagoe, I. The World of Oral Cancer and Its Risk Factors Viewed from the Aspect of MicroRNA Expression Patterns. Genes 2022, 13, 594. [Google Scholar] [CrossRef]
- Zygogianni, A.G.; Kyrgias, G.; Karakitsos, P.; Psyrri, A.; Kouvaris, J.; Kelekis, N.; Kouloulias, V. Oral squamous cell cancer: Early detection and the role of alcohol and smoking. Head Neck Oncol. 2011, 3, 2. [Google Scholar] [CrossRef]
- Koyfman, S.A.; Ismaila, N.; Crook, D.; D’Cruz, A.; Rodriguez, C.P.; Sher, D.J.; Silbermins, D.; Sturgis, E.M.; Tsue, T.T.; Weiss, J.; et al. Management of the Neck in Squamous Cell Carcinoma of the Oral Cavity and Oropharynx: ASCO Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 1753–1774. [Google Scholar] [CrossRef]
- Ukpo, O.C.; Flanagan, J.J.; Ma, X.-J.; Luo, Y.; Thorstad, W.L.; Lewis, J.S. High-Risk Human Papillomavirus E6/E7 mRNA Detection by a Novel In Situ Hybridization Assay Strongly Correlates With p16 Expression and Patient Outcomes in Oropharyngeal Squamous Cell Carcinoma. Am. J. Surg. Pathol. 2011, 35, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.P.; Thomas, G.R. Animal models for the study of squamous cell carcinoma of the upper aerodigestive tract: A historical perspective with review of their utility and limitations. Part A. Chemically-induced de novo cancer, syngeneic animal models of HNSCC, animal models of transplanted xenogeneic human tumors. Int. J. Cancer 2006, 118, 2111–2122. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.K.; Hildreth, B.E., 3rd; Supsavhad, W.; Elshafae, S.M.; Hassan, B.B.; Dirksen, W.P.; Toribio, R.E.; Rosol, T.J. Animal Models of Bone Metastasis. Vet. Pathol. 2015, 52, 827–841. [Google Scholar] [CrossRef]
- Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Cell Culture Based in vitro Test Systems for Anticancer Drug Screening. Front. Bioeng. Biotechnol. 2020, 8, 322. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Chang, J.T.-C.; Liao, C.-T.; Wang, H.-M.; Yen, T.-C.; Chiu, C.-C.; Lu, Y.-C.; Li, H.-F.; Cheng, A.-J. Head and neck cancer in the betel quid chewing area: Recent advances in molecular carcinogenesis. Cancer Sci. 2008, 99, 1507–1514. [Google Scholar] [CrossRef]
- Mun, J.-Y.; Leem, S.-H.; Lee, J.H.; Kim, H.S. Dual Relationship Between Stromal Cells and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2022, 13, 864739. [Google Scholar] [CrossRef]
- Lv, C.; Li, S.; Zhao, J.; Yang, P.; Yang, C. M1 Macrophages Enhance Survival and Invasion of Oral Squamous Cell Carcinoma by Inducing GDF15-Mediated ErbB2 Phosphorylation. ACS Omega 2022, 7, 11405–11414. [Google Scholar] [CrossRef]
- Hadjigol, S.; Shah, B.; O’Brien-Simpson, N. The ‘Danse Macabre’—Neutrophils the Interactive Partner Affecting Oral Cancer Outcomes. Front. Immunol. 2022, 13, 894021. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular Aspects and Future Perspectives of Cytokine-Based Anti-cancer Immunotherapy. Front. Cell Dev. Biol. 2020, 8, 402. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef]
- Ribatti, D. The concept of immune surveillance against tumors. The first theories. Oncotarget 2017, 8, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Naimi, A.; Mohammed, R.N.; Raji, A.; Chupradit, S.; Yumashev, A.V.; Suksatan, W.; Shalaby, M.N.; Thangavelu, L.; Kamrava, S.; Shomali, N.; et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun. Signal. 2022, 20, 44. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Lin, L.; Chen, C.-W.; Ou, D.-L. Mouse Models for Immunotherapy in Hepatocellular Carcinoma. Cancers 2019, 11, 1800. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Mouse Tumor Models for Advanced Cancer Immunotherapy. Int. J. Mol. Sci. 2020, 21, 4118. [Google Scholar] [CrossRef]
- Luo, J.J.; Young, C.D.; Zhou, H.M.; Wang, X.J. Mouse Models for Studying Oral Cancer: Impact in the Era of Cancer Immunotherapy. J. Dent. Res. 2018, 97, 683–690. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Ballegeer, E.A.; Madrill, N.J.; Berger, K.L.; Agnew, D.W.; McNiel, E.A. Evaluation of hypoxia in a feline model of head and neck cancer using 64 Cu-ATSM positron emission tomography/computed tomography. BMC cancer 2013, 13, 218. [Google Scholar] [CrossRef]
- Wypij, J.M. A naturally occurring feline model of head and neck squamous cell carcinoma. Pathol. Res. Int. 2013, 2013, 502197. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulières, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.J.; Soria, A.; Machiels, J.P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Haddad, R.; Even, C.; Tahara, M.; Dvorkin, M.; Ciuleanu, T.E.; Clement, P.M.; Mesia, R.; Kutukova, S.; Zholudeva, L.; et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: EAGLE, a randomized, open-label phase III study. Ann. Oncol. 2020, 31, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Saba, N.F.; Gitlitz, B.J.; Haddad, R.; Sukari, A.; Neupane, P.; Morris, J.C.; Misiukiewicz, K.; Bauman, J.E.; Fenton, M.; et al. Effect of Adding Motolimod to Standard Combination Chemotherapy and Cetuximab Treatment of Patients With Squamous Cell Carcinoma of the Head and Neck: The Active8 Randomized Clinical Trial. JAMA Oncol. 2018, 4, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Harrington, K.; Tahara, M.; Ferris, R.L.; Gillison, M.; Fayette, J.; Daste, A.; Koralewski, P.; Mesia Nin, R.; Saba, N.F.; et al. LBA36 Nivolumab (N) + ipilimumab (I) vs EXTREME as first-line (1L) treatment (tx) for recurrent/metastatic squamous cell carcinoma of the head and neck (R/M SCCHN): Final results of CheckMate 651. Ann. Oncol. 2021, 32, S1310–S1311. [Google Scholar] [CrossRef]
- Adachi, M.; Mizuno-Kamiya, M.; Takayama, E.; Kawaki, H.; Inagaki, T.; Sumi, S.; Motohashi, M.; Muramatsu, Y.; Sumitomo, S.I.; Shikimori, M.; et al. Gene expression analyses associated with malignant phenotypes of metastatic sub-clones derived from a mouse oral squamous cell carcinoma Sq-1979 cell line. Oncol. Lett. 2018, 15, 3350–3356. [Google Scholar] [CrossRef]
- Su, H.; Luo, Q.; Xie, H.; Huang, X.; Ni, Y.; Mou, Y.; Hu, Q. Therapeutic antitumor efficacy of tumor-derived autophagosome (DRibble) vaccine on head and neck cancer. Int. J. Nanomed. 2015, 10, 1921. [Google Scholar]
- Dong, H.; Su, H.; Chen, L.; Liu, K.; Hu, H.-m.; Yang, W.; Mou, Y. Immunocompetence and mechanism of the DRibble-DCs vaccine for oral squamous cell carcinoma. Cancer Manag. Res. 2018, 10, 493. [Google Scholar] [CrossRef]
- Moroishi, T.; Hayashi, T.; Pan, W.-W.; Fujita, Y.; Holt, M.V.; Qin, J.; Carson, D.A.; Guan, K.-L. The Hippo pathway kinases LATS1/2 suppress cancer immunity. Cell 2016, 167, 1525–1539.e1517. [Google Scholar] [CrossRef]
- Nagaya, T.; Nakamura, Y.; Okuyama, S.; Ogata, F.; Maruoka, Y.; Choyke, P.L.; Allen, C.; Kobayashi, H. Syngeneic Mouse Models of Oral Cancer Are Effectively Targeted by Anti-CD44-Based NIR-PIT. Mol. Cancer Res. 2017, 15, 1667–1677. [Google Scholar] [CrossRef]
- Chung, M.K.; Jung, Y.H.; Lee, J.K.; Cho, S.Y.; Murillo-Sauca, O.; Uppaluri, R.; Shin, J.H.; Sunwoo, J.B. CD271 Confers an Invasive and Metastatic Phenotype of Head and Neck Squamous Cell Carcinoma through the Upregulation of Slug. Clin. Cancer Res. 2018, 24, 674. [Google Scholar] [CrossRef] [PubMed]
- Judd, N.P.; Allen, C.T.; Winkler, A.E.; Uppaluri, R. Comparative Analysis of Tumor-Infiltrating Lymphocytes in a Syngeneic Mouse Model of Oral Cancer. Otolaryngol.–Head Neck Surg. 2012, 147, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Chang, K.-W.; Yang, I.T.; Tu, H.-F.; Lin, S.-C. Establishment of syngeneic murine model for oral cancer therapy. Oral Oncol. 2019, 95, 194–201. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Liu, C.-J.; Lin, L.-H.; Chou, C.-H.; Yeh, L.-Y.; Lin, S.-C.; Chang, K.-W. Establishing of mouse oral carcinoma cell lines derived from transgenic mice and their use as syngeneic tumorigenesis models. BMC Cancer 2019, 19, 281. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Liu, K.-J.; Jang, C.-W.; Hsu, C.-C.; Yen, Y.-C.; Liu, Y.-L.; Chuang, T.-H.; Wang, S.-H.; Fu, Y.-K.; Kuo, C.-C.; et al. ERK Activation Modulates Cancer Stemness and Motility of a Novel Mouse Oral Squamous Cell Carcinoma Cell Line. Cancers 2019, 12, 61. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, V.H.; Allevato, M.M.; Gilardi, M.; He, Y.; Luis Callejas-Valera, J.; Vitale-Cross, L.; Martin, D.; Amornphimoltham, P.; McDermott, J.; et al. Syngeneic animal models of tobacco-associated oral cancer reveal the activity of in situ anti-CTLA-4. Nat. Commun. 2019, 10, 5546. [Google Scholar] [CrossRef] [PubMed]
- Scheff, N.N.; Ye, Y.; Bhattacharya, A.; MacRae, J.; Hickman, D.N.; Sharma, A.K.; Dolan, J.C.; Schmidt, B.L. Tumor necrosis factor alpha secreted from oral squamous cell carcinoma contributes to cancer pain and associated inflammation. Pain 2017, 158, 2396–2409. [Google Scholar] [CrossRef]
- Maji, S.; Samal, S.K.; Pattanaik, L.; Panda, S.; Quinn, B.A.; Das, S.K.; Sarkar, D.; Pellecchia, M.; Fisher, P.B.; Dash, R. Mcl-1 is an important therapeutic target for oral squamous cell carcinomas. Oncotarget 2015, 6, 16623. [Google Scholar] [CrossRef]
- Chen, W.-C.; Lai, C.-H.; Chuang, H.-C.; Lin, P.-Y.; Chen, M.-F. Inflammation-induced myeloid-derived suppressor cells associated with squamous cell carcinoma of the head and neck. Head Neck 2017, 39, 347–355. [Google Scholar] [CrossRef]
- Vincent-Chong, V.K.; DeJong, H.; Attwood, K.; Hershberger, P.A.; Seshadri, M. Preclinical prevention trial of calcitriol: Impact of stage of intervention and duration of treatment on oral carcinogenesis. Neoplasia 2019, 21, 376–388. [Google Scholar] [CrossRef]
- Droguett, D.; Castillo, C.; Leiva, E.; Theoduloz, C.; Schmeda-Hirschmann, G.; Kemmerling, U. Efficacy of quercetin against chemically induced murine oral squamous cell carcinoma. Oncol. Lett. 2015, 10, 2432–2438. [Google Scholar] [CrossRef] [PubMed]
- Foy, J.-P.; Tortereau, A.; Caulin, C.; Le Texier, V.; Lavergne, E.; Thomas, E.; Chabaud, S.; Perol, D.; Lachuer, J.; Lang, W. The dynamics of gene expression changes in a mouse model of oral tumorigenesis may help refine prevention and treatment strategies in patients with oral cancer. Oncotarget 2016, 7, 35932. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, R.; Wu, Y.; Diao, P.; Zhang, W.; Li, J.; Li, Z.; Wang, Y.; Cheng, J.; Yang, J. Overexpression of CDK7 is associated with unfavourable prognosis in oral squamous cell carcinoma. Pathology 2019, 51, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-S.; Zheng, M.; Zhang, M.; Pang, X.; Li, L.; Wang, S.-S.; Yang, X.; Wu, J.-B.; Tang, Y.-J.; Tang, Y.-L.; et al. Porphyromonas gingivalis Promotes 4-Nitroquinoline-1-Oxide-Induced Oral Carcinogenesis With an Alteration of Fatty Acid Metabolism. Front. Microbiol. 2018, 9, 2081. [Google Scholar] [CrossRef]
- Liu, Y.C.; Ho, H.C.; Lee, M.R.; Yeh, C.M.; Tseng, H.C.; Lin, Y.C.; Chung, J.G. Cortactin is a prognostic marker for oral squamous cell carcinoma and its overexpression is involved in oral carcinogenesis. Environ. Toxicol. 2017, 32, 799–812. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, X.; Zhang, X.; Sun, Z.; Chen, X. Ethanol promotes chemically induced oral cancer in mice through activation of the 5-lipoxygenase pathway of arachidonic acid metabolism. Cancer Prev. Res. 2011, 4, 1863–1872. [Google Scholar] [CrossRef]
- Chang, N.-W.; Pei, R.-J.; Tseng, H.-C.; Yeh, K.-T.; Chan, H.-C.; Lee, M.-R.; Lin, C.; Hsieh, W.-T.; Kao, M.-C.; Tsai, M.-H. Co-treating with arecoline and 4-nitroquinoline 1-oxide to establish a mouse model mimicking oral tumorigenesis. Chem. Biol. Interact. 2010, 183, 231–237. [Google Scholar] [CrossRef]
- Guttenplan, J.B.; Kosinska, W.; Zhao, Z.L.; Chen, K.M.; Aliaga, C.; DelTondo, J.; Cooper, T.; Sun, Y.W.; Zhang, S.M.; Jiang, K. Mutagenesis and carcinogenesis induced by dibenzo [a, l] pyrene in the mouse oral cavity: A potential new model for oral cancer. Int. J. Cancer 2012, 130, 2783–2790. [Google Scholar] [CrossRef]
- Culp, S.J.; Gaylor, D.W.; Sheldon, W.G.; Goldstein, L.S.; Beland, F.A. A comparison of the tumors induced by coal tar and benzo [a] pyrene in a 2-year bioassay. Carcinogenesis 1998, 19, 117–124. [Google Scholar] [CrossRef]
- Guttenplan, J.B.; Chen, K.-M.; Sun, Y.-W.; Shalaby, N.A.; Kosinska, W.; Desai, D.; Gowda, K.; Amin, S.; El-Bayoumy, K. Effects of the Tobacco Carcinogens N′-Nitrosonornicotine and Dibenzo [a, l] pyrene Individually and in Combination on DNA Damage in Human Oral Leukoplakia and on Mutagenicity and Mutation Profiles in lacI Mouse Tongue. Chem. Res. Toxicol. 2019, 32, 1893–1899. [Google Scholar] [CrossRef]
- Opitz, O.G.; Harada, H.; Suliman, Y.; Rhoades, B.; Sharpless, N.E.; Kent, R.; Kopelovich, L.; Nakagawa, H.; Rustgi, A.K. A mouse model of human oral-esophageal cancer. J. Clin. Investig. 2002, 110, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Caulin, C.; Nguyen, T.; Longley, M.A.; Zhou, Z.; Wang, X.-J.; Roop, D.R. Inducible activation of oncogenic K-ras results in tumor formation in the oral cavity. Cancer Res. 2004, 64, 5054–5058. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Hall, B.; Sun, Z.-J.; Molinolo, A.; Chen, W.; Gutkind, J.S.; Waes, C.; Kulkarni, A.B. Loss of TGF-β signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012, 31, 3322–3332. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gonzalez, C.L.; Wang, B.; Zhang, Y.; Mejia, O.; Katsonis, P.; Lichtarge, O.; Myers, J.N.; El-Naggar, A.K.; Caulin, C. Cdkn2a suppresses metastasis in squamous cell carcinomas induced by the gain-of-function mutant p53R172H. J. Pathol. 2016, 240, 224–234. [Google Scholar] [CrossRef]
- Zhong, R.; Pytynia, M.; Pelizzari, C.; Spiotto, M. Bioluminescent imaging of HPV-positive oral tumor growth and its response to image-guided radiotherapy. Cancer Res. 2014, 74, 2073–2081. [Google Scholar] [CrossRef]
- Carper, M.B.; Troutman, S.; Wagner, B.L.; Byrd, K.M.; Selitsky, S.R.; Parag-Sharma, K.; Henry, E.C.; Li, W.; Parker, J.S.; Montgomery, S.A. An Immunocompetent mouse model of HPV16 (+) head and neck squamous cell carcinoma. Cell Rep. 2019, 29, 1660–1674.e1667. [Google Scholar] [CrossRef]
- Morton, J.J.; Bird, G.; Keysar, S.B.; Astling, D.P.; Lyons, T.R.; Anderson, R.T.; Glogowska, M.J.; Estes, P.; Eagles, J.R.; Le, P.N. XactMice: Humanizing mouse bone marrow enables microenvironment reconstitution in a patient-derived xenograft model of head and neck cancer. Oncogene 2016, 35, 290–300. [Google Scholar] [CrossRef]
- Morton, J.J.; Keysar, S.B.; Perrenoud, L.; Chimed, T.S.; Reisinger, J.; Jackson, B.; Le, P.N.; Nieto, C.; Gomez, K.; Miller, B. Dual use of hematopoietic and mesenchymal stem cells enhances engraftment and immune cell trafficking in an allogeneic humanized mouse model of head and neck cancer. Mol. Carcinog. 2018, 57, 1651–1663. [Google Scholar] [CrossRef]
- Kenney, L.; Shultz, L.D.; Greiner, D.; Brehm, M. Humanized mouse models for transplant immunology. Am. J. Transplant. 2016, 16, 389–397. [Google Scholar] [CrossRef]
- Li, Q.; Dong, H.; Yang, G.; Song, Y.; Mou, Y.; Ni, Y. Mouse Tumor-Bearing Models as Preclinical Study Platforms for Oral Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 212. [Google Scholar] [CrossRef]
- Kim, S.-S.; Harford, J.B.; Moghe, M.; Slaughter, T.; Doherty, C.; Chang, E.H. A tumor-targeting nanomedicine carrying the p53 gene crosses the blood–brain barrier and enhances anti-PD-1 immunotherapy in mouse models of glioblastoma. Int. J. Cancer 2019, 145, 2535–2546. [Google Scholar] [CrossRef] [PubMed]
- Jiao, R.; Allen, K.J.H.; Malo, M.E.; Rickles, D.; Dadachova, E. Evaluating the Combination of Radioimmunotherapy and Immunotherapy in a Melanoma Mouse Model. Int. J. Mol. Sci. 2020, 21, 773. [Google Scholar] [CrossRef] [PubMed]
- Ngiow, S.F.; Loi, S.; Thomas, D.; Smyth, M.J. Mouse Models of Tumor Immunotherapy. Adv. Immunol. 2016, 130, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Drake, C.G. Immunotherapy for prostate cancer: Recent advances, lessons learned, and areas for further research. Clin. Cancer Res. 2011, 17, 3884–3891. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Chen, X.; Chan, C.y.; Li, D.; Yuan, C.; Yu, F.; Lin, M.C.; Yew, D.T.; Kung, H.F.; Lai, L. Two-dimensional differential gel electrophoresis/analysis of diethylnitrosamine induced rat hepatocellular carcinoma. Int. J. Cancer 2008, 122, 2682–2688. [Google Scholar] [CrossRef]
- Enoch, S.J.; Cronin, M.T. A review of the electrophilic reaction chemistry involved in covalent DNA binding. Crit. Rev. Toxicol. 2010, 40, 728–748. [Google Scholar] [CrossRef]
- Oliveira, P.A.; Colaço, A.; Chaves, R.; Guedes-Pinto, H.; De-La-Cruz, P.L.; Lopes, C. Chemical carcinogenesis. An. Acad. Bras. Cienc. 2007, 79, 593–616. [Google Scholar] [CrossRef]
- Golan, D.E.; Tashjian, A.H.; Armstrong, E.J. Principles of Pharmacology: The Pathophysiologic Basis of Drug Therapy; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011. [Google Scholar]
- Williams, G.M. Mechanisms of chemical carcinogenesis and application to human cancer risk assessment. Toxicology 2001, 166, 3–10. [Google Scholar] [CrossRef]
- Neumann, H.-G. The role of DNA damage in chemical carcinogenesis of aromatic amines. J. Cancer Res. Clin. Oncol. 1986, 112, 100–106. [Google Scholar] [CrossRef]
- Hassan, A.; Alam, S.; Abdel-Aziem, S.; Ahmed, K. Benzo-a-pyrene induced genotoxicity and cytotoxicity in germ cells of mice: Intervention of radish and cress. J. Genet. Eng. Biotechnol. 2011, 9, 65–72. [Google Scholar] [CrossRef]
- Bukowska, B.; Duchnowicz, P. Molecular Mechanisms of Action of Selected Substances Involved in the Reduction of Benzo[a]pyrene-Induced Oxidative Stress. Molecules 2022, 27, 1379. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? 1. Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.A.O. The Role of MicroRNA in Chemical Carcinogenesis. J. Environ. Sci. Health Part C 2010, 28, 89–124. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.; Christensen, J.; Preston, R.; Goldsworthy, T.; Tlsty, T.; Fox, T. Attenuation of G1 checkpoint function by the non-genotoxic carcinogen phenobarbital. Carcinogenesis 1998, 19, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Luch, A. Nature and nurture–lessons from chemical carcinogenesis. Nat. Rev. Cancer 2005, 5, 113–125. [Google Scholar] [CrossRef]
- Walker, N.J.; Tritscher, A.M.; Sills, R.C.; Lucier, G.W.; Portier, C.J. Hepatocarcinogenesis in female Sprague-Dawley rats following discontinuous treatment with 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2000, 54, 330–337. [Google Scholar] [CrossRef]
- Yang, H.; Bocchetta, M.; Kroczynska, B.; Elmishad, A.G.; Chen, Y.; Liu, Z.; Bubici, C.; Mossman, B.T.; Pass, H.I.; Testa, J.R. TNF-α inhibits asbestos-induced cytotoxicity via a NF-κB-dependent pathway, a possible mechanism for asbestos-induced oncogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 10397–10402. [Google Scholar] [CrossRef]
- Trosko, J.E. The role of stem cells and gap junctional intercellular communication in carcinogenesis. BMB Rep. 2003, 36, 43–48. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chow, M.T.; Sceneay, J.; Paget, C.; Wong, C.S.; Duret, H.; Tschopp, J.; Möller, A.; Smyth, M.J. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 2012, 72, 5721–5732. [Google Scholar] [CrossRef] [PubMed]
- Levingston, C.A.; Young, M.R. Transient immunological and clinical effectiveness of treating mice bearing premalignant oral lesions with PD-1 antibodies. Int. J. Cancer 2017, 140, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xie, T.; Wang, B.; William, W.N., Jr.; Heymach, J.V.; El-Naggar, A.K.; Myers, J.N.; Caulin, C. PD-1 Blockade Prevents the Development and Progression of Carcinogen-Induced Oral Premalignant Lesions. Cancer Prev. Res. 2017, 10, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Neville, B.W.; Day, T.A. Oral cancer and precancerous lesions. CA. Cancer J. Clin. 2002, 52, 195–215. [Google Scholar] [CrossRef]
- Johnson, N. Tobacco use and oral cancer: A global perspective. J. Dent. Educ. 2001, 65, 328–339. [Google Scholar] [CrossRef]
- Hecht, S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 2003, 3, 733–744. [Google Scholar] [CrossRef]
- Jin, F.; Thaiparambil, J.; Donepudi, S.R.; Vantaku, V.; Piyarathna, D.W.B.; Maity, S.; Krishnapuram, R.; Putluri, V.; Gu, F.; Purwaha, P. Tobacco-specific carcinogens induce hypermethylation, DNA adducts, and DNA damage in bladder cancer. Cancer Prev. Res. 2017, 10, 588–597. [Google Scholar] [CrossRef]
- Ludwig, S.; Hong, C.-S.; Razzo, B.M.; Fabian, K.P.; Chelvanambi, M.; Lang, S.; Storkus, W.J.; Whiteside, T.L. Impact of combination immunochemotherapies on progression of 4NQO-induced murine oral squamous cell carcinoma. Cancer Immunol. Immunother. 2019, 68, 1133–1141. [Google Scholar] [CrossRef]
- Vincent-Chong, V.; DeJong, H.; Rich, L.; Patti, A.; Merzianu, M.; Hershberger, P.; Seshadri, M. Impact of age on disease progression and microenvironment in oral cancer. J. Dent. Res. 2018, 97, 1268–1276. [Google Scholar] [CrossRef]
- Tang, X.-H.; Knudsen, B.; Bemis, D.; Tickoo, S.; Gudas, L.J. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin. Cancer Res. 2004, 10, 301–313. [Google Scholar] [CrossRef]
- Vitale-Cross, L.; Czerninski, R.; Amornphimoltham, P.; Patel, V.; Molinolo, A.A.; Gutkind, J.S. Chemical carcinogenesis models for evaluating molecular-targeted prevention and treatment of oral cancer. Cancer Prev. Res. 2009, 2, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Schoop, R.A.; Noteborn, M.H.; Baatenburg de Jong, R.J. A mouse model for oral squamous cell carcinoma. J. Mol. Histol. 2009, 40, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Bisetto, S.; Whitaker-Menezes, D.; Wilski, N.A.; Tuluc, M.; Curry, J.; Zhan, T.; Snyder, C.M.; Martinez-Outschoorn, U.E.; Philp, N.J. Monocarboxylate transporter 4 (MCT4) knockout mice have attenuated 4NQO induced carcinogenesis; a role for MCT4 in driving oral squamous cell cancer. Front. Oncol. 2018, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, Y.; Liao, L.; Zhu, X.; Tang, S.; Yang, Q.; Sun, L.; Li, Y.; Gao, S.; Xie, Z. Inhibition of 4NQO-induced oral carcinogenesis by dietary oyster shell calcium. Integr. Cancer Ther. 2016, 15, 96–101. [Google Scholar] [CrossRef]
- Fujino, H.; Chino, T.; Imai, T. Experimental production of labial and lingual carcinoma by local application of 4-nitroquinoline N-oxide. J. Natl. Cancer Inst. 1965, 35, 907–918. [Google Scholar] [PubMed]
- Khandelwal, A.R.; Moore-Medlin, T.; Ekshyyan, O.; Gu, X.; Abreo, F.; Nathan, C.-A.O. Local and systemic Curcumin C3 complex inhibits 4NQO-induced oral tumorigenesis via modulating FGF-2/FGFR-2 activation. Am. J. Cancer Res. 2018, 8, 2538. [Google Scholar]
- Tamura, T.; Ichikawa, T.; Nakahata, S.; Kondo, Y.; Tagawa, Y.; Yamamoto, K.; Nagai, K.; Baba, T.; Yamaguchi, R.; Futakuchi, M. Loss of NDRG2 expression confers oral squamous cell carcinoma with enhanced metastatic potential. Cancer Res. 2017, 77, 2363–2374. [Google Scholar] [CrossRef]
- Wu, T.; Hong, Y.; Jia, L.; Wu, J.; Xia, J.; Wang, J.; Hu, Q.; Cheng, B. Modulation of IL-1β reprogrammes the tumor microenvironment to interrupt oral carcinogenesis. Sci. Rep. 2016, 6, 20208. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Fang, J.; Song, J.; Ma, D.; Luo, L.; He, B.; Xia, J.; Lui, V.W.Y.; Cheng, B. Mesenchymal stem cells participate in oral mucosa carcinogenesis by regulating T cell proliferation. Clin. Immunol. 2019, 198, 46–53. [Google Scholar] [CrossRef]
- Young, M.R.I. Use of carcinogen-induced premalignant oral lesions in a dendritic cell-based vaccine to stimulate immune reactivity against both premalignant oral lesions and oral cancer. J. Immunother. 2008, 31, 148. [Google Scholar] [CrossRef]
- Li, J.; Qiu, G.; Fang, B.; Dai, X.; Cai, J. Deficiency of IL-18 aggravates esophageal carcinoma through inhibiting IFN-γ production by CD8+ T cells and NK cells. Inflammation 2018, 41, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Stashenko, P.; Yost, S.; Choi, Y.; Danciu, T.; Chen, T.; Yoganathan, S.; Kressirer, C.; Ruiz-Tourrella, M.; Das, B.; Kokaras, A. The oral mouse microbiome promotes tumorigenesis in oral squamous cell carcinoma. Msystems 2019, 4, e00323-19. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Yerneni, S.S.; Razzo, B.M.; Whiteside, T.L. Exosomes from HNSCC promote angiogenesis through reprogramming of endothelial cells. Mol. Cancer Res. 2018, 16, 1798–1808. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Fujii, T.; Ugaki, Y.; Yasui, H.; Watanabe, M.; Dateoka, S.; Kakudo, K. Usefulness of a fluorescence visualization system for the detection of oral precancerous and early cancerous lesions. Oncol. Rep. 2016, 36, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Q.; Li, X.; Ma, D.; Fang, J.; Luo, L.; Liu, X.; Wang, X.; Lui, V.W.Y.; Xia, J. Blockade of PD-1 effectively inhibits in vivo malignant transformation of oral mucosa. Oncoimmunology 2018, 7, e1388484. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Su, Y.X.; Wang, L.; Zhang, T.H.; Liang, Y.J.; Liang, L.Z.; Liao, G.Q. Myeloid-derived suppressor cells contribute to oral cancer progression in 4NQO-treated mice. Oral Dis. 2012, 18, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Oghumu, S.; Knobloch, T.J.; Terrazas, C.; Varikuti, S.; Ahn-Jarvis, J.; Bollinger, C.E.; Iwenofu, H.; Weghorst, C.M.; Satoskar, A.R. Deletion of macrophage migration inhibitory factor inhibits murine oral carcinogenesis: Potential role for chronic pro-inflammatory immune mediators. Int. J. Cancer 2016, 139, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.S.; Li, L.; Wang, S.S.; Pang, X.; Wu, J.B.; Sheng, S.R.; Tang, Y.J.; Tang, Y.L.; Zheng, M.; Liang, X.H. Autophagy is positively associated with the accumulation of myeloid-derived suppressor cells in 4-nitroquinoline-1-oxide-induced oral cancer. Oncol. Rep. 2018, 40, 3381–3391. [Google Scholar] [CrossRef]
- Wen, L.; Lu, H.; Li, Q.; Li, Q.; Wen, S.; Wang, D.; Wang, X.; Fang, J.; Cui, J.; Cheng, B. Contributions of T cell dysfunction to the resistance against anti-PD-1 therapy in oral carcinogenesis. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef]
- De Costa, A.-M.A.; Justis, D.N.; Schuyler, C.A.; Young, M.R.I. Administration of a vaccine composed of dendritic cells pulsed with premalignant oral lesion lysate to mice bearing carcinogen-induced premalignant oral lesions stimulates a protective immune response. Int. Immunopharmacol. 2012, 13, 322–330. [Google Scholar] [CrossRef]
- Prahalad, A.K.; Ross, J.A.; Nelson, G.B.; Roop, B.C.; King, L.C.; Nesnow, S.; Mass, M.J. Dibenzo [a, l] pyrene-induced DNA adduction, tumorigenicity, and Ki-ras oncogene mutations in strain A/J mouse lung. Carcinogenesis 1997, 18, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.L.; Rogan, E.G.; Higginbotham, S.; Cremonesi, P.; Salmasi, S. Tumor-initiating activity in mouse skin and carcinogenicity in rat mammary gland of dibenzo [a] pyrenes: The very potent environmental carcinogen dibenzo [a, l] pyrene. J. Cancer Res. Clin. Oncol. 1989, 115, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, S.; RamaKrishna, N.; Johansson, S.L.; Rogan, E.G.; Cavalieri, E.L. Tumor-initiating activity and carcinogenicity of dibenzo [a, l] pyrene versus 7, 12-dimethylbenz [a] anthracene and benzo [a] pyrene at low doses in mouse skin. Carcinogenesis 1993, 14, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H. Polycyclic aromatic hydrocarbons in the diet. Mutat. Res. 1999, 443, 139–147. [Google Scholar] [CrossRef]
- Tu, H.F.; Chen, M.Y.; Lai, J.C.Y.; Chen, Y.L.; Wong, Y.W.; Yang, C.C.; Chen, H.Y.; Hsia, S.M.; Shih, Y.H.; Shieh, T.M. Arecoline-regulated ataxia telangiectasia mutated expression level in oral cancer progression. Head Neck 2019, 41, 2525–2537. [Google Scholar] [CrossRef]
- Kuo, T.M.; Nithiyanantham, S.; Lee, C.P.; Hsu, H.T.; Luo, S.Y.; Lin, Y.Z.; Yeh, K.T.; Ko, Y.C. Arecoline N-oxide regulates oral squamous cell carcinoma development through NOTCH1 and FAT1 expressions. J. Cell. Physiol. 2019, 234, 13984–13993. [Google Scholar] [CrossRef]
- Lo, W.-Y.; Tsai, M.-H.; Tsai, Y.; Hua, C.-H.; Tsai, F.-J.; Huang, S.-Y.; Tsai, C.-H.; Lai, C.-C. Identification of over-expressed proteins in oral squamous cell carcinoma (OSCC) patients by clinical proteomic analysis. Clin. Chim. Acta 2007, 376, 101–107. [Google Scholar] [CrossRef]
- Walrath, J.C.; Hawes, J.J.; Van Dyke, T.; Reilly, K.M. Genetically engineered mouse models in cancer research. Adv. Cancer Res. 2010, 106, 113–164. [Google Scholar]
- Lampreht Tratar, U.; Horvat, S.; Cemazar, M. Transgenic mouse models in cancer research. Front. Oncol. 2018, 8, 268. [Google Scholar] [CrossRef]
- Vitale-Cross, L.; Amornphimoltham, P.; Fisher, G.; Molinolo, A.A.; Gutkind, J.S. Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res. 2004, 64, 8804–8807. [Google Scholar] [CrossRef]
- Nakagawa, H.; Wang, T.C.; Zukerberg, L.; Odze, R.; Togawa, K.; May, G.H.; Wilson, J.; Rustgi, A.K. The targeting of the cyclin D1 oncogene by an Epstein-Barr virus promoter in transgenic mice causes dysplasia in the tongue, esophagus and forestomach. Oncogene 1997, 14, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Munirajan, A.K.; Mohanprasad, B.; Shanmugam, G.; Tsuchida, N. Detection of a rare point mutation at codon 59 and relatively high incidence of H-ras mutation in Indian oral cancer. Int. J. Oncol. 1998, 13, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Saranath, D.; Chang, S.; Bhoite, L.; Panchal, R.; Kerr, I.; Mehta, A.; Johnson, N.; Deo, M. High frequency mutation in codons 12 and 61 of H-ras oncogene in chewing tobacco-related human oral carcinoma in India. Br. J. Cancer 1991, 63, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Holzapfel, B.M.; Wagner, F.; Thibaudeau, L.; Levesque, J.P.; Hutmacher, D.W. Concise review: Humanized models of tumor immunology in the 21st century: Convergence of cancer research and tissue engineering. Stem Cells 2015, 33, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J.; Miller, P. Trial watch: Phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef]
- Jung, J. Human tumor xenograft models for preclinical assessment of anticancer drug development. Toxicol. Res. 2014, 30, 1–5. [Google Scholar] [CrossRef]
- Hsu, C.; Lin, L.I.; Cheng, Y.C.; Feng, Z.R.; Shao, Y.Y.; Cheng, A.L.; Ou, D.L. Cyclin E1 Inhibition can Overcome Sorafenib Resistance in Hepatocellular Carcinoma Cells Through Mcl-1 Suppression. Clin. Cancer Res. 2016, 22, 2555–2564. [Google Scholar] [CrossRef]
- Goldman, J.P.; Blundell, M.P.; Lopes, L.; Kinnon, C.; DI Santo, J.P.; Thrasher, A.J. Enhanced human cell engraftment in mice deficient in RAG2 and the common cytokine receptor γ chain. Br. J. Haematol. 1998, 103, 335–342. [Google Scholar] [CrossRef]
- Bosma, M.J.; Carroll, A.M. The SCID mouse mutant: Definition, characterization, and potential uses. Annu. Rev. Immunol. 1991, 9, 323–350. [Google Scholar] [CrossRef]
- Taghian, A.; Budach, W.; Zietman, A.; Freeman, J.; Gioioso, D.; Ruka, W.; Suit, H.D. Quantitative comparison between the transplantability of human and murine tumors into the subcutaneous tissue of NCr/Sed-nu/nu nude and severe combined immunodeficient mice. Cancer Res. 1993, 53, 5012–5017. [Google Scholar]
- McCune, J.; Namikawa, R.; Kaneshima, H.; Shultz, L.; Lieberman, M.; Weissman, I. The SCID-hu mouse: Murine model for the analysis of human hematolymphoid differentiation and function. Science 1988, 241, 1632–1639. [Google Scholar] [CrossRef]
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 1988, 335, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef]
- Mombaerts, P.; Iacomini, J.; Johnson, R.S.; Herrup, K.; Tonegawa, S.; Papaioannou, V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992, 68, 869–877. [Google Scholar] [CrossRef]
- Shinkai, Y.; Lam, K.-P.; Oltz, E.M.; Stewart, V.; Mendelsohn, M.; Charron, J.; Datta, M.; Young, F.; Stall, A.M.; Alt, F.W. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V (D) J rearrangement. Cell 1992, 68, 855–867. [Google Scholar] [CrossRef]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.; Greiner, D.L. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K. NOD/SCID/γ c null mouse: An excellent recipient mouse model for engraftment of human cells. Blood J. Am. Soc. Hematol. 2002, 100, 3175–3182. [Google Scholar]
- Traggiai, E.; Chicha, L.; Mazzucchelli, L.; Bronz, L.; Piffaretti, J.-C.; Lanzavecchia, A.; Manz, M.G. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 2004, 304, 104–107. [Google Scholar] [CrossRef]
- Brehm, M.A.; Cuthbert, A.; Yang, C.; Miller, D.M.; DiIorio, P.; Laning, J.; Burzenski, L.; Gott, B.; Foreman, O.; Kavirayani, A. Parameters for establishing humanized mouse models to study human immunity: Analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rγnull mutation. Clin. Immunol. 2010, 135, 84–98. [Google Scholar] [CrossRef]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2Rγnull mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized mice for the study of immuno-oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gu, H.; Li, H.; Gao, S.; Shi, X.; Shen, J.; Li, B.; Wang, H.; Zheng, K.; Shao, Z.; et al. Large-cohort humanized NPI mice reconstituted with CD34+ hematopoietic stem cells are feasible for evaluating preclinical cancer immunotherapy. FASEB J. 2022, 36, e22244. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, T.; Iizuka, A.; Nonomura, C.; Kondou, R.; Maeda, C.; Miyata, H.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; et al. Antitumor Effect of Programmed Death-1 (PD-1) Blockade in Humanized the NOG-MHC Double Knockout Mouse. Clin. Cancer Res. 2017, 23, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Rodriguez, I.; Schalper, K.A.; Oñate, C.; Azpilikueta, A.; Rodriguez-Ruiz, M.E.; Morales-Kastresana, A.; Labiano, S.; Pérez-Gracia, J.L.; Martín-Algarra, S.; et al. Nivolumab and Urelumab Enhance Antitumor Activity of Human T Lymphocytes Engrafted in Rag2−/−IL2Rγnull Immunodeficient Mice. Cancer Res. 2015, 75, 3466–3478. [Google Scholar] [CrossRef]
- Ma, S.-D.; Xu, X.; Jones, R.; Delecluse, H.-J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef]
- Morton, J.J.; Bird, G.; Refaeli, Y.; Jimeno, A. Humanized mouse xenograft models: Narrowing the tumor–microenvironment gap. Cancer Res. 2016, 76, 6153–6158. [Google Scholar] [CrossRef]
- Greenblatt, M.B.; Vbranac, V.; Tivey, T.; Tsang, K.; Tager, A.M.; Aliprantis, A.O. Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. PLoS ONE 2012, 7, e44664. [Google Scholar] [CrossRef]
- BIOCYTOGEN. Humanized Immune-Checkpoint Mice. Available online: https://biocytogen.com/products/humanized-immune-checkpoint-mice/ (accessed on 9 May 2022).
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vély, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbé, C. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961. [Google Scholar] [CrossRef]
- Applied StemCell, Inc. Immune Checkpoint Humanized Mouse Models. Available online: https://www.appliedstemcell.com/research/animal-models/mouse-repository/immune-checkpoint-mouse-models (accessed on 9 May 2022).
- Horowitz, N.B.; Mohammad, I.; Moreno-Nieves, U.Y.; Koliesnik, I.; Tran, Q.; Sunwoo, J.B. Humanized Mouse Models for the Advancement of Innate Lymphoid Cell-Based Cancer Immunotherapies. Front. Immunol. 2021, 12, 648580. [Google Scholar] [CrossRef]
- Sternberg-Simon, M.; Brodin, P.; Pickman, Y.; Önfelt, B.; Kärre, K.; Malmberg, K.-J.; Höglund, P.; Mehr, R. Natural killer cell inhibitory receptor expression in humans and mice: A closer look. Front. Immunol. 2013, 4, 65. [Google Scholar] [CrossRef]
- Du, X.; Liu, M.; Su, J.; Zhang, P.; Tang, F.; Ye, P.; Devenport, M.; Wang, X.; Zhang, Y.; Liu, Y. Uncoupling therapeutic from immunotherapy-related adverse effects for safer and effective anti-CTLA-4 antibodies in CTLA4 humanized mice. Cell Res. 2018, 28, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Lute, K.D.; May, K.F., Jr.; Lu, P.; Zhang, H.; Kocak, E.; Mosinger, B.; Wolford, C.; Phillips, G.; Caligiuri, M.A.; Zheng, P. Human CTLA4 knock-in mice unravel the quantitative link between tumor immunity and autoimmunity induced by anti–CTLA-4 antibodies. Blood 2005, 106, 3127–3133. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T. Manipulating the microbiome to improve the efficacy of immunotherapy. Clin. Adv. Hematol. Oncol. HO 2016, 14, 424–426. [Google Scholar]
- Uno, T.; Takeda, K.; Kojima, Y.; Yoshizawa, H.; Akiba, H.; Mittler, R.S.; Gejyo, F.; Okumura, K.; Yagita, H.; Smyth, M.J. Eradication of established tumors in mice by a combination antibody-based therapy. Nat. Med. 2006, 12, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Blake, S.J.; Harjunpää, H.; Fairfax, K.A.; Yong, M.C.; Allen, S.; Kohrt, H.E.; Takeda, K.; Smyth, M.J.; Teng, M.W. Assessing immune-related adverse events of efficacious combination immunotherapies in preclinical models of cancer. Cancer Res. 2016, 76, 5288–5301. [Google Scholar] [CrossRef]
- Liu, J.; Blake, S.J.; Smyth, M.J.; Teng, M.W. Improved mouse models to assess tumour immunity and irAEs after combination cancer immunotherapies. Clin. Transl. Immunol. 2014, 3, e22. [Google Scholar] [CrossRef]

{kind=link}
| Trial (Code) | Drugs (Brand Name) Interventions | Patients Number | Immune Checkpoint Tested | |
|---|---|---|---|---|
| KEYNOTE-048 NCT02358031 | Pembrolizumab ± chemotherapy vs. chemotherapy + cetuximab | 882 [30] | Pembrolizumab: PD-1 | First line treatment for R/M HNSCC |
| KEYNOTE-040 NCT02252042 | Pembrolizumab vs. methotrexate, docetaxel or cetuximab | 495 [31] | Pembrolizumab: PD-1 | Second line treatment for R/M HNSCC |
| CheckMate 141 NCT02105636 | Nivolumab vs. chemotherapy | 361 [32] | Nivolumab: PD-1 | Second line treatment for R/M HNSCC |
| EAGLE NCT02369874 | Durvalumab ± tremelimumab vs. chemotherapy | 736 [33] | Durvalumab: PD-L1 Tremelimumab: CTLA-4 | Second line treatment for R/M HNSCC |
| NCT01836029 | Pembrolizumab | 195 [34] | Pembrolizumab: PD-1 | R/M HNSCC |
| CheckMate 651 NCT02741570 | Ipilumumab and nivolumab vs. cetuximab with platinum and fluorouracil | 947 [35] | Ipilimumab: CTLA-4 Nivolumab: PD-1 | First line treatment for HNSCC |
| KESTREL NCT02551159 | Durvalumab and tremelimumab vs. Durvalumab monotherapy | 823 | Durvalumab: PD-L1 Tremelimumab: CTLA-4 | R/M HNSCC |
| NCT03673735 | Durvalumab before CRT and every four weeks for six months after CRT. Control: placebo before CRT and six months every four weeks after CRT Radiotherapy | 650 | Durvalumab: PD-L1 | R/M HNSCC |
| Model | Pros | Cons |
|---|---|---|
| Syngeneic | Low in cost. Easy to set up. Rapid tumor development. Fully functional mouse immune system. | Tumors do not develop a natural microenvironment. Tumor heterogeneity is low. There are major differences between the immune systems of mice and humans. May cause vaccination effects. |
| Chemotoxic agent | Development of precancerous lesions. Host-tumor cell interactions are conserved. Tumors form a natural microenvironment. Fully functional mouse immune system. Tumor heterogeneity is high. Easy to use. Sporadic cancer development. It can be used in combination with other tumor induction methods. | High in cost. Difficult to set up. Time-consuming. Laborious. Tumor formation is not initiated by chronic inflammation. Chemical hazards. Metastasis and bone invasion are rare. Difficult to monitor tumors. Variability in tumor progression time. Due to high heterogeneity, larger sample sizes were required for data interpretation. |
| Genetically engineered | Gene expression can be manipulated. Useful for the study of genetic alterations. Encompasses natural tumor microenvironments. The tumor forms a natural microenvironment. Fully functional mouse immune system. The genetic and histopathological aspects of all stages of cancer can be recapitulated. Heterogeneity of the tumor is higher than in syngeneic models, depending on the production method. | High in cost. Overexpression of transgene. Tumors unrelated to the oral cavity may develop. Labor intensive. Limited accessibility. Difficult to monitor a tumor. Low immunogenicity. Challenging for breeding and gene manipulation. Homogeneous in the genomic aspect. |
| Humanized | Highly reflect promoter methylation in tumors and reproduced tumor heterogeneity. Heterogeneity of the tumor is high. | High in cost. Difficult to set up. A partial TME is transplanted from the patient and is dependent on the transplantation site and donor immune cells. |
| Hu-PBMC | Simple to construct, the T cell transplantation efficiency is high and it is stable. | Limited study time due to short survival as well as the occurrence of GvHD. |
| Hu-HSC | Development of multilineage hematopoietic cells, including T cells, B cells, NK cells, and myeloid cells. | T cells are educated by the mouse thymus; T cells are few and non-functional, not HLA-restricted. |
| Humanized Immune Checkpoint Knock-In Mice | Human gene knock-in mice strains allow for a robust expansion of human immune cells in the mouse TME. Precise humanized gene loci. Fully functional and potent immune system. | Often the transplant is subcutaneous, resulting in the surrounding environment lacking the chronic inflammatory milieu and organ-specific factors of the tumor. |
| Model | Animal Background | Inducer | Dosage/Treatment Period | Tumor Harvest/Formation/End Point/Conclusions/Development Period (Weeks) | References |
|---|---|---|---|---|---|
| Syngeneic | C3H | SQ-1979 (Subcutaneous; Orthotopic); SCC7 (Subcutaneous) | 5 × 106~1 × 107 cells; 1 × 105~1 × 106 cells | 3 weeks | [36,37,38,39] |
| C57BL/6 (Orthotopic)(Subcutaneous) | MTCQ1, MTCQ2, MOC-L1, MOC-L2, MOC-L3, MOC-L4, NHRI-HN1, NHRI-HN2, MOC1, MOC2-luc, MOC2 mKate2 | 1.5 × 105~5 × 106 cells | 2~5 weeks | [40,41,42,43,44,45] | |
| BALB/cAnN.Cg-Foxn1nu/CrlNarl (Subcutaneous) | NHRI-HN1, NHRI-HN2 | 1 × 106 cells | 6 weeks | [45] | |
| Chemotoxic agent | C3H (Orthotopic); C57BL/6 (Orthotopic); BALB/c CF-1 (Orthotopic); CBA (Orthotopic) | 4-NQO | 50~100 μg/mL/4~20 weeks | 4~70 weeks | [46,47,48,49,50,51,52,53,54,55] |
| C57BL/6J (Orthotopic) (male wild-type) 5-Lox knockout | 4NQO + Alcohol | 100 μg/mL + 8%/8 weeks + 16 weeks | [56] | ||
| C57BL/6JNarl (Orthotopic) | 4NQO + Arecoline | 200 μg/mL + 500 μg/mL/8 weeks | [57] | ||
| B6C3F1 (Orthotopic) | Tobacco-related (1) DB(a,l)P (2) B(a)P (3)N’-nitrosonornicotine (NNN) | 24 nmol 100 ppm/2 years 8.46 μmoL/2 times/week, 4 weeks | [58,59,60] | ||
| Genetically engineered | L2D1+ /p53+/− and L2D1+ /p53−/− (Orthotopic) | Formation of invasive oral–esophageal SCC at 6 months. | [61] | ||
| LSL-KrasG12D (Orthotopic) | K5 or K14-CrePR1 CrePR1 | Oncogenic K-ras G12D overexpression induced in oral epithelium of mice by16–24 weeks administration of RU486. Formation of squamous papilloma in the oral cavity. | [62] | ||
| Tgfbr1/Pten 2cKO mice (Orthotopic) | Tgfbr1/Pten 2cKO mice induced with 10-week administration of tamoxifen (tam). Formation of cancer and precancerous lesions in the oral epithelium. | [63] | |||
| p53R172H; K5-CrePR1 and p53flflox/flflox; K5-CrePR1 (Orthotopic) | Formation of OSCC at 15–16 months: p53flflox/flflox; K5-CrePR1 (25%) and p53 R172H; K5-CrePR1 (16%) mice | [64] | |||
| LSL-Kras (Orthotopic); iHPV-Luc (Orthotopic); K14-CreERtam mice (KHR mice) and LSL-Kras (Orthotopic); K14-CreERtam mice (KR mice) (Orthotopic) | Formation of oral tumors in KR and KHR mice using tamoxifen. Bioluminescence signal of KHR mice 74.8 times higher than control mice. | [65] | |||
| HPV16 E7iresE6 (Orthotopic); PIK3CA E545K (Orthotopic); KRT14-CreERtam mice (Orthotopic) | After the administration of tamoxifen for 6–8 weeks, oropharyngeal tumors developed with about 40% penetrance (1–2 tumors/tongue). | [66] | |||
| Humanized | XactMice (Orthotopic) | SHPCs, MSCs | [67,68] | ||
| (1) BALB/c-Rag2null (Orthotopic); IL2rgnull (BRG) (Orthotopic) (2) NOD.Cg-Rag1tm1Mom IL2rgtm1Wjl Ins2Akita (NRG-Akita) (Orthotopic) (3) NOD.Cg-Prkdcscid Il2rgtm1Wjl (NSG) (Orthotopic) | CD34+ umbilical cord blood cells | (1) 1 × 105 cells (2) 5 × 104 cells (3) 2 × 105 cells | (1) Total of 300–500 human islets transplanted subrenal capsule 8–26 weeks post CD34 HSC engraftment into normoglycemic BRG mice. (2) Total of 4000 human islets transplanted subrenal capsule into diabetic NRG-Akita mice. (3) Total of 3000–4000 human islets transplanted subrenal capsule into diabetic NSG mice, mice treated with streptozotocin to induce diabetes (timing not reported in relation to HSC engraftment or islet transplantation). | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, W.-C.; Ou, D.-L.; Tan, C.-T. Mouse Models for Immune Checkpoint Blockade Therapeutic Research in Oral Cancer. Int. J. Mol. Sci. 2022, 23, 9195. https://doi.org/10.3390/ijms23169195
Chiu W-C, Ou D-L, Tan C-T. Mouse Models for Immune Checkpoint Blockade Therapeutic Research in Oral Cancer. International Journal of Molecular Sciences. 2022; 23(16):9195. https://doi.org/10.3390/ijms23169195
Chicago/Turabian StyleChiu, Wei-Chiao, Da-Liang Ou, and Ching-Ting Tan. 2022. "Mouse Models for Immune Checkpoint Blockade Therapeutic Research in Oral Cancer" International Journal of Molecular Sciences 23, no. 16: 9195. https://doi.org/10.3390/ijms23169195