MicroRNA-223 Suppresses Human Hepatic Stellate Cell Activation Partly via Regulating the Actin Cytoskeleton and Alleviates Fibrosis in Organoid Models of Liver Injury

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
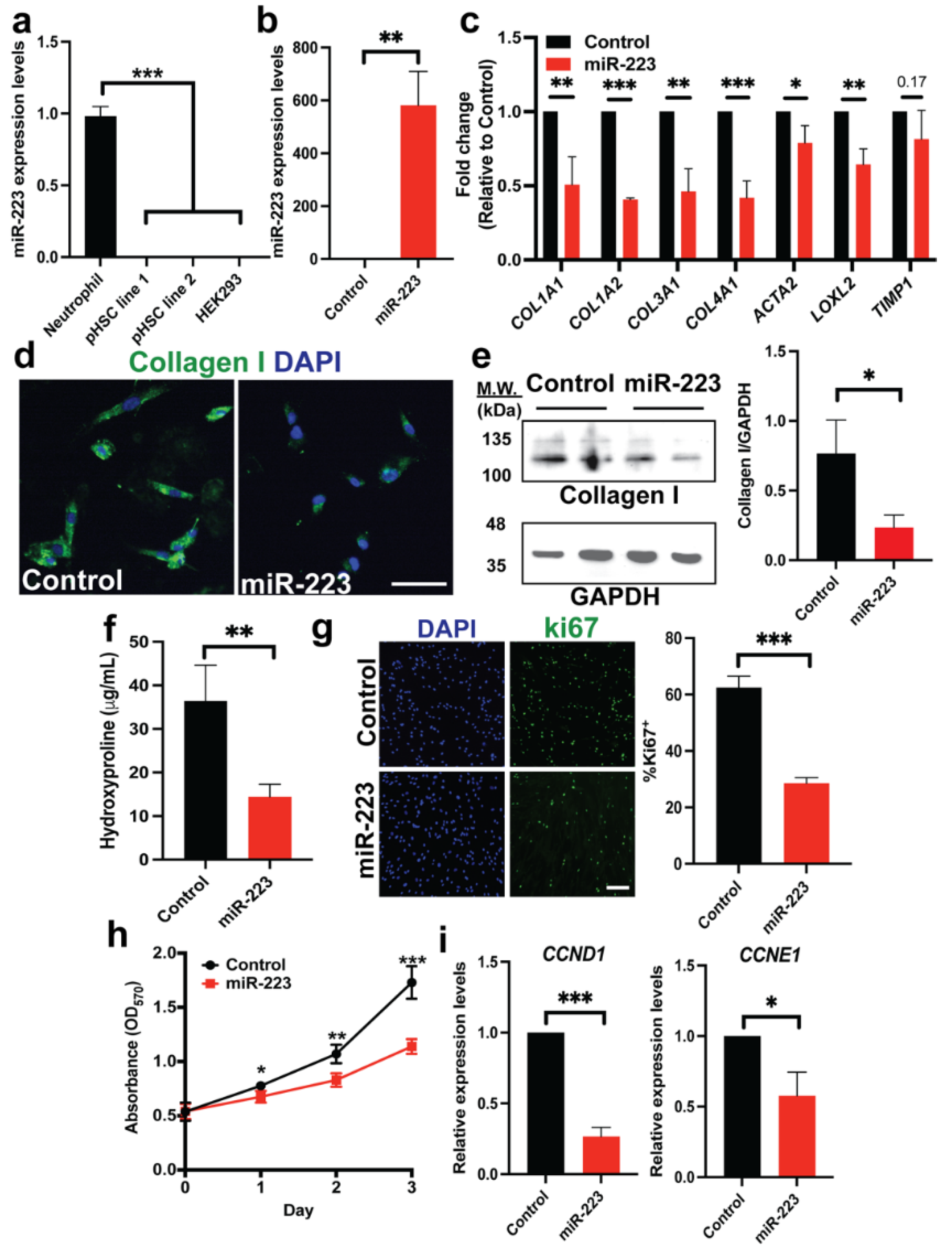
2.1. Ectopic miR-223 Expression Suppresses Activation of Human HSCs
2.2. miR-223 Promotes Quiescent Features of Human HSCs
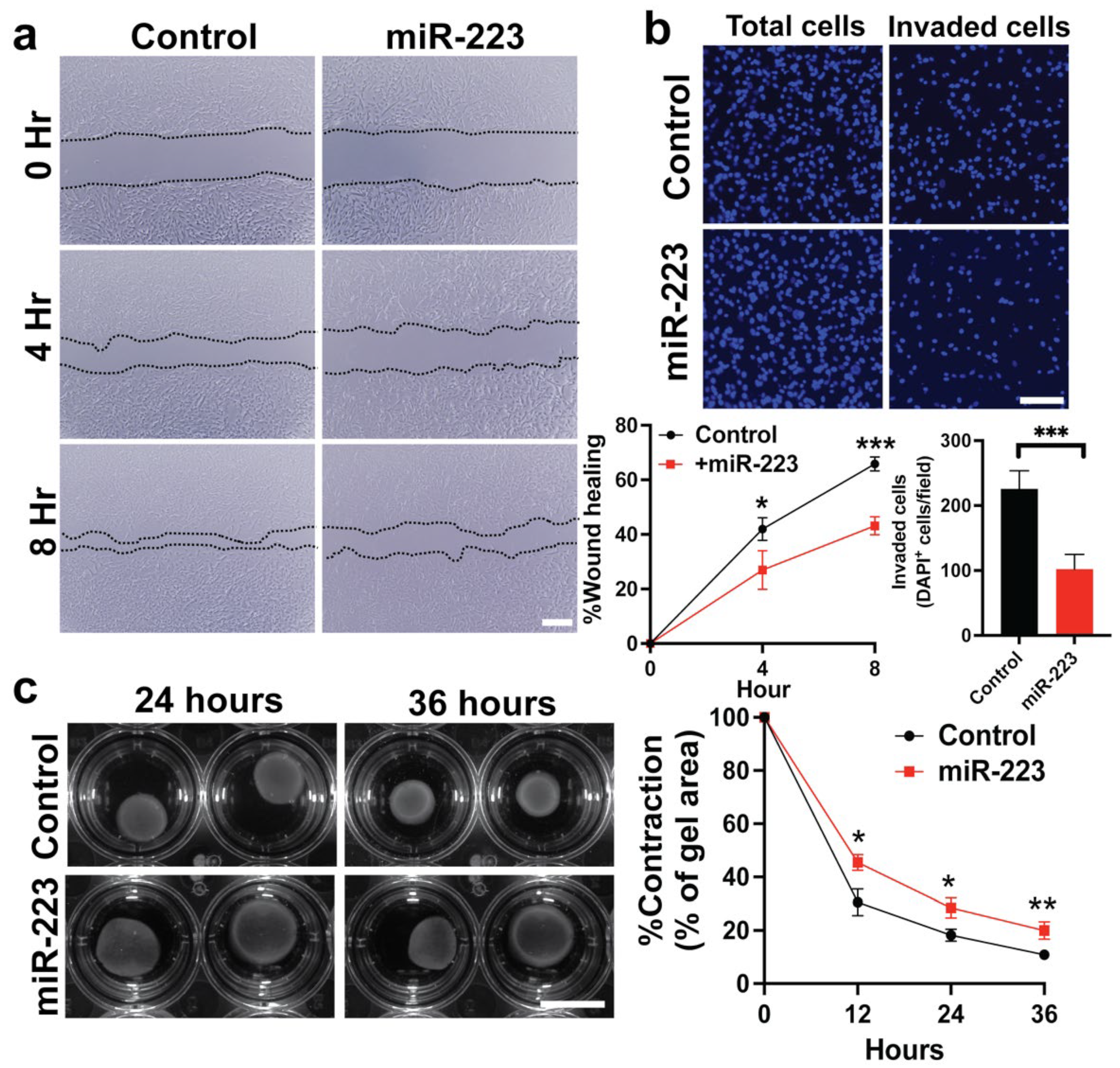
2.3. miR-223 Suppressed Cytoskeletal Activity of Primary Human HSCs
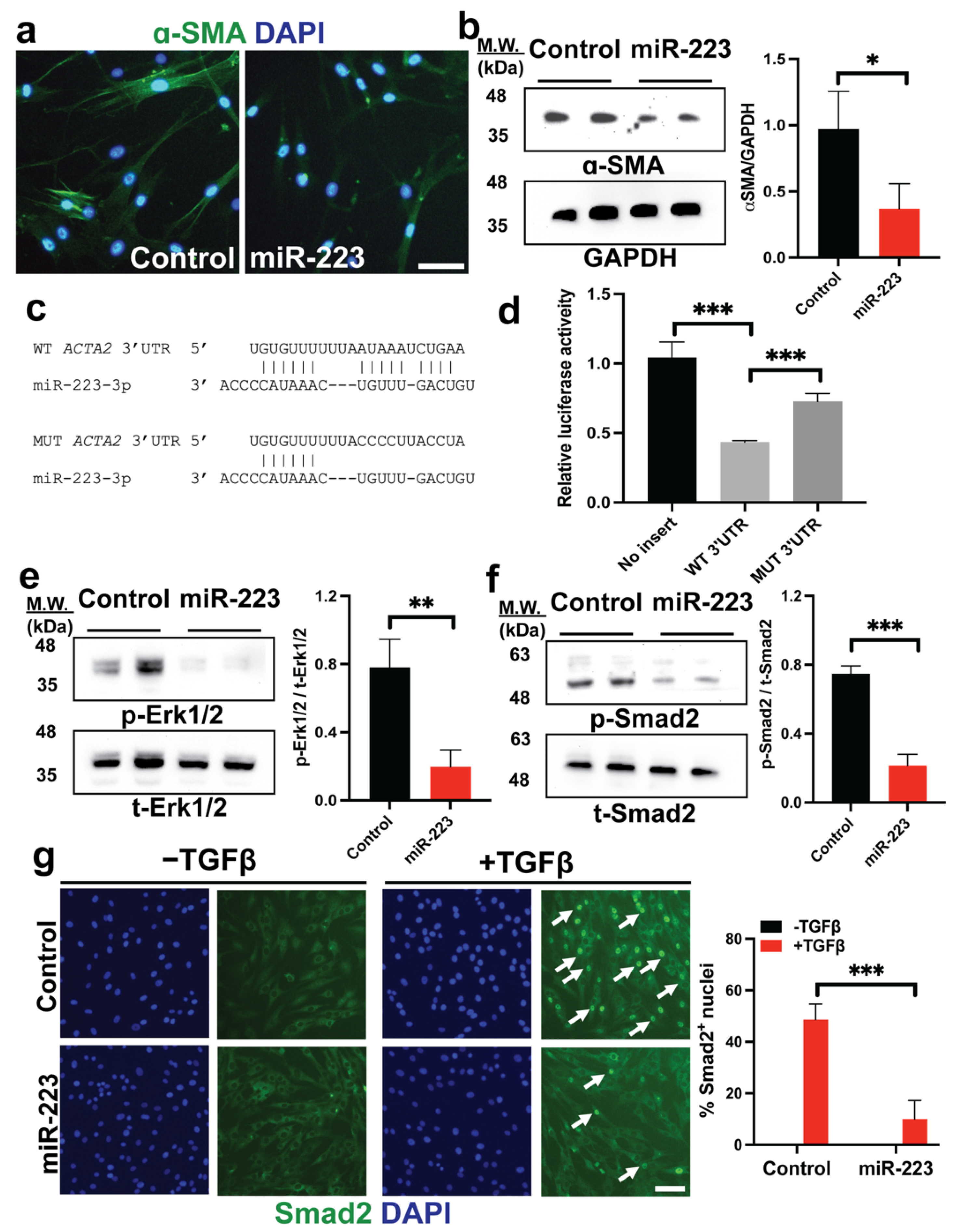
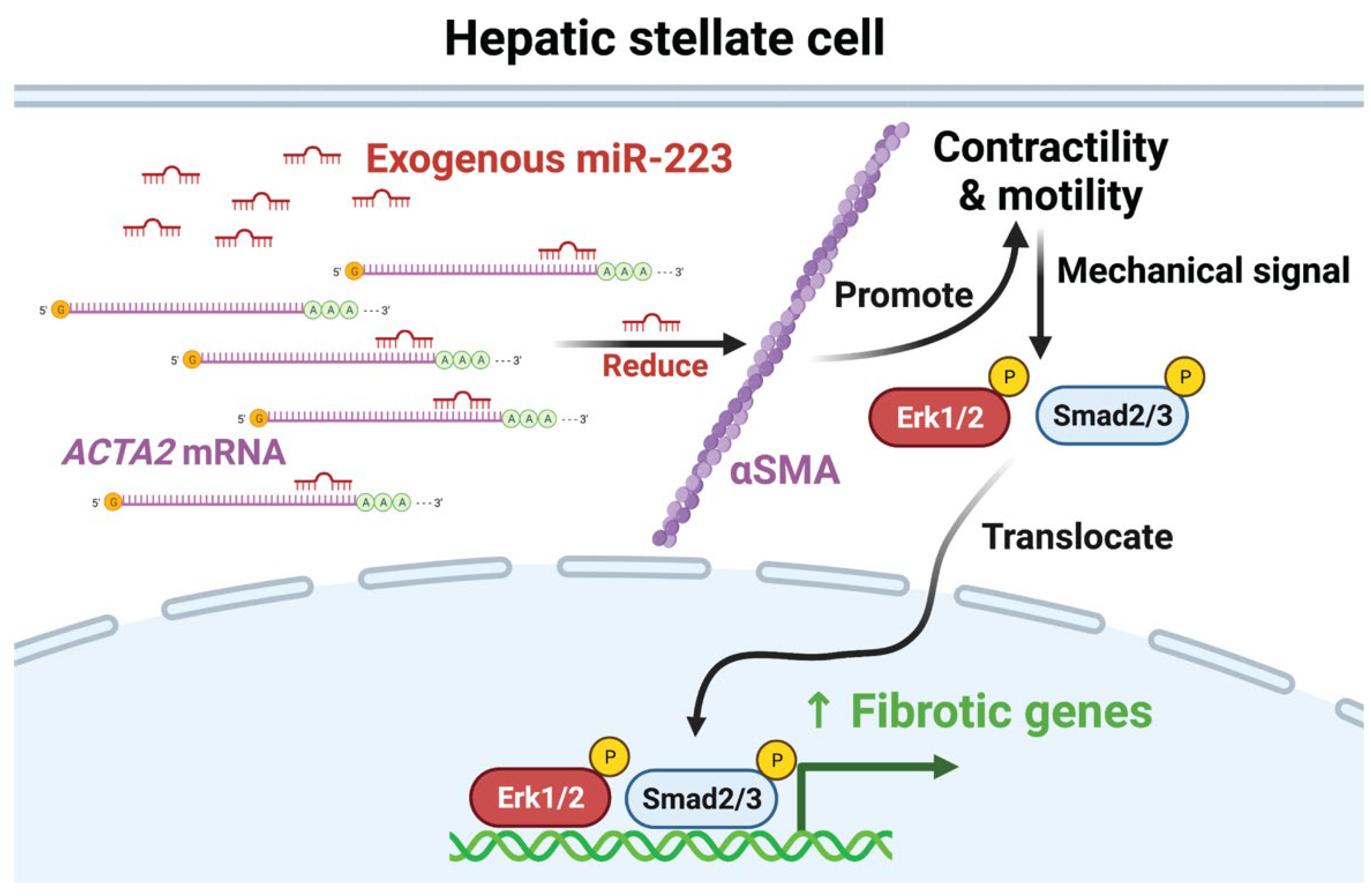
2.4. miR-223 Directly Targets ACTA2 and Inhibits Erk1/2 and Smad2/3 Phosphorylation
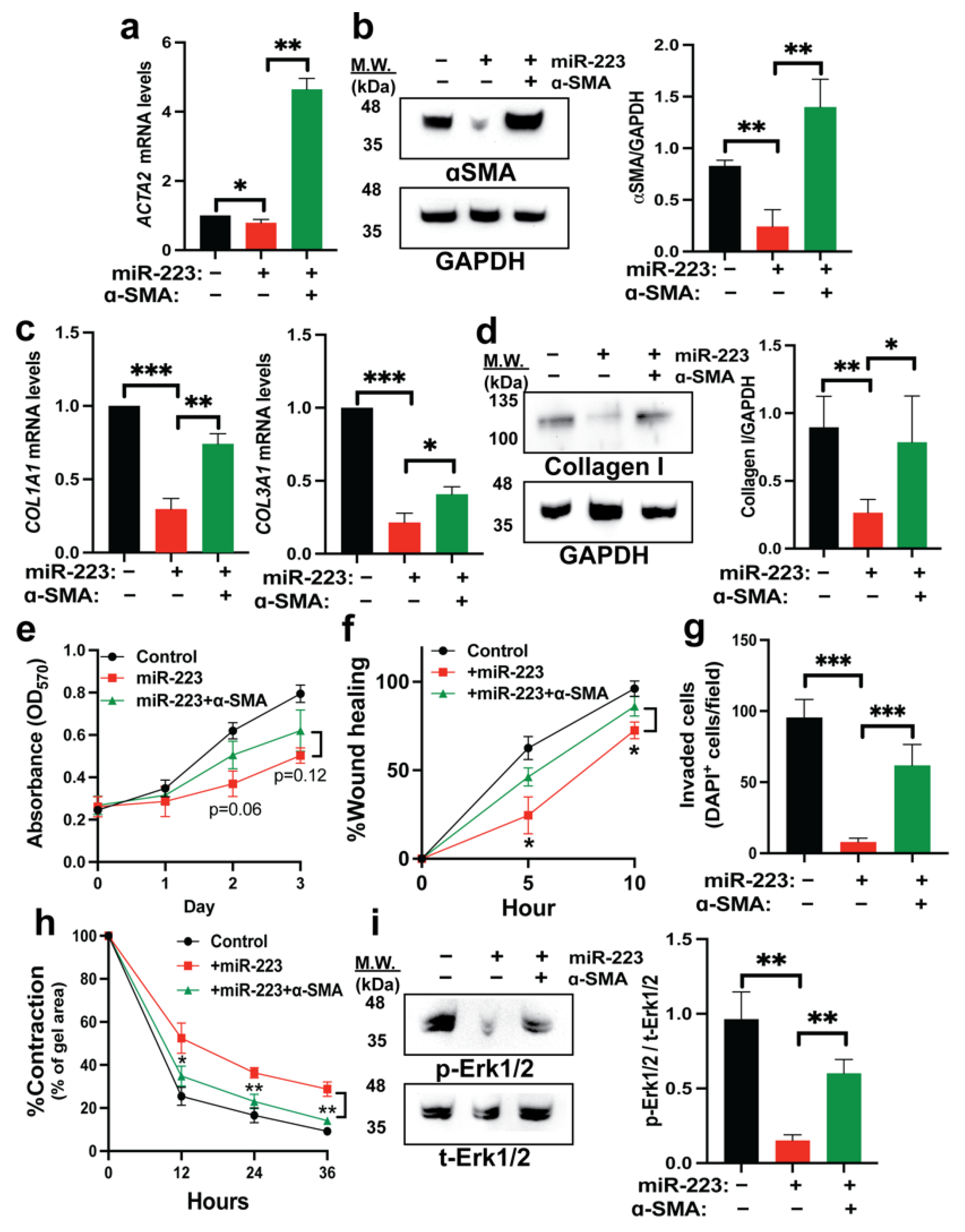
2.5. Restoration of α-SMA Inhibits the Antifibrotic Effects of miR-223
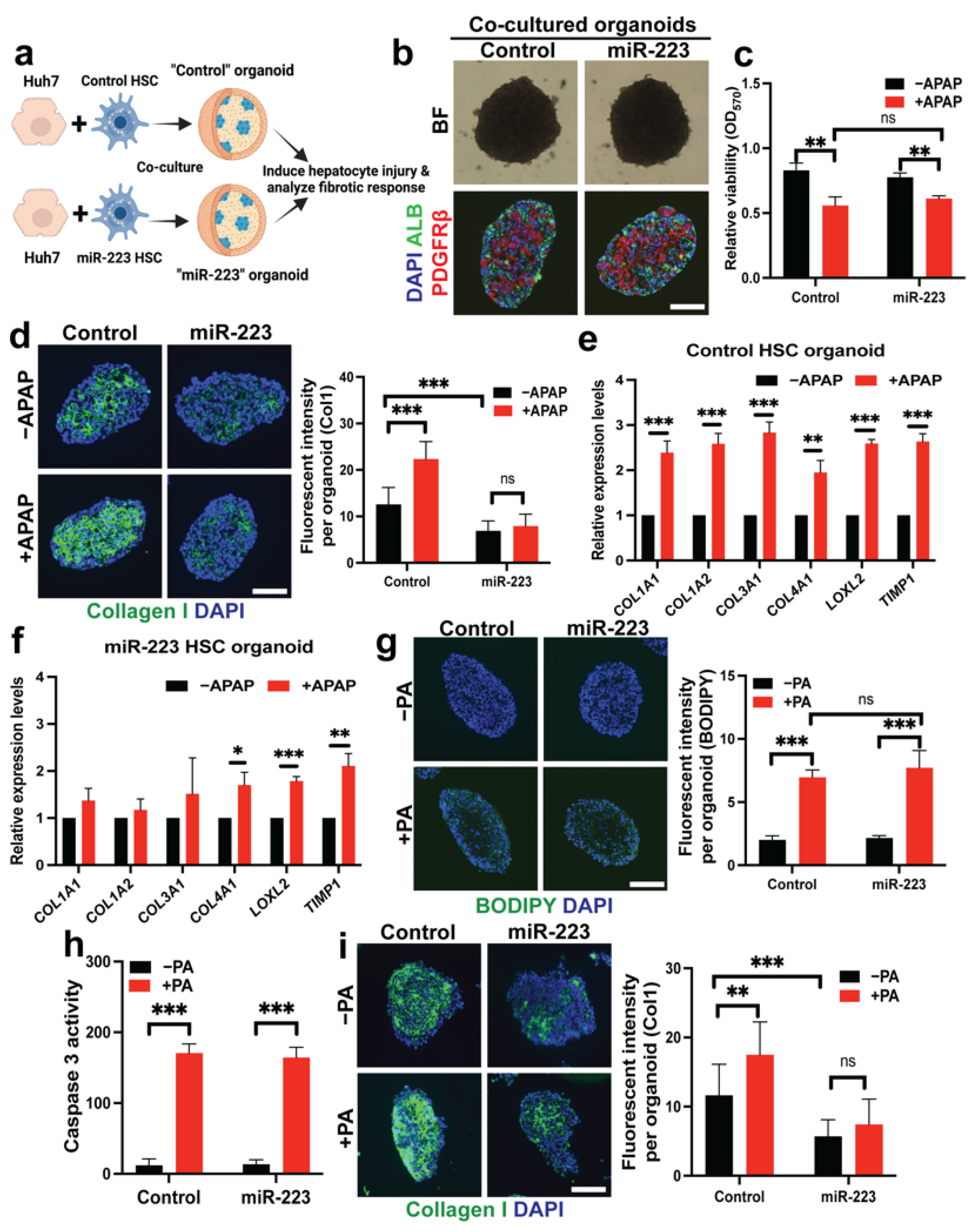
2.6. miR-223 Blocks Fibrogenesis Induced by Hepatotoxicity in Liver Organoid Models
3. Discussion
4. Materials and Methods
4.1. DNA Constructs and Lentiviral Production
4.2. Cells and Cell Culture
4.3. Organoid Culture
4.4. Immunofluorescence (IF) Staining and Microscopy
4.5. Analysis of mRNA Expression
4.6. Western Blot Analysis
4.7. Analysis of Cell Proliferation and Apoptosis
4.8. Cell Motility and Contraction Assays
4.9. Hydroxyproline Assay
4.10. Prediction of MiRNA Targets and Dual-Luciferase® Reporter Assay
4.11. Lipid Droplet and Vitamin A Storage Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Friedman, S.L.; Pinzani, M. Hepatic fibrosis 2022: Unmet needs and a blueprint for the future. Hepatology 2022, 75, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Ellis, E.L.; Mann, D.A. Clinical evidence for the regression of liver fibrosis. J. Hepatol. 2012, 56, 1171–1180. [Google Scholar] [CrossRef]
- Tacke, F.; Trautwein, C. Mechanisms of liver fibrosis resolution. J. Hepatol. 2015, 63, 1038–1039. [Google Scholar] [CrossRef]
- Wang, X.; He, Y.; Mackowiak, B.; Gao, B. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut 2021, 70, 784–795. [Google Scholar] [CrossRef]
- Jing, F.; Geng, Y.; Xu, X.-Y.; Xu, H.-Y.; Shi, J.-S.; Xu, Z.-H. MicroRNA29a Reverts the Activated Hepatic Stellate Cells in the Regression of Hepatic Fibrosis through Regulation of ATPase H+ Transporting V1 Subunit C1. Int. J. Mol. Sci. 2019, 20, 796. [Google Scholar] [CrossRef]
- Hamada-Tsutsumi, S.; Onishi, M.; Matsuura, K.; Isogawa, M.; Kawashima, K.; Sato, Y.; Tanaka, Y. Inhibitory Effect of a Human MicroRNA, miR-6133-5p, on the Fibrotic Activity of Hepatic Stellate Cells in Culture. Int. J. Mol. Sci. 2020, 21, 7251. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, Z.; Shen, X.; Wu, Z.; Dong, Y.; Cheng, J.C. MicroRNA-146a-5p Negatively Regulates Pro-Inflammatory Cytokine Secretion and Cell Activation in Lipopolysaccharide Stimulated Human Hepatic Stellate Cells through Inhibition of Toll-Like Receptor 4 Signaling Pathways. Int. J. Mol. Sci. 2016, 17, 1076. [Google Scholar] [CrossRef]
- Bushati, N.; Cohen, S.M. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Mohr, A.M.; Mott, J.L. Overview of microRNA biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef]
- Coll, M.; El Taghdouini, A.; Perea, L.; Mannaerts, I.; Vila-Casadesús, M.; Blaya, D.; Rodrigo-Torres, D.; Affò, S.; Morales-Ibanez, O.; Graupera, I. Integrative miRNA and gene expression profiling analysis of human quiescent hepatic stellate cells. Sci. Rep. 2015, 5, 11549. [Google Scholar] [CrossRef]
- Calvente, C.J.; Tameda, M.; Johnson, C.D.; Del Pilar, H.; Lin, Y.C.; Adronikou, N.; Du Jeu, X.D.M.; Llorente, C.; Boyer, J.; Feldstein, A.E. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J. Clin. Investig. 2019, 129, 4091–4109. [Google Scholar] [CrossRef]
- Wang, X.; Seo, W.; Park, S.H.; Fu, Y.; Hwang, S.; Rodrigues, R.M.; Feng, D.; Gao, B.; He, Y. MicroRNA-223 restricts liver fibrosis by inhibiting the TAZ-IHH-GLI2 and PDGF signaling pathways via the crosstalk of multiple liver cell types. Int. J. Biol. Sci. 2021, 17, 1153–1167. [Google Scholar] [CrossRef]
- Ye, D.; Zhang, T.; Lou, G.; Liu, Y. Role of miR-223 in the pathophysiology of liver diseases. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef]
- Gu, J.; Xu, H.; Chen, Y.; Li, N.; Hou, X. miR-223 as a Regulator and Therapeutic Target in Liver Diseases. Front. Immunol. 2022, 13, 860661. [Google Scholar] [CrossRef]
- Haneklaus, M.; Gerlic, M.; O’Neill, L.A.J.; Masters, S.L. miR-223: Infection, inflammation and cancer. J. Intern. Med. 2013, 274, 215–226. [Google Scholar] [CrossRef]
- Zhuang, G.; Meng, C.; Guo, X.; Cheruku, P.S.; Shi, L.; Xu, H.; Li, H.; Wang, G.; Evans, A.R.; Safe, S. A novel regulator of macrophage activation: miR-223 in obesity-associated adipose tissue inflammation. Circulation 2012, 125, 2892–2903. [Google Scholar] [CrossRef]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar] [CrossRef]
- Jimenez Calvente, C.; Del Pilar, H.; Tameda, M.; Johnson, C.D.; Feldstein, A.E. MicroRNA 223 3p Negatively Regulates the NLRP3 Inflammasome in Acute and Chronic Liver Injury. Mol. Ther. 2020, 28, 653–663. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Quan, J. Exosomal miR-223 derived from natural killer cells inhibits hepatic stellate cell activation by suppressing autophagy. Mol. Med. 2020, 26, 81. [Google Scholar] [CrossRef]
- Cai, X.; Wang, J.; Wang, J.; Zhou, Q.; Yang, B.; He, Q.; Weng, Q. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New insights into therapy. Pharmacol. Res. 2020, 155, 104720. [Google Scholar] [CrossRef]
- Yang, F.; Li, H.; Li, Y.; Hao, Y.; Wang, C.; Jia, P.; Chen, X.; Ma, S.; Xiao, Z. Crosstalk between hepatic stellate cells and surrounding cells in hepatic fibrosis. Int. Immunopharmacol. 2021, 99, 108051. [Google Scholar] [CrossRef]
- Prior, N.; Inacio, P.; Huch, M. Liver organoids: From basic research to therapeutic applications. Gut 2019, 68, 2228–2237. [Google Scholar] [CrossRef]
- Nuciforo, S.; Heim, M.H. Organoids to model liver disease. JHEP Rep. 2021, 3, 100198. [Google Scholar] [CrossRef]
- Sun, L.; Hui, L. Progress in human liver organoids. J. Mol. Cell Biol. 2020, 12, 607–617. [Google Scholar] [CrossRef]
- Harrison, S.P.; Baumgarten, S.F.; Verma, R.; Lunov, O.; Dejneka, A.; Sullivan, G.J. Liver organoids: Recent developments, limitations and potential. Front. Med. 2021, 8, 574047. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell Metab. 2019, 30, 374–384. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P. Human primary liver cancer–derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424. [Google Scholar] [CrossRef]
- Sun, L.; Wang, Y.; Cen, J.; Ma, X.; Cui, L.; Qiu, Z.; Zhang, Z.; Li, H.; Yang, R.-Z.; Wang, C. Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat. Cell Biol. 2019, 21, 1015–1026. [Google Scholar] [CrossRef]
- Buchholz, M.; Kestler, H.A.; Holzmann, K.; Ellenrieder, V.; Schneiderhan, W.; Siech, M.; Adler, G.; Bachem, M.G.; Gress, T.M. Transcriptome analysis of human hepatic and pancreatic stellate cells: Organ-specific variations of a common transcriptional phenotype. J. Mol. Med. 2005, 83, 795–805. [Google Scholar] [CrossRef]
- Delgado, I.; Carrasco, M.; Cano, E.; Carmona, R.; García-Carbonero, R.; Marín-Gómez, L.M.; Soria, B.; Martín, F.; Cano, D.A.; Muñoz-Chápuli, R. GATA4 loss in the septum transversum mesenchyme promotes liver fibrosis in mice. Hepatology 2014, 59, 2358–2370. [Google Scholar] [CrossRef]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.Y.; Hahn, W.C.; Sharp, P.A. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef]
- Coll, M.; Perea, L.; Boon, R.; Leite, S.B.; Vallverdú, J.; Mannaerts, I.; Smout, A.; El Taghdouini, A.; Blaya, D.; Rodrigo-Torres, D.; et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem Cell 2018, 23, 101–113.e7. [Google Scholar] [CrossRef]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2009, 1791, 467–473. [Google Scholar] [CrossRef]
- Molenaar, M.R.; Vaandrager, A.B.; Helms, J.B. Some Lipid Droplets Are More Equal Than Others: Different Metabolic Lipid Droplet Pools in Hepatic Stellate Cells. Lipid Insights 2017, 10, 1178635317747281. [Google Scholar] [CrossRef]
- Mederacke, I.; Dapito, D.H.; Affò, S.; Uchinami, H.; Schwabe, R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015, 10, 305–315. [Google Scholar] [CrossRef]
- El Taghdouini, A.; Najimi, M.; Sancho-Bru, P.; Sokal, E.; van Grunsven, L.A. In vitro reversion of activated primary human hepatic stellate cells. Fibrogenesis Tissue Repair 2015, 8, 14. [Google Scholar] [CrossRef]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth muscle α actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS ONE 2013, 8, e77166. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Du, Q.; Shi, Z. Smooth muscle α-actin deficiency leads to decreased liver fibrosis via impaired cytoskeletal signaling in hepatic stellate cells. Am. J. Pathol. 2019, 189, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Caliari, S.R.; Perepelyuk, M.; Cosgrove, B.D.; Tsai, S.J.; Lee, G.Y.; Mauck, R.L.; Wells, R.G.; Burdick, J.A. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Sci. Rep. 2016, 6, 21387. [Google Scholar] [CrossRef] [PubMed]
- Karvar, S.; Ansa-Addo, E.A.; Suda, J.; Singh, S.; Zhu, L.; Li, Z.; Rockey, D.C. Moesin, an Ezrin/Radixin/Moesin Family Member, Regulates Hepatic Fibrosis. Hepatology 2020, 72, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, I.; Eysackers, N.; Anne van Os, E.; Verhulst, S.; Roosens, T.; Smout, A.; Hierlemann, A.; Frey, O.; Leite, S.B.; van Grunsven, L.A. The fibrotic response of primary liver spheroids recapitulates in vivo hepatic stellate cell activation. Biomaterials 2020, 261, 120335. [Google Scholar] [CrossRef] [PubMed]
- Leite, S.B.; Roosens, T.; El Taghdouini, A.; Mannaerts, I.; Smout, A.J.; Najimi, M.; Sokal, E.; Noor, F.; Chesne, C.; van Grunsven, L.A. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 2016, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.; Babar, A.; Choudhary, M.; Kutner, M.; Pyrsopoulos, N. Acetaminophen-induced hepatotoxicity: A comprehensive update. J. Clin. Transl. Hepatol. 2016, 4, 131. [Google Scholar]
- Macanas-Pirard, P.; Yaacob, N.-S.; Lee, P.C.; Holder, J.C.; Hinton, R.H.; Kass, G.E.N. Glycogen Synthase Kinase-3 Mediates Acetaminophen-Induced Apoptosis in Human Hepatoma Cells. J. Pharmacol. Exp. Ther. 2005, 313, 780–789. [Google Scholar] [CrossRef]
- Bandi, S.; Viswanathan, P.; Gupta, S. Evaluation of cytotoxicity and DNA damage response with analysis of intracellular ATM signaling pathways. Assay Drug Dev. Technol. 2014, 12, 272–281. [Google Scholar] [CrossRef]
- Sjogren, A.-K.M.; Liljevald, M.; Glinghammar, B.; Sagemark, J.; Li, X.-Q.; Jonebring, A.; Cotgreave, I.; Brolén, G.; Andersson, T.B. Critical differences in toxicity mechanisms in induced pluripotent stem cell-derived hepatocytes, hepatic cell lines and primary hepatocytes. Arch. Toxicol. 2014, 88, 1427–1437. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Weiskirchen, R. Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)-related liver fibrosis: Mechanisms, treatment and prevention. Ann. Transl. Med. 2021, 9, 729. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.-J.; Rifkin, D.B.; Meister, J.-J.; Hinz, B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef]
- Lindahl, G.E.; Chambers, R.C.; Papakrivopoulou, J.; Dawson, S.J.; Jacobsen, M.C.; Bishop, J.E.; Laurent, G.J. Activation of Fibroblast Procollagen α1(I) Transcription by Mechanical Strain Is Transforming Growth Factor-β-dependent and Involves Increased Binding of CCAAT-binding Factor (CBF/NF-Y) at the Proximal Promoter. J. Biol. Chem. 2002, 277, 6153–6161. [Google Scholar] [CrossRef]
- Shi, Z.; Rockey, D.C. Upregulation of the actin cytoskeleton via myocardin leads to increased expression of type 1 collagen. Lab. Investig. 2017, 97, 1412–1426. [Google Scholar] [CrossRef]
- Liu, W.; Feng, R.; Li, X.; Li, D.; Zhai, W. TGF-β- and lipopolysaccharide-induced upregulation of circular RNA PWWP2A promotes hepatic fibrosis via sponging miR-203 and miR-223. Aging 2019, 11, 9569–9580. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ariyachet, C.; Chuaypen, N.; Kaewsapsak, P.; Chantaravisoot, N.; Jindatip, D.; Potikanond, S.; Tangkijvanich, P. MicroRNA-223 Suppresses Human Hepatic Stellate Cell Activation Partly via Regulating the Actin Cytoskeleton and Alleviates Fibrosis in Organoid Models of Liver Injury. Int. J. Mol. Sci. 2022, 23, 9380. https://doi.org/10.3390/ijms23169380
Ariyachet C, Chuaypen N, Kaewsapsak P, Chantaravisoot N, Jindatip D, Potikanond S, Tangkijvanich P. MicroRNA-223 Suppresses Human Hepatic Stellate Cell Activation Partly via Regulating the Actin Cytoskeleton and Alleviates Fibrosis in Organoid Models of Liver Injury. International Journal of Molecular Sciences. 2022; 23(16):9380. https://doi.org/10.3390/ijms23169380
Chicago/Turabian StyleAriyachet, Chaiyaboot, Nattaya Chuaypen, Pornchai Kaewsapsak, Naphat Chantaravisoot, Depicha Jindatip, Saranyapin Potikanond, and Pisit Tangkijvanich. 2022. "MicroRNA-223 Suppresses Human Hepatic Stellate Cell Activation Partly via Regulating the Actin Cytoskeleton and Alleviates Fibrosis in Organoid Models of Liver Injury" International Journal of Molecular Sciences 23, no. 16: 9380. https://doi.org/10.3390/ijms23169380