
Excitatory Synaptic Transmission in Ischemic Stroke: A New Outlet for Classical Neuroprotective Strategies

Abstract
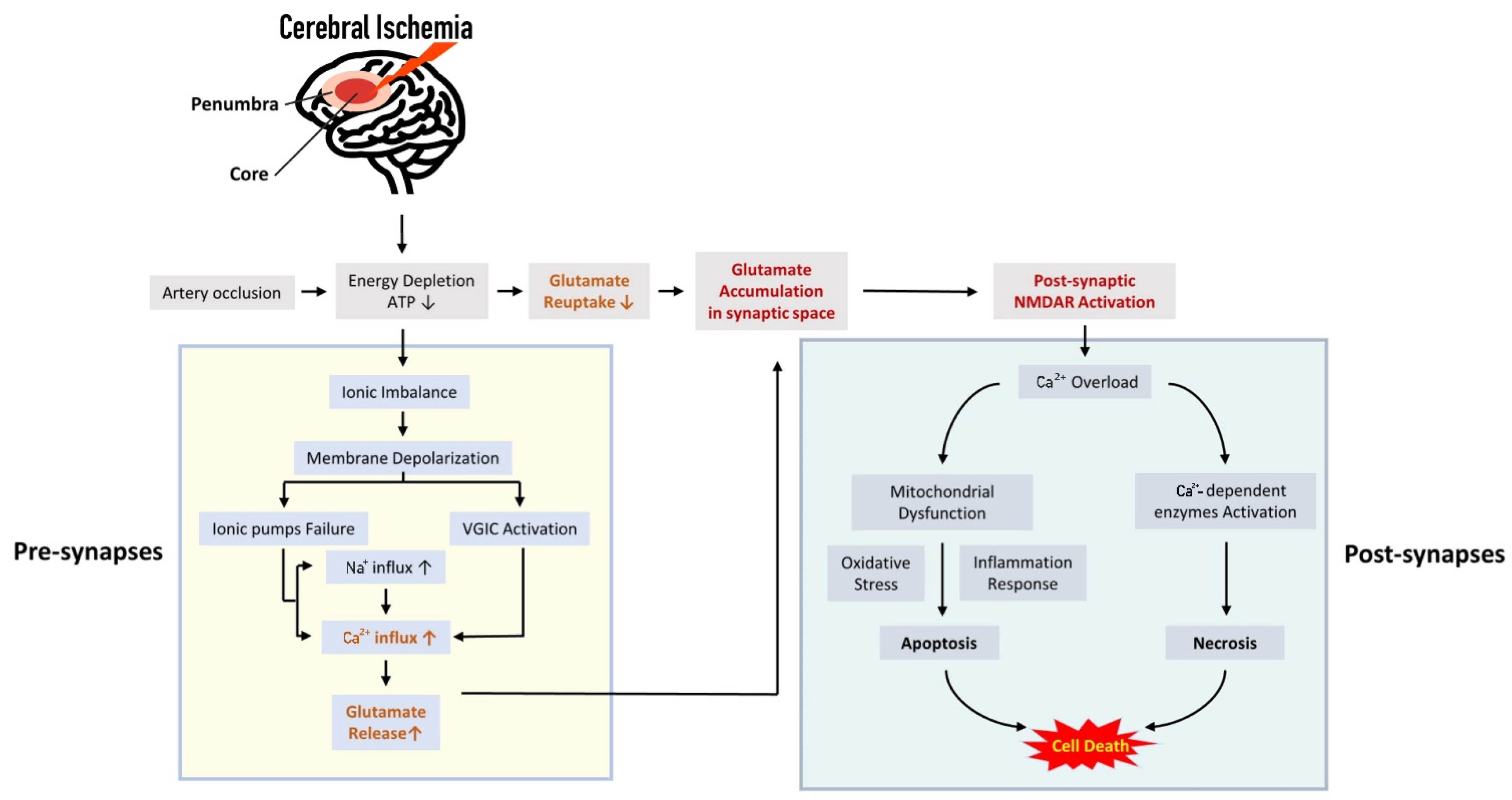
1. Introduction
2. Morphological and Functional Changes of Synapse in Stroke
2.1. Ischemia-Induced Synaptic Plasticity Damage
2.2. Causal Relationship between Neuronal Death, Synaptic Loss, and Transmission Disorder
2.3. The Role of Excitatory Synaptic Transmission for the Ischemic Cascade
3. Excessive Synaptic or Extra-Synaptic Glutamate Release Results in Early Consequence of Cerebral Ischemia
3.1. Targeted the Calcium-Dependent Presynaptic Exocytotic Release of Glutamate
3.1.1. Modulation of Ion Imbalance-Induced Depolarization upon Extracellular Calcium Entry
Sodium Calcium Exchanger (NCX)
Voltage-Gated Calcium Channels (VGCC)
3.1.2. Modulation of Presynaptic Store Calcium from the Endoplasmic Reticulum (ER)
CICR/SOCE Mechanism
Ryanodine Receptor
STIM-Orai1 Pathway
3.2. Targeted the Extra-Synaptic Release of Glutamate
3.2.1. Modulation of Exocytosis from Astrocytes
3.2.2. Modulation of VRAC
3.2.3. Modulation of Reverse Glutamate Transporter
4. Postsynaptic Effect of Glutamate as the Main Mechanism of Neuronal Death
4.1. NMDA Receptor: The Most Effective Neurotoxic Agonist
4.2. NMDAR Mediates the Dual Effects of Neuronal Survival and Death
4.2.1. Neuronal Survival Signal Complex Downstream of NMDAR
PI3K/Akt Complex
PI3K/Akt-GSK3
PI3K/Akt-BDNF
PI3K/Akt-PTEN
PI3K/Akt-APPL1
4.2.2. Neuronal Death-Signaling Complexes Downstream of NMDAR
GluN2B-DAPK1 Complex
GluN2B-PSD95-nNOS Complex
NMDAR-PSD93-SynGAP Complex
NMDAR-TRPM Complex
5. Glutamate Uptake and Metabolic Inhibition Aggravating Synaptotoxicity
5.1. Glutamate Uptake by High-Affinity Transporter
5.2. Glutamate Metabolism by Glutamate–Glutamine–Glutathione Cycle
5.3. Glutamate Grabbing by Blood–Brain Endothelium Regulation
6. Regulation of Glutamatergic Transmission by Inhibitory Synapse
6.1. The Role of GABA Receptors in Ischemic Stroke
6.2. Microbiota–Gut–Brain Axis: A New Target for Intervention of Ischemic Stroke
7. Challenges and Prospects
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, S.; Wu, B.; Liu, M.; Chen, Z.; Wang, W.; Anderson, C.S.; Sandercock, P.; Wang, Y.; Huang, Y.; Cui, L.; et al. Stroke in China: Advances and challenges in epidemiology, prevention, and management. Lancet Neurol. 2019, 18, 394–405. [Google Scholar] [CrossRef]
- Saini, V.; Guada, L.; Yavagal, D.R. Global Epidemiology of Stroke and Access to Acute Ischemic Stroke Interventions. Neurology 2021, 97, S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, S.J.; Prabhakaran, S. Diagnosis and Management of Transient Ischemic Attack and Acute Ischemic Stroke: A Review. Jama 2021, 325, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lu, J.; Liu, W.W.; Manaenko, A.; Hou, X.; Mei, Q.; Huang, J.L.; Tang, J.; Zhang, J.H.; Yao, H.; et al. Advances in stroke pharmacology. Pharmacol. Ther. 2018, 191, 23–42. [Google Scholar] [CrossRef]
- Xiong, Y.; Wakhloo, A.K.; Fisher, M. Advances in Acute Ischemic Stroke Therapy. Circul. Res. 2022, 130, 1230–1251. [Google Scholar] [CrossRef]
- Qin, C.; Yang, S.; Chu, Y.H.; Zhang, H.; Pang, X.W.; Chen, L.; Zhou, L.Q.; Chen, M.; Tian, D.S.; Wang, W. Signaling pathways involved in ischemic stroke: Molecular mechanisms and therapeutic interventions. Signal. Transduct. Target Ther. 2022, 7, 215. [Google Scholar] [CrossRef]
- Shen, Z.; Xiang, M.; Chen, C.; Ding, F.; Wang, Y.; Shang, C.; Xin, L.; Zhang, Y.; Cui, X. Glutamate excitotoxicity: Potential therapeutic target for ischemic stroke. Biomed. Pharmacother. 2022, 151, 113125. [Google Scholar] [CrossRef]
- Yang, Q.; Huang, Q.; Hu, Z.; Tang, X. Potential Neuroprotective Treatment of Stroke: Targeting Excitotoxicity, Oxidative Stress, and Inflammation. Front. Neurosci. 2019, 13, 1036. [Google Scholar] [CrossRef]
- Kullmann, D.M.; Asztely, F. Extrasynaptic glutamate spillover in the hippocampus: Evidence and implications. Trends Neurosci. 1998, 21, 8–14. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Zhang, X.; Peng, K.; Zhang, X. The Function of the NMDA Receptor in Hypoxic-Ischemic Encephalopathy. Front. Neurosci. 2020, 14, 567665. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Girouard, H.; Wang, G.; Gallo, E.F.; Anrather, J.; Zhou, P.; Pickel, V.M.; Iadecola, C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci. 2009, 29, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Waring, P. Redox active calcium ion channels and cell death. Arch. Biochem. Biophys. 2005, 434, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Lees, K.R.; Dyker, A.G.; Sharma, A.; Ford, G.A.; Ardron, M.E.; Grosset, D.G. Tolerability of the low-affinity, use-dependent NMDA antagonist AR-R15896AR in stroke patients: A dose-ranging study. Stroke 2001, 32, 466–472. [Google Scholar] [CrossRef]
- Zhou, S.; Yu, Y. Synaptic E-I Balance Underlies Efficient Neural Coding. Front. Neurosci. 2018, 12, 46. [Google Scholar] [CrossRef]
- Mayor, D.; Tymianski, M. Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology 2018, 134, 178–188. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, L.; Ju, F.; Ran, Y.; Wang, C.; Zhang, S. Transient global cerebral ischemia induces rapid and sustained reorganization of synaptic structures. J. Cereb. Blood Flow Metab. 2017, 37, 2756–2767. [Google Scholar] [CrossRef]
- Michiels, L.; Mertens, N.; Thijs, L.; Radwan, A.; Sunaert, S.; Vandenbulcke, M.; Verheyden, G.; Koole, M.; Van Laere, K.; Lemmens, R. Changes in synaptic density in the subacute phase after ischemic stroke: A (11)C-UCB-J PET/MR study. J. Cereb. Blood Flow Metab. 2022, 42, 303–314. [Google Scholar] [CrossRef]
- Chen, Z.H.; Han, Y.Y.; Shang, Y.J.; Zhuang, S.Y.; Huang, J.N.; Wu, B.Y.; Li, C.H. Cordycepin Ameliorates Synaptic Dysfunction and Dendrite Morphology Damage of Hippocampal CA1 via A1R in Cerebral Ischemia. Front. Cell. Neurosci. 2021, 15, 783478. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, M.; Fan, J.; Yan, W.; Zha, X.; Song, H.; Wan, R.; Yin, Y.; Wang, W. Ischemia-induced upregulation of autophagy preludes dysfunctional lysosomal storage and associated synaptic impairments in neurons. Autophagy 2021, 17, 1519–1542. [Google Scholar] [CrossRef] [PubMed]
- Fedorovich, S.; Hofmeijer, J.; van Putten, M.J.; Le Feber, J. Reduced Synaptic Vesicle Recycling during Hypoxia in Cultured Cortical Neurons. Front. Cell. Neurosci. 2017, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Fan, J.; Zhang, X.; Song, H.; Wan, R.; Wang, W.; Yin, Y. Decreased neuronal synaptosome associated protein 29 contributes to poststroke cognitive impairment by disrupting presynaptic maintenance. Theranostics 2021, 11, 4616–4636. [Google Scholar] [CrossRef] [PubMed]
- Rizalar, F.S.; Roosen, D.A.; Haucke, V. A Presynaptic Perspective on Transport and Assembly Mechanisms for Synapse Formation. Neuron 2021, 109, 27–41. [Google Scholar] [CrossRef]
- Hofmeijer, J.; van Putten, M.J. Ischemic cerebral damage: An appraisal of synaptic failure. Stroke 2012, 43, 607–615. [Google Scholar] [CrossRef]
- Sigler, A.; Murphy, T.H. In vivo 2-photon imaging of fine structure in the rodent brain: Before, during, and after stroke. Stroke 2010, 41, S117–S123. [Google Scholar] [CrossRef]
- Saber, H.; Liebeskind, D.S. Infarct Progression in the Early and Late Phases of Acute Ischemic Stroke. Neurology 2021, 97, S60–S67. [Google Scholar] [CrossRef]
- Baron, J.C.; Yamauchi, H.; Fujioka, M.; Endres, M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J. Cereb. Blood Flow Metab. 2014, 34, 2–18. [Google Scholar] [CrossRef]
- Chao, N.; Li, S.T. Synaptic and extrasynaptic glutamate signaling in ischemic stroke. Curr. Med. Chem. 2014, 21, 2043–2064. [Google Scholar] [CrossRef]
- MartInez-Coria, H.; Arrieta-Cruz, I.; Cruz, M.E.; López-Valdés, H.E. Physiopathology of ischemic stroke and its modulation using memantine: Evidence from preclinical stroke. Neural. Regen. Res. 2021, 16, 433–439. [Google Scholar] [CrossRef]
- Tymianski, M. Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat. Neurosci. 2011, 14, 1369–1373. [Google Scholar] [CrossRef] [PubMed]
- Dhir, N.; Medhi, B.; Prakash, A.; Goyal, M.K.; Modi, M.; Mohindra, S. Pre-clinical to Clinical Translational Failures and Current Status of Clinical Trials in Stroke Therapy: A Brief Review. Curr. Neuropharmacol. 2020, 18, 596–612. [Google Scholar] [CrossRef] [PubMed]
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Kan, J.; Zhang, J.; Zhou, L.; Huang, Y.; Zhang, Y. Chinese Herbal Medicine Interventions in Neurological Disorder Therapeutics by Regulating Glutamate Signaling. Curr. Neuropharmacol. 2020, 18, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.Z.; Zhang, S.T.; Lei, P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med. Res. Rev. 2022, 42, 259–305. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.; Gorini, A.; Hoyer, S.; Villa, R.F. Glutamate metabolism in cerebral mitochondria after ischemia and post-ischemic recovery during aging: Relationships with brain energy metabolism. J. Neurochem. 2018, 146, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity from the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef]
- Mira, R.G.; Cerpa, W. Building a Bridge between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2021, 41, 1413–1430. [Google Scholar] [CrossRef]
- Beeraka, N.M.; Vikram, P.R.H.; Greeshma, M.V.; Uthaiah, C.A.; Huria, T.; Liu, J.; Kumar, P.; Nikolenko, V.N.; Bulygin, K.V.; Sinelnikov, M.Y.; et al. Recent Investigations on Neurotransmitters’ Role in Acute White Matter Injury of Perinatal Glia and Pharmacotherapies-Glia Dynamics in Stem Cell Therapy. Mol. Neurobiol. 2022, 59, 2009–2026. [Google Scholar] [CrossRef]
- González-Nieto, D.; Fernández-Serra, R.; Pérez-Rigueiro, J.; Panetsos, F.; Martinez-Murillo, R.; Guinea, G.V. Biomaterials to Neuroprotect the Stroke Brain: A Large Opportunity for Narrow Time Windows. Cells 2020, 9, 1074. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Lu, M.H.; Yuan, D.J.; Xu, D.E.; Yao, P.P.; Ji, W.L.; Chen, H.; Liu, W.L.; Yan, C.X.; Xia, Y.Y.; et al. Mitochondrial Dysfunction in Neural Injury. Front. Neurosci. 2019, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, E.; Chen, F.; Xiao, J.; Wang, M. Neuroprotective Phytochemicals in Experimental Ischemic Stroke: Mechanisms and Potential Clinical Applications. Oxid. Med. Cell. Longev. 2021, 2021, 6687386. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kearns, K.N.; Eli, I.; Sharifi, K.A.; Soldozy, S.; Carlson, E.W.; Scott, K.W.; Sluzewski, M.F.; Acton, S.T.; Stauderman, K.A.; et al. Microglial Calcium Waves during the Hyperacute Phase of Ischemic Stroke. Stroke 2021, 52, 274–283. [Google Scholar] [CrossRef]
- Hernández, I.H.; Villa-González, M.; Martín, G.; Soto, M.; Pérez-Álvarez, M.J. Glial Cells as Therapeutic Approaches in Brain Ischemia-Reperfusion Injury. Cells 2021, 10, 1639. [Google Scholar] [CrossRef]
- Zhu, T.; Wang, L.; Wang, L.P.; Wan, Q. Therapeutic targets of neuroprotection and neurorestoration in ischemic stroke: Applications for natural compounds from medicinal herbs. Biomed. Pharmacother. 2022, 148, 112719. [Google Scholar] [CrossRef]
- da Silva-Candal, A.; Pérez-Díaz, A.; Santamaría, M.; Correa-Paz, C.; Rodríguez-Yáñez, M.; Ardá, A.; Pérez-Mato, M.; Iglesias-Rey, R.; Brea, J.; Azuaje, J.; et al. Clinical validation of blood/brain glutamate grabbing in acute ischemic stroke. Ann. Neurol. 2018, 84, 260–273. [Google Scholar] [CrossRef]
- Chabriat, H.; Bassetti, C.L.; Marx, U.; Audoli-Inthavong, M.L.; Sors, A.; Lambert, E.; Wattez, M.; Hermann, D.M. Safety and efficacy of GABA(A) α5 antagonist S44819 in patients with ischaemic stroke: A multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2020, 19, 226–233. [Google Scholar] [CrossRef]
- Xu, Q.; Hu, M.; Li, J.; Ma, X.; Chu, Z.; Zhu, Q.; Zhang, Y.; Zhu, P.; Huang, Y.; He, G. Discovery of novel brain-penetrant GluN2B NMDAR antagonists via pharmacophore-merging strategy as anti-stroke therapeutic agents. Eur. J. Med. Chem. 2022, 227, 113876. [Google Scholar] [CrossRef]
- Chanaday, N.L.; Kavalali, E.T. Presynaptic origins of distinct modes of neurotransmitter release. Curr. Opin. Neurobiol. 2018, 51, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural. Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Vitery, M.D.C.; Chen, J.; Osei-Owusu, J.; Chu, J.; Qiu, Z. Glutamate-Releasing SWELL1 Channel in Astrocytes Modulates Synaptic Transmission and Promotes Brain Damage in Stroke. Neuron 2019, 102, 813–827.e6. [Google Scholar] [CrossRef] [PubMed]
- Silver, I.A.; Erecińska, M. Intracellular and extracellular changes of [Ca2+] in hypoxia and ischemia in rat brain in vivo. J. Gen. Physiol. 1990, 95, 837–866. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.L.; Rossi, D.J. Simulated ischaemia induces Ca2+-independent glutamatergic vesicle release through actin filament depolymerization in area CA1 of the hippocampus. J. Physiol. 2010, 588, 1499–1514. [Google Scholar] [CrossRef]
- Nizami, S.; Lee, V.W.; Davies, J.; Long, P.; Jovanovic, J.N.; Sihra, T.S. Presynaptic roles of intracellular Ca(2+) stores in signalling and exocytosis. Biochem. Soc. Trans. 2010, 38, 529–535. [Google Scholar] [CrossRef]
- Silva, M.; Tran, V.; Marty, A. Calcium-dependent docking of synaptic vesicles. Trends Neurosci. 2021, 44, 579–592. [Google Scholar] [CrossRef]
- Weilinger, N.L.; Maslieieva, V.; Bialecki, J.; Sridharan, S.S.; Tang, P.L.; Thompson, R.J. Ionotropic receptors and ion channels in ischemic neuronal death and dysfunction. Acta Pharmacol. Sin. 2013, 34, 39–48. [Google Scholar] [CrossRef]
- Shenoda, B. The role of Na+/Ca2+ exchanger subtypes in neuronal ischemic injury. Transl. Stroke Res. 2015, 6, 181–190. [Google Scholar] [CrossRef]
- Stys, P.K. Anoxic and ischemic injury of myelinated axons in CNS white matter: From mechanistic concepts to therapeutics. J. Cereb. Blood Flow Metab. 1998, 18, 2–25. [Google Scholar] [CrossRef]
- Nishizawa, Y. Glutamate release and neuronal damage in ischemia. Life Sci. 2001, 69, 369–381. [Google Scholar] [CrossRef]
- Kiedrowski, L. NCX and NCKX operation in ischemic neurons. Ann. N. Y. Acad. Sci. 2007, 1099, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, G.; Sirabella, R.; Anzilotti, S.; Di Renzo, G.; Annunziato, L. Does Na⁺/Ca²⁺ exchanger, NCX, represent a new druggable target in stroke intervention? Transl. Stroke Res. 2014, 5, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, J.H. Mechanisms underlying presynaptic Ca2+ transient and vesicular glutamate release at a CNS nerve terminal during in vitro ischaemia. J. Physiol. 2015, 593, 2793–2806. [Google Scholar] [CrossRef]
- Pilitsis, J.G.; Diaz, F.G.; O’Regan, M.H.; Phillis, J.W. Inhibition of Na(+)/Ca(2+) exchange by KB-R7943, a novel selective antagonist, attenuates phosphoethanolamine and free fatty acid efflux in rat cerebral cortex during ischemia-reperfusion injury. Brain Res. 2001, 916, 192–198. [Google Scholar] [CrossRef]
- Matsuda, T.; Arakawa, N.; Takuma, K.; Kishida, Y.; Kawasaki, Y.; Sakaue, M.; Takahashi, K.; Takahashi, T.; Suzuki, T.; Ota, T.; et al. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J. Pharmacol. Exp. Ther. 2001, 298, 249–256. [Google Scholar]
- Tortiglione, A.; Pignataro, G.; Minale, M.; Secondo, A.; Scorziello, A.; Di Renzo, G.F.; Amoroso, S.; Caliendo, G.; Santagada, V.; Annunziato, L. Na+/Ca2+ exchanger in Na+ efflux-Ca2+ influx mode of operation exerts a neuroprotective role in cellular models of in vitro anoxia and in vivo cerebral ischemia. Ann. N. Y. Acad. Sci. 2002, 976, 408–412. [Google Scholar] [CrossRef]
- Cerullo, P.; Brancaccio, P.; Anzilotti, S.; Vinciguerra, A.; Cuomo, O.; Fiorino, F.; Severino, B.; Di Vaio, P.; Di Renzo, G.; Annunziato, L.; et al. Acute and long-term NCX activation reduces brain injury and restores behavioral functions in mice subjected to neonatal brain ischemia. Neuropharmacology 2018, 135, 180–191. [Google Scholar] [CrossRef]
- Magli, E.; Fattorusso, C.; Persico, M.; Corvino, A.; Esposito, G.; Fiorino, F.; Luciano, P.; Perissutti, E.; Santagada, V.; Severino, B.; et al. New Insights into the Structure-Activity Relationship and Neuroprotective Profile of Benzodiazepinone Derivatives of Neurounina-1 as Modulators of the Na(+)/Ca(2+) Exchanger Isoforms. J. Med. Chem. 2021, 64, 17901–17919. [Google Scholar] [CrossRef]
- Molinaro, P.; Sirabella, R.; Pignataro, G.; Petrozziello, T.; Secondo, A.; Boscia, F.; Vinciguerra, A.; Cuomo, O.; Philipson, K.D.; De Felice, M.; et al. Neuronal NCX1 overexpression induces stroke resistance while knockout induces vulnerability via Akt. J. Cereb. Blood Flow Metab. 2016, 36, 1790–1803. [Google Scholar] [CrossRef]
- Fern, R. Intracellular calcium and cell death during ischemia in neonatal rat white matter astrocytes in situ. J. Neurosci. 1998, 18, 7232–7243. [Google Scholar] [CrossRef]
- Osuga, H.; Hakim, A.M. Relationship between extracellular glutamate concentration and voltage-sensitive calcium channel function in focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 1996, 16, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, S.; Matsushima, K.; Fujita, H.; Nanri, K.; Ogawa, S.; Shinohara, Y. A selective N-type calcium channel antagonist reduces extracellular glutamate release and infarct volume in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Pringle, A.K.; Benham, C.D.; Sim, L.; Kennedy, J.; Iannotti, F.; Sundstrom, L.E. Selective N-type calcium channel antagonist omega conotoxin MVIIA is neuroprotective against hypoxic neurodegeneration in organotypic hippocampal-slice cultures. Stroke 1996, 27, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Valentino, K.; Newcomb, R.; Gadbois, T.; Singh, T.; Bowersox, S.; Bitner, S.; Justice, A.; Yamashiro, D.; Hoffman, B.B.; Ciaranello, R.; et al. A selective N-type calcium channel antagonist protects against neuronal loss after global cerebral ischemia. Proc. Natl. Acad. Sci. USA 1993, 90, 7894–7897. [Google Scholar] [CrossRef] [PubMed]
- Carlson, A.P.; Hänggi, D.; Macdonald, R.L.; Shuttleworth, C.W. Nimodipine Reappraised: An Old Drug with a Future. Curr. Neuropharmacol. 2020, 18, 65–82. [Google Scholar] [CrossRef]
- Buendia, I.; Tenti, G.; Michalska, P.; Méndez-López, I.; Luengo, E.; Satriani, M.; Padín-Nogueira, F.; López, M.G.; Ramos, M.T.; García, A.G.; et al. ITH14001, a CGP37157-Nimodipine Hybrid Designed to Regulate Calcium Homeostasis and Oxidative Stress, Exerts Neuroprotection in Cerebral Ischemia. ACS Chem. Neurosci. 2017, 8, 67–81. [Google Scholar] [CrossRef]
- Huang, S.; Huang, Z.; Fu, Z.; Shi, Y.; Dai, Q.; Tang, S.; Gu, Y.; Xu, Y.; Chen, J.; Wu, X.; et al. A Novel Drug Delivery Carrier Comprised of Nimodipine Drug Solution and a Nanoemulsion: Preparation, Characterization, in vitro, and in vivo Studies. Int. J. Nanomed. 2020, 15, 1161–1172. [Google Scholar] [CrossRef]
- Matsuda, S.; Nishikawa, H.; Fukatsu, A.; Kurokawa, Y.; Tsubota, M.; Sekiguchi, F.; Tokuyama, S.; Kawabata, A. NNC 55-0396, a T-type calcium channel blocker, protects against the brain injury induced by middle cerebral artery occlusion and reperfusion in mice. J. Pharmacol. Sci. 2019, 140, 193–196. [Google Scholar] [CrossRef]
- Nikonenko, I.; Bancila, M.; Bloc, A.; Muller, D.; Bijlenga, P. Inhibition of T-type calcium channels protects neurons from delayed ischemia-induced damage. Mol. Pharmacol. 2005, 68, 84–89. [Google Scholar] [CrossRef]
- Katchman, A.N.; Hershkowitz, N. Early anoxia-induced vesicular glutamate release results from mobilization of calcium from intracellular stores. J. Neurophysiol. 1993, 70, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Li, D.; Zhang, C.; Liu, M. Calcium antagonists for acute ischemic stroke. Cochrane Database Syst. Rev. 2019, 2, Cd001928. [Google Scholar] [CrossRef] [PubMed]
- Chanaday, N.L.; Kavalali, E.T. Role of the endoplasmic reticulum in synaptic transmission. Curr. Opin. Neurobiol. 2022, 73, 102538. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Kavalali, E.T. Presynaptic endoplasmic reticulum and neurotransmission. Cell Calcium 2020, 85, 102133. [Google Scholar] [CrossRef]
- Chanaday, N.L.; Nosyreva, E.; Shin, O.H.; Zhang, H.; Aklan, I.; Atasoy, D.; Bezprozvanny, I.; Kavalali, E.T. Presynaptic store-operated Ca(2+) entry drives excitatory spontaneous neurotransmission and augments endoplasmic reticulum stress. Neuron 2021, 109, 1314–1332.e1315. [Google Scholar] [CrossRef]
- Sharma, G.; Vijayaraghavan, S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 2003, 38, 929–939. [Google Scholar] [CrossRef]
- Emptage, N.J.; Reid, C.A.; Fine, A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 2001, 29, 197–208. [Google Scholar] [CrossRef]
- de Juan-Sanz, J.; Holt, G.T.; Schreiter, E.R.; de Juan, F.; Kim, D.S.; Ryan, T.A. Axonal Endoplasmic Reticulum Ca(2+) Content Controls Release Probability in CNS Nerve Terminals. Neuron 2017, 93, 867–881.e6. [Google Scholar] [CrossRef]
- Qin, Z.; Zhou, X.; Gomez-Smith, M.; Pandey, N.R.; Lee, K.F.; Lagace, D.C.; Béïque, J.C.; Chen, H.H. LIM domain only 4 (LMO4) regulates calcium-induced calcium release and synaptic plasticity in the hippocampus. J. Neurosci. 2012, 32, 4271–4283. [Google Scholar] [CrossRef]
- Grillo, M.A.; Grillo, S.L.; Gerdes, B.C.; Kraus, J.G.; Koulen, P. Control of Neuronal Ryanodine Receptor-Mediated Calcium Signaling by Calsenilin. Mol. Neurobiol. 2019, 56, 525–534. [Google Scholar] [CrossRef]
- Bull, R.; Finkelstein, J.P.; Gálvez, J.; Sánchez, G.; Donoso, P.; Behrens, M.I.; Hidalgo, C. Ischemia enhances activation by Ca2+ and redox modification of ryanodine receptor channels from rat brain cortex. J. Neurosci. 2008, 28, 9463–9472. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, H.; Tanaka, K.; Gomi, S.; Mihara, B.; Nogawa, S.; Nagata, E.; Kondo, T.; Fukuuchi, Y. Role of the ryanodine receptor in ischemic brain damage--localized reduction of ryanodine receptor binding during ischemia in hippocampus CA1. Cell. Mol. Neurobiol. 1999, 19, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ransom, B.R.; Brown, A.M. Intracellular Ca2+ release and ischemic axon injury: The Trojan horse is back. Neuron 2003, 40, 2–4. [Google Scholar] [CrossRef]
- Ohashi, M.; Hirano, T.; Watanabe, K.; Katsumi, K.; Ohashi, N.; Baba, H.; Endo, N.; Kohno, T. Hydrogen peroxide modulates synaptic transmission in ventral horn neurons of the rat spinal cord. J. Physiol. 2016, 594, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Ovcjak, A.; Xiao, A.; Kim, J.S.; Xu, B.; Szeto, V.; Turlova, E.; Abussaud, A.; Chen, N.H.; Miller, S.P.; Sun, H.S.; et al. Ryanodine receptor inhibitor dantrolene reduces hypoxic-ischemic brain injury in neonatal mice. Exp. Neurol. 2022, 351, 113985. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Maruyama, E.; Miyamoto, O.; Okabe, N.; Himi, N.; Feng, L.; Narita, K.; Keep, R.F.; Yamamoto, T.; Nakamura, T. Ryanodine receptors contribute to the induction of ischemic tolerance. Brain Res. Bull. 2016, 122, 45–53. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef]
- Secondo, A.; Petrozziello, T.; Tedeschi, V.; Boscia, F.; Vinciguerra, A.; Ciccone, R.; Pannaccione, A.; Molinaro, P.; Pignataro, G.; Annunziato, L. ORAI1/STIM1 Interaction Intervenes in Stroke and in Neuroprotection Induced by Ischemic Preconditioning Through Store-Operated Calcium Entry. Stroke 2019, 50, 1240–1249. [Google Scholar] [CrossRef]
- La Russa, D.; Frisina, M.; Secondo, A.; Bagetta, G.; Amantea, D. Modulation of Cerebral Store-operated Calcium Entry-Regulatory Factor (SARAF) and Peripheral Orai1 Following Focal Cerebral Ischemia and Preconditioning in Mice. Neuroscience 2020, 441, 8–21. [Google Scholar] [CrossRef]
- Rao, W.; Peng, C.; Zhang, L.; Su, N.; Wang, K.; Hui, H.; Dai, S.H.; Yang, Y.F.; Luo, P.; Fei, Z. Homer1a attenuates glutamate-induced oxidative injury in HT-22 cells through regulation of store-operated calcium entry. Sci. Rep. 2016, 6, 33975. [Google Scholar] [CrossRef]
- Song, Q.; Gou, W.L.; Zou, Y.L. FAM3A Protects against Glutamate-Induced Toxicity by Preserving Calcium Homeostasis in Differentiated PC12 Cells. Cell. Physiol. Biochem. 2017, 44, 2029–2041. [Google Scholar] [CrossRef] [PubMed]
- Jalini, S.; Ye, H.; Tonkikh, A.A.; Charlton, M.P.; Carlen, P.L. Raised Intracellular Calcium Contributes to Ischemia-Induced Depression of Evoked Synaptic Transmission. PLoS ONE 2016, 11, e0148110. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.B.; Attwell, D. Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 2010, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Gundersen, V.; Galbete, J.L.; Seifert, G.; Steinhäuser, C.; Pilati, E.; Volterra, A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat. Neurosci. 2004, 7, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Mendoza, E.; Burguete, M.C.; Castelló-Ruiz, M.; González, M.P.; Roncero, C.; Salom, J.B.; Arce, C.; Cañadas, S.; Torregrosa, G.; Alborch, E.; et al. Transient focal cerebral ischemia significantly alters not only EAATs but also VGLUTs expression in rats: Relevance of changes in reactive astroglia. J. Neurochem. 2010, 113, 1343–1355. [Google Scholar] [CrossRef]
- Llorente, I.L.; Pérez-Rodríguez, D.; Burgin, T.C.; Gonzalo-Orden, J.M.; Martínez-Villayandre, B.; Fernández-López, A. Age and meloxicam modify the response of the glutamate vesicular transporters (VGLUTs) after transient global cerebral ischemia in the rat brain. Brain Res. Bull. 2013, 94, 90–97. [Google Scholar] [CrossRef]
- Lobo, A.C.; Gomes, J.R.; Catarino, T.; Mele, M.; Fernandez, P.; Inácio, A.R.; Bahr, B.A.; Santos, A.E.; Wieloch, T.; Carvalho, A.L.; et al. Cleavage of the vesicular glutamate transporters under excitotoxic conditions. Neurobiol. Dis. 2011, 44, 292–303. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Amrani, A.; Gris, D. NLRX1 Enhances Glutamate Uptake and Inhibits Glutamate Release by Astrocytes. Cells 2019, 8, 400. [Google Scholar] [CrossRef]
- Zhou, J.J.; Luo, Y.; Chen, S.R.; Shao, J.Y.; Sah, R.; Pan, H.L. LRRC8A-dependent volume-regulated anion channels contribute to ischemia-induced brain injury and glutamatergic input to hippocampal neurons. Exp. Neurol. 2020, 332, 113391. [Google Scholar] [CrossRef]
- Lauderdale, K.; Murphy, T.; Tung, T.; Davila, D.; Binder, D.K.; Fiacco, T.A. Osmotic Edema Rapidly Increases Neuronal Excitability through Activation of NMDA Receptor-Dependent Slow Inward Currents in Juvenile and Adult Hippocampus. ASN Neuro 2015, 7, 1759091415605115. [Google Scholar] [CrossRef]
- Dong, Q.P.; He, J.Q.; Chai, Z. Astrocytic Ca(2+) waves mediate activation of extrasynaptic NMDA receptors in hippocampal neurons to aggravate brain damage during ischemia. Neurobiol. Dis. 2013, 58, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.S.; Bach, M.D.; Ashkavand, Z.; Norman, K.R.; Martino, N.; Adam, A.P.; Mongin, A.A. Metabolic constraints of swelling-activated glutamate release in astrocytes and their implication for ischemic tissue damage. J. Neurochem. 2019, 151, 255–272. [Google Scholar] [CrossRef]
- Alibrahim, A.; Zhao, L.Y.; Bae, C.Y.; Barszczyk, A.; Sun, C.L.; Wang, G.L.; Sun, H.S. Neuroprotective effects of volume-regulated anion channel blocker DCPIB on neonatal hypoxic-ischemic injury. Acta Pharmacol. Sin. 2013, 34, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Grewer, C.; Gameiro, A.; Rauen, T. SLC1 glutamate transporters. Pflugers Arch. 2014, 466, 3–24. [Google Scholar] [CrossRef]
- Kanai, Y.; Clémençon, B.; Simonin, A.; Leuenberger, M.; Lochner, M.; Weisstanner, M.; Hediger, M.A. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol. Aspects Med. 2013, 34, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Borisova, T. Permanent dynamic transporter-mediated turnover of glutamate across the plasma membrane of presynaptic nerve terminals: Arguments in favor and against. Rev. Neurosci. 2016, 27, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Borisova, T.; Kucherenko, D.; Soldatkin, O.; Kucherenko, I.; Pastukhov, A.; Nazarova, A.; Galkin, M.; Borysov, A.; Krisanova, N.; Soldatkin, A.; et al. An amperometric glutamate biosensor for monitoring glutamate release from brain nerve terminals and in blood plasma. Anal. Chim. Acta 2018, 1022, 113–123. [Google Scholar] [CrossRef]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef]
- Soria, F.N.; Pérez-Samartín, A.; Martin, A.; Gona, K.B.; Llop, J.; Szczupak, B.; Chara, J.C.; Matute, C.; Domercq, M. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Investig. 2014, 124, 3645–3655. [Google Scholar] [CrossRef]
- Colleoni, S.; Jensen, A.A.; Landucci, E.; Fumagalli, E.; Conti, P.; Pinto, A.; De Amici, M.; Pellegrini-Giampietro, D.E.; De Micheli, C.; Mennini, T.; et al. Neuroprotective effects of the novel glutamate transporter inhibitor (-)-3-hydroxy-4,5,6,6a-tetrahydro-3aH-pyrrolo[3,4-d]-isoxazole-4-carboxylic acid, which preferentially inhibits reverse transport (glutamate release) compared with glutamate reuptake. J. Pharmacol. Exp. Ther. 2008, 326, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Puyal, J.; Ginet, V.; Clarke, P.G. Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: A challenge for neuroprotection. Prog. Neurobiol. 2013, 105, 24–48. [Google Scholar] [CrossRef] [PubMed]
- Bolay, H.; Gürsoy-Ozdemir, Y.; Sara, Y.; Onur, R.; Can, A.; Dalkara, T. Persistent defect in transmitter release and synapsin phosphorylation in cerebral cortex after transient moderate ischemic injury. Stroke 2002, 33, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Goenka, L.; Uppugunduri Satyanarayana, C.R.; George, M. Neuroprotective Agents in Acute Ischemic Stroke-A Reality Check. Biomed. Pharmacother. 2019, 109, 2539–2547. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol. Rev. 2021, 73, 298–487. [Google Scholar] [CrossRef] [PubMed]
- Hazell, A.S. Excitotoxic mechanisms in stroke: An update of concepts and treatment strategies. Neurochem. Int. 2007, 50, 941–953. [Google Scholar] [CrossRef]
- Glasgow, N.G.; Siegler Retchless, B.; Johnson, J.W. Molecular bases of NMDA receptor subtype-dependent properties. J. Physiol. 2015, 593, 83–95. [Google Scholar] [CrossRef]
- Hardingham, G.E. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem. Soc. Trans. 2009, 37, 1147–1160. [Google Scholar] [CrossRef]
- Papadia, S.; Hardingham, G.E. The dichotomy of NMDA receptor signaling. Neuroscientist 2007, 13, 572–579. [Google Scholar] [CrossRef]
- Ge, Y.; Chen, W.; Axerio-Cilies, P.; Wang, Y.T. NMDARs in Cell Survival and Death: Implications in Stroke Pathogenesis and Treatment. Trends Mol. Med. 2020, 26, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Wu, J.; Zhu, H.; Yan, H.; Guo, Y.; Cai, Y.; Yan, H.; Shi, Y.; Shu, S.; Pei, L.; et al. Genetic Mutation of GluN2B Protects Brain Cells against Stroke Damages. Mol. Neurobiol. 2018, 55, 2979–2990. [Google Scholar] [CrossRef] [PubMed]
- Hannan, M.A.; Haque, M.N.; Mohibbullah, M.; Dash, R.; Hong, Y.K.; Moon, I.S. Gelidium amansii Attenuates Hypoxia/Reoxygenation-Induced Oxidative Injury in Primary Hippocampal Neurons through Suppressing GluN2B Expression. Antioxidants 2020, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, C.; Yang, Q.; Jiao, M.; Qiu, S. Endocytosis of GluN2B-containing NMDA receptors mediates NMDA-induced excitotoxicity. Mol. Pain 2017, 13, 1744806917701921. [Google Scholar] [CrossRef] [PubMed]
- Gonda, S.; Giesen, J.; Sieberath, A.; West, F.; Buchholz, R.; Klatt, O.; Ziebarth, T.; Räk, A.; Kleinhubbert, S.; Riedel, C.; et al. GluN2B but Not GluN2A for Basal Dendritic Growth of Cortical Pyramidal Neurons. Front. Neuroanat. 2020, 14, 571351. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhao, S.J.; Wang, H.B.; Hou, Y.J.; Mi, Q.J.; Yang, M.F.; Yuan, H.; Ni, Q.B.; Sun, B.L.; Zhang, Z.Y. Ifenprodil Improves Long-Term Neurologic Deficits through Antagonizing Glutamate-Induced Excitotoxicity after Experimental Subarachnoid Hemorrhage. Transl. Stroke Res. 2021, 12, 1067–1080. [Google Scholar] [CrossRef]
- Yao, H.; Liu, W.; Liao, H.; Sheng, T.; Chen, P.; Zhou, H.; Pan, Y.; Xie, J.; Zhang, Q.; Zou, Z.; et al. Geniposide attenuates postischemic long-term potentiation via GluN2A. Pak. J. Pharm. Sci. 2021, 34, 909–914. [Google Scholar]
- Eyo, U.B.; Bispo, A.; Liu, J.; Sabu, S.; Wu, R.; DiBona, V.L.; Zheng, J.; Murugan, M.; Zhang, H.; Tang, Y.; et al. The GluN2A Subunit Regulates Neuronal NMDA receptor-Induced Microglia-Neuron Physical Interactions. Sci. Rep. 2018, 8, 828. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Khatri, A.; Swanger, S.A.; DiRaddo, J.O.; Yi, F.; Hansen, K.B.; Yuan, H.; Traynelis, S.F. Triheteromeric GluN1/GluN2A/GluN2C NMDARs with Unique Single-Channel Properties Are the Dominant Receptor Population in Cerebellar Granule Cells. Neuron 2018, 99, 315–328.e315. [Google Scholar] [CrossRef]
- Andrews, W.T.; Donahue, D.; Holmes, A.; Balsara, R.; Castellino, F.J.; Hummon, A.B. In situ metabolite and lipid analysis of GluN2D(-/-) and wild-type mice after ischemic stroke using MALDI MSI. Anal. Bioanal. Chem. 2020, 412, 6275–6285. [Google Scholar] [CrossRef]
- Holmes, A.; Zhou, N.; Donahue, D.L.; Balsara, R.; Castellino, F.J. A deficiency of the GluN2C subunit of the N-methyl-D-aspartate receptor is neuroprotective in a mouse model of ischemic stroke. Biochem. Biophys. Res. Commun. 2018, 495, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Jullienne, A.; Montagne, A.; Orset, C.; Lesept, F.; Jane, D.E.; Monaghan, D.T.; Maubert, E.; Vivien, D.; Ali, C. Selective inhibition of GluN2D-containing N-methyl-D-aspartate receptors prevents tissue plasminogen activator-promoted neurotoxicity both in vitro and in vivo. Mol. Neurodegener. 2011, 6, 68. [Google Scholar] [CrossRef]
- Doyle, S.; Hansen, D.B.; Vella, J.; Bond, P.; Harper, G.; Zammit, C.; Valentino, M.; Fern, R. Vesicular glutamate release from central axons contributes to myelin damage. Nat. Commun. 2018, 9, 1032. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Marson, J.D.; Zhang, Q.G.; Kim, J.; Wu, W.H.; Brann, D.W.; Chen, B.S. Neuroprotection Mediated through GluN2C-Containing N-methyl-D-aspartate (NMDA) Receptors Following Ischemia. Sci. Rep. 2016, 6, 37033. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Feng, X.; Niu, M.; Wang, L.; Xie, Y.; Wang, L.; Ha, J.; Cheng, X.; Gao, Z.; Sun, Y. Therapeutic time windows of compounds against NMDA receptors signaling pathways for ischemic stroke. J. Neurosci. Res. 2021, 99, 3204–3221. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; McLaughlin, N.; Edbauer, D.; Phillips, M.; Bolton, A.; Constantine-Paton, M.; Sheng, M. Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J. Neurosci. 2010, 30, 2676–2685. [Google Scholar] [CrossRef]
- Martel, M.A.; Ryan, T.J.; Bell, K.F.; Fowler, J.H.; McMahon, A.; Al-Mubarak, B.; Komiyama, N.H.; Horsburgh, K.; Kind, P.C.; Grant, S.G.; et al. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron 2012, 74, 543–556. [Google Scholar] [CrossRef]
- Kim, J.I.; Lee, H.R.; Sim, S.E.; Baek, J.; Yu, N.K.; Choi, J.H.; Ko, H.G.; Lee, Y.S.; Park, S.W.; Kwak, C.; et al. PI3Kγ is required for NMDA receptor-dependent long-term depression and behavioral flexibility. Nat. Neurosci. 2011, 14, 1447–1454. [Google Scholar] [CrossRef]
- Xifró, X.; Miñano-Molina, A.J.; Saura, C.A.; Rodríguez-Álvarez, J. Ras protein activation is a key event in activity-dependent survival of cerebellar granule neurons. J. Biol. Chem. 2014, 289, 8462–8472. [Google Scholar] [CrossRef]
- Barrio, E.; Vecino, R.; Sánchez-Morán, I.; Rodríguez, C.; Suárez-Pindado, A.; Bolaños, J.P.; Almeida, A.; Delgado-Esteban, M. Preconditioning-Activated AKT Controls Neuronal Tolerance to Ischemia through the MDM2-p53 Pathway. Int. J. Mol. Sci. 2021, 22, 7275. [Google Scholar] [CrossRef]
- Kourti, M.; Liaropoulou, D.; Paschou, M.; Giagklisi, I.; Paschalidi, M.; Petani, E.; Papazafiri, P. Enhanced Ca(2+) Entry Sustains the Activation of Akt in Glucose Deprived SH-SY5Y Cells. Int. J. Mol. Sci. 2022, 23, 1386. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, A.; Constantine-Paton, M. BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat. Neurosci. 2007, 10, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Wang, L.; Xie, W.; Meng, X.; Feng, Y.; Sun, G.; Sun, X. Notoginsenoside R1 Improves Cerebral Ischemia/Reperfusion Injury by Promoting Neurogenesis via the BDNF/Akt/CREB Pathway. Front. Pharmacol. 2021, 12, 615998. [Google Scholar] [CrossRef] [PubMed]
- Ning, K.; Pei, L.; Liao, M.; Liu, B.; Zhang, Y.; Jiang, W.; Mielke, J.G.; Li, L.; Chen, Y.; El-Hayek, Y.H.; et al. Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J. Neurosci. 2004, 24, 4052–4060. [Google Scholar] [CrossRef]
- Xin, H.; Liu, Z.; Buller, B.; Li, Y.; Golembieski, W.; Gan, X.; Wang, F.; Lu, M.; Ali, M.M.; Zhang, Z.G.; et al. MiR-17-92 enriched exosomes derived from multipotent mesenchymal stromal cells enhance axon-myelin remodeling and motor electrophysiological recovery after stroke. J. Cereb. Blood Flow Metab. 2021, 41, 1131–1144. [Google Scholar] [CrossRef]
- Hou, K.; Li, G.; Zhao, J.; Xu, B.; Zhang, Y.; Yu, J.; Xu, K. Bone mesenchymal stem cell-derived exosomal microRNA-29b-3p prevents hypoxic-ischemic injury in rat brain by activating the PTEN-mediated Akt signaling pathway. J. Neuroinflamm. 2020, 17, 46. [Google Scholar] [CrossRef]
- Wang, J.; Lu, W.; Chen, L.; Zhang, P.; Qian, T.; Cao, W.; Luoo, J. Serine 707 of APPL1 is Critical for the Synaptic NMDA Receptor-Mediated Akt Phosphorylation Signaling Pathway. Neurosci. Bull. 2016, 32, 323–330. [Google Scholar] [CrossRef][Green Version]
- Wang, Y.B.; Wang, J.J.; Wang, S.H.; Liu, S.S.; Cao, J.Y.; Li, X.M.; Qiu, S.; Luo, J.H. Adaptor protein APPL1 couples synaptic NMDA receptor with neuronal prosurvival phosphatidylinositol 3-kinase/Akt pathway. J. Neurosci. 2012, 32, 11919–11929. [Google Scholar] [CrossRef]
- Nair, S.; Hagberg, H.; Krishnamurthy, R.; Thornton, C.; Mallard, C. Death associated protein kinases: Molecular structure and brain injury. Int. J. Mol. Sci. 2013, 14, 13858–13872. [Google Scholar] [CrossRef]
- Tu, W.; Xu, X.; Peng, L.; Zhong, X.; Zhang, W.; Soundarapandian, M.M.; Balel, C.; Wang, M.; Jia, N.; Zhang, W.; et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 2010, 140, 222–234. [Google Scholar] [CrossRef]
- McQueen, J.; Ryan, T.J.; McKay, S.; Marwick, K.; Baxter, P.; Carpanini, S.M.; Wishart, T.M.; Gillingwater, T.H.; Manson, J.C.; Wyllie, D.J.A.; et al. Pro-death NMDA receptor signaling is promoted by the GluN2B C-terminus independently of Dapk1. Elife 2017, 6, e17161. [Google Scholar] [CrossRef] [PubMed]
- Tullis, J.E.; Buonarati, O.R.; Coultrap, S.J.; Bourke, A.M.; Tiemeier, E.L.; Kennedy, M.J.; Herson, P.S.; Bayer, K.U. GluN2B S1303 phosphorylation by CaMKII or DAPK1: No indication for involvement in ischemia or LTP. iScience 2021, 24, 103214. [Google Scholar] [CrossRef] [PubMed]
- Buonarati, O.R.; Cook, S.G.; Goodell, D.J.; Chalmers, N.E.; Rumian, N.L.; Tullis, J.E.; Restrepo, S.; Coultrap, S.J.; Quillinan, N.; Herson, P.S.; et al. CaMKII versus DAPK1 Binding to GluN2B in Ischemic Neuronal Cell Death after Resuscitation from Cardiac Arrest. Cell Rep. 2020, 30, 1–8.e4. [Google Scholar] [CrossRef] [PubMed]
- DeGregorio-Rocasolano, N.; Guirao, V.; Ponce, J.; Melià-Sorolla, M.; Aliena-Valero, A.; García-Serran, A.; Salom, J.B.; Dávalos, A.; Martí-Sistac, O.; Gasull, T. Comparative Proteomics Unveils LRRFIP1 as a New Player in the DAPK1 Interactome of Neurons Exposed to Oxygen and Glucose Deprivation. Antioxidants 2020, 9, 1202. [Google Scholar] [CrossRef]
- Wu, S.; Yue, Y.; Tian, H.; Tao, L.; Wang, Y.; Xiang, J.; Wang, S.; Ding, H. Tramiprosate protects neurons against ischemic stroke by disrupting the interaction between PSD95 and nNOS. Neuropharmacology 2014, 83, 107–117. [Google Scholar] [CrossRef]
- Bach, A.; Clausen, B.H.; Møller, M.; Vestergaard, B.; Chi, C.N.; Round, A.; Sørensen, P.L.; Nissen, K.B.; Kastrup, J.S.; Gajhede, M.; et al. A high-affinity, dimeric inhibitor of PSD-95 bivalently interacts with PDZ1-2 and protects against ischemic brain damage. Proc. Natl. Acad. Sci. USA 2012, 109, 3317–3322. [Google Scholar] [CrossRef]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in.n stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 15. [Google Scholar] [CrossRef]
- Cao, J.; Viholainen, J.I.; Dart, C.; Warwick, H.K.; Leyland, M.L.; Courtney, M.J. The PSD95-nNOS interface: A target for inhibition of excitotoxic p38 stress-activated protein kinase activation and cell death. J. Cell Biol. 2005, 168, 117–126. [Google Scholar] [CrossRef]
- Ni, H.Y.; Song, Y.X.; Lin, Y.H.; Cao, B.; Wang, D.L.; Zhang, Y.; Dong, J.; Liang, H.Y.; Xu, K.; Li, T.Y.; et al. Dissociating nNOS (Neuronal NO Synthase)-CAPON (Carboxy-Terminal Postsynaptic Density-95/Discs Large/Zona Occludens-1 Ligand of nNOS) Interaction Promotes Functional Recovery after Stroke via Enhanced Structural Neuroplasticity. Stroke 2019, 50, 728–737. [Google Scholar] [CrossRef]
- Ballarin, B.; Tymianski, M. Discovery and development of NA-1 for the treatment of acute ischemic stroke. Acta Pharmacol. Sin. 2018, 39, 661–668. [Google Scholar] [CrossRef]
- Zhang, M.; Li, Q.; Chen, L.; Li, J.; Zhang, X.; Chen, X.; Zhang, Q.; Shao, Y.; Xu, Y. PSD-93 deletion inhibits Fyn-mediated phosphorylation of NR2B and protects against focal cerebral ischemia. Neurobiol. Dis. 2014, 68, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, H.; Gao, H.; Liu, X.; Li, Q.; Rong, R.; Liu, Z.; Wei, X.E.; Kong, L.; Xu, Y.; et al. PSD-93 Interacts with SynGAP and Promotes SynGAP Ubiquitination and Ischemic Brain Injury in Mice. Transl. Stroke Res. 2020, 11, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Zong, P.; Feng, J.; Yue, Z.; Li, Y.; Wu, G.; Sun, B.; He, Y.; Miller, B.; Yu, A.S.; Su, Z.; et al. Functional coupling of TRPM2 and extrasynaptic NMDARs exacerbates excitotoxicity in ischemic brain injury. Neuron 2022, 110, 1944–1958.e8. [Google Scholar] [CrossRef]
- Yan, J.; Bengtson, C.P.; Buchthal, B.; Hagenston, A.M.; Bading, H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 2020, 370, eaay3302. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Herde, M.K.; Bohmbach, K.; Domingos, C.; Vana, N.; Komorowska-Müller, J.A.; Passlick, S.; Schwarz, I.; Jackson, C.J.; Dietrich, D.; Schwarz, M.K.; et al. Local Efficacy of Glutamate Uptake Decreases with Synapse Size. Cell Rep. 2020, 32, 108182. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, S.; Chen, Y.; Li, L.; Li, X.; Qu, Z.; Huang, J.; Fan, L.; Yuan, C.; Song, N.; et al. Smoothened is a therapeutic target for reducing glutamate toxicity in ischemic stroke. Sci. Transl. Med. 2021, 13, eaba3444. [Google Scholar] [CrossRef]
- Sugiyama, K.; Aida, T.; Nomura, M.; Takayanagi, R.; Zeilhofer, H.U.; Tanaka, K. Calpain-Dependent Degradation of Nucleoporins Contributes to Motor Neuron Death in a Mouse Model of Chronic Excitotoxicity. J. Neurosci. 2017, 37, 8830–8844. [Google Scholar] [CrossRef]
- Lee, M.; Ko, D.G.; Hong, D.K.; Lim, M.S.; Choi, B.Y.; Suh, S.W. Role of Excitatory Amino Acid Carrier 1 (EAAC1) in Neuronal Death and Neurogenesis after Ischemic Stroke. Int. J. Mol. Sci. 2020, 21, 5676. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, T.W.; Paul, B.D.; Parker, G.M.; Hester, L.D.; Snowman, A.M.; Taniguchi, Y.; Kamiya, A.; Snyder, S.H.; Sawa, A. The glutathione cycle shapes synaptic glutamate activity. Proc. Natl. Acad. Sci. USA 2019, 116, 2701–2706. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Gong, Z.; Liu, K.; Kou, J.; Liu, B.; Liu, K. Baicalin combats glutamate excitotoxicity via protecting glutamine synthetase from ROS-induced 20S proteasomal degradation. Redox Biol. 2020, 34, 101559. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, A.; Boyko, M.; Shapira, Y.; Zlotnik, A. Blood glutamate scavenging: Insight into neuroprotection. Int. J. Mol. Sci. 2012, 13, 10041–10066. [Google Scholar] [CrossRef] [PubMed]
- Zaghmi, A.; Pérez-Mato, M.; Dopico-López, A.; Candamo-Lourido, M.; Campos, F.; Gauthier, M.A. New Perspectives for Developing Therapeutic Bioconjugates of Metabolite-Depleting Enzymes: Lessons Learned Combating Glutamate Excitotoxicity. Biomacromolecules 2022, 23, 1864–1872. [Google Scholar] [CrossRef]
- Gomez-Castro, F.; Zappettini, S.; Pressey, J.C.; Silva, C.G.; Russeau, M.; Gervasi, N.; Figueiredo, M.; Montmasson, C.; Renner, M.; Canas, P.M.; et al. Convergence of adenosine and GABA signaling for synapse stabilization during development. Science 2021, 374, eabk2055. [Google Scholar] [CrossRef]
- Schwartz-Bloom, R.D.; Sah, R. Gamma-Aminobutyric acid(A) neurotransmission and cerebral ischemia. J. Neurochem. 2001, 77, 353–371. [Google Scholar] [CrossRef]
- Zhan, R.Z.; Nadler, J.V.; Schwartz-Bloom, R.D. Depressed responses to applied and synaptically-released GABA in CA1 pyramidal cells, but not in CA1 interneurons, after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2006, 26, 112–124. [Google Scholar] [CrossRef]
- Wu, K.; Castellano, D.; Tian, Q.; Lu, W. Distinct regulation of tonic GABAergic inhibition by NMDA receptor subtypes. Cell Rep. 2021, 37, 109960. [Google Scholar] [CrossRef]
- Han, W.; Shepard, R.D.; Lu, W. Regulation of GABA(A)Rs by Transmembrane Accessory Proteins. Trends Neurosci. 2021, 44, 152–165. [Google Scholar] [CrossRef]
- Sears, S.M.; Hewett, S.J. Influence of glutamate and GABA transport on brain excitatory/inhibitory balance. Exp. Biol. Med. 2021, 246, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Pandey, S.; Li, T.; Castellano, D.; Gu, X.; Li, J.; Tian, Q.; Lu, W. Genetic Deletion of GABA(A) Receptors Reveals Distinct Requirements of Neurotransmitter Receptors for GABAergic and Glutamatergic Synapse Development. Front. Cell. Neurosci. 2019, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Leone, G.; Saulle, E.; Pisani, F.; Bernardi, G.; Calabresi, P. Coactivation of GABA(A) and GABA(B) receptor results in neuroprotection during in vitro ischemia. Stroke 2004, 35, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, A.N.; Boothman-Burrell, L.; Dósa, Z.; Nagaraja, R.Y.; Jin, L.; Parker, K.; van Nieuwenhuijzen, P.S.; Neumann, S.; Gowing, E.K.; Gavande, N.; et al. The flavonoid, 2′-methoxy-6-methylflavone, affords neuroprotection following focal cerebral ischaemia. J. Cereb. Blood Flow Metab. 2019, 39, 1266–1282. [Google Scholar] [CrossRef]
- Mittal, R.; Debs, L.H.; Patel, A.P.; Nguyen, D.; Patel, K.; O’Connor, G.; Grati, M.; Mittal, J.; Yan, D.; Eshraghi, A.A.; et al. Neurotransmitters: The Critical Modulators Regulating Gut-Brain Axis. J. Cell. Physiol. 2017, 232, 2359–2372. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eaat5236. [Google Scholar] [CrossRef]
- Zareian, M.; Ebrahimpour, A.; Bakar, F.A.; Mohamed, A.K.; Forghani, B.; Ab-Kadir, M.S.; Saari, N. A glutamic acid-producing lactic acid bacteria isolated from Malaysian fermented foods. Int. J. Mol. Sci. 2012, 13, 5482–5497. [Google Scholar] [CrossRef]
- Tsai, M.F.; Miller, C. Substrate selectivity in arginine-dependent acid resistance in enteric bacteria. Proc. Natl. Acad. Sci. USA 2013, 110, 5893–5897. [Google Scholar] [CrossRef]
- Janik, R.; Thomason, L.A.M.; Stanisz, A.M.; Forsythe, P.; Bienenstock, J.; Stanisz, G.J. Magnetic resonance spectroscopy reveals oral Lactobacillus promotion of increases in brain GABA, N-acetyl aspartate and glutamate. Neuroimage 2016, 125, 988–995. [Google Scholar] [CrossRef]
- Mazzoli, R.; Pessione, E. The Neuro-endocrinological Role of Microbial Glutamate and GABA Signaling. Front. Microbiol. 2016, 7, 1934. [Google Scholar] [CrossRef]
- Baj, A.; Moro, E.; Bistoletti, M.; Orlandi, V.; Crema, F.; Giaroni, C. Glutamatergic Signaling along The Microbiota-Gut-Brain Axis. Int. J. Mol. Sci. 2019, 20, 1482. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112, 399–412. [Google Scholar] [CrossRef]
- Xu, K.; Gao, X.; Xia, G.; Chen, M.; Zeng, N.; Wang, S.; You, C.; Tian, X.; Di, H.; Tang, W.; et al. Rapid gut dysbiosis induced by stroke exacerbates brain infarction in turn. Gut 2021, 70, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. The Role of Gut Microbiota in an Ischemic Stroke. Int. J. Mol. Sci. 2021, 22, 915. [Google Scholar] [CrossRef]
- Dumitrescu, L.; Popescu-Olaru, I.; Cozma, L.; Tulbă, D.; Hinescu, M.E.; Ceafalan, L.C.; Gherghiceanu, M.; Popescu, B.O. Oxidative Stress and the Microbiota-Gut-Brain Axis. Oxid. Med. Cell. Longev. 2018, 2018, 2406594. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Yao, R.; Yu, H.; Liu, Y. Neuroprotection of Ro25-6981 against Ischemia/Reperfusion-Induced Brain Injury via Inhibition of Autophagy. Cell. Mol. Neurobiol. 2017, 37, 743–752. [Google Scholar] [CrossRef]
- Hong, J.M.; Choi, M.H.; Sohn, S.I.; Hwang, Y.H.; Ahn, S.H.; Lee, Y.B.; Shin, D.I.; Chamorro, Á.; Choi, D.W. Safety and Optimal Neuroprotection of neu2000 in acute Ischemic stroke with reCanalization: Study protocol for a randomized, double-blinded, placebo-controlled, phase-II trial. Trials 2018, 19, 375. [Google Scholar] [CrossRef]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Qin, Y.; Feng, L.; Fan, X.; Zheng, L.; Zhang, Y.; Chang, L.; Li, T. Neuroprotective Effect of N-Cyclohexylethyl-[A/G]-[D/E]-X-V Peptides on Ischemic Stroke by Blocking nNOS-CAPON Interaction. ACS Chem. Neurosci. 2021, 12, 244–255. [Google Scholar] [CrossRef]
- Zaghmi, A.; Dopico-López, A.; Pérez-Mato, M.; Iglesias-Rey, R.; Hervella, P.; Greschner, A.A.; Bugallo-Casal, A.; da Silva, A.; Gutiérrez-Fernández, M.; Castillo, J.; et al. Sustained blood glutamate scavenging enhances protection in ischemic stroke. Commun. Biol. 2020, 3, 729. [Google Scholar] [CrossRef]
- Xu, J.; Wang, A.; Meng, X.; Yalkun, G.; Xu, A.; Gao, Z.; Chen, H.; Ji, Y.; Xu, J.; Geng, D.; et al. Edaravone Dexborneol Versus Edaravone Alone for the Treatment of Acute Ischemic Stroke: A Phase III, Randomized, Double-Blind, Comparative Trial. Stroke 2021, 52, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Hogins, J.; Crawford, D.C.; Jiang, X.; Mennerick, S. Presynaptic silencing is an endogenous neuroprotectant during excitotoxic insults. Neurobiol. Dis. 2011, 43, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Farsi, Z.; Preobraschenski, J.; van den Bogaart, G.; Riedel, D.; Jahn, R.; Woehler, A. Single-vesicle imaging reveals different transport mechanisms between glutamatergic and GABAergic vesicles. Science 2016, 351, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qin, L.; Li, Y.; Yu, H.; Zhang, Z.; Tao, C.; Liu, Y.; Xue, Y.; Zhang, X.; Xu, Z.; et al. Presynaptic Endosomal Cathepsin D Regulates the Biogenesis of GABAergic Synaptic Vesicles. Cell Rep. 2019, 28, 1015–1028.e1015. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Therapy | Targeting Pathway | Therapeutic Effects/Mechanisms | References | Applications |
|---|---|---|---|---|
| Dantrolene | Inhibition of Ryanodine receptor | Reducing infarction volume and morphological damage induced by HI and cell death induced by OGD via restraining the intracellular calcium levels, apoptosis, and elevating pro-survival protein levels | [95] | Mice HI/In vitro OGD |
| DCPIB | Selective block of VRAC | Attenuating cell death via blocking the decrease in Cl− in PC12 cells OGD model, as well as lessening infarct volume and promoting functional recovery in the mice HI model | [114] | Mice HI/In vitro OGD |
| HIP-A | Inhibition of EAAT | Suppressing selectively the reverse transport of glutamate upon the low concentration, thus alleviating ischemic damage | [121] | Rat hippocampal slices/Mice brain cortical cultures |
| ifenprodil | Selective block of GluN2B | Improving apoptosis, cytosolic Ca2+ overload, BBB damage, and permeability in HBMEC, resulting in declined neurological deficits, cerebral edema, and death | [136] | Phase IV clinical |
| Ro25-6981 | Selective block of GluN2B | Suppressing ischemic brain injury via enhancing the expression of NSE and regulating autophagy-related proteins | [206] | Rat 4-VO/In vitro |
| Neu2000 | Selective block of GluN2B | A multi-target neuroprotectant and scavenging for free radicals | [207] | Phase II clinical |
| Notoginsenoside R1 | Stimulation of Akt-CREB-BDNF | Activating BDNF/Akt/CREB signaling in the rat MCAO/R model, exerting neuroprotective and pro-neurogenic effects | [153] | Rat MCAO/R |
| NA-1 | Selective block of PSD95-nNOS | Combating excitotoxicity via reducing the efficiency of Ca2+-induced excitotoxic NO production both in cortical cells and animal IS models | [170] | Phase III clinical |
| Nerinetide | Selective block of PSD95-nNOS | Inhibiting the protein-protein interaction of PSD-95. | [208] | Phase III clinical |
| N-Cyclohexylethyl-[A/G]-[D/E]-X-V Peptides | Selective block of nNOS- CAPON | Reducing infarct size in rats via blocking nNOS-CAPON interaction upon cerebral I/R models | [209] | Mice MCAO/R |
| Tat-SynGAP | Selective block of PSD93- SynGAP | Attenuating ischemic brain damage in mice | [171] | Mice MCAO/R |
| TAT-EE3 | Selective block of NMDAR-TRPM2 | Uncoupling TRPM2-NMDARs interaction, thus alleviating neuron ischemic injury in vitro and in vivo | [173] | Mice MCAO/In vitro OGD |
| TwinF/Compound 8/19 | Selective block of NMDAR-TRPM4 | Disrupting the NMDAR-TRPM4 interaction, thereby stripping off the toxicity of extrasynaptic NMDARs | [174] | Mice MCAO/In vitro OGD |
| NVP-LDE225 | Inhibition of EAAT2 | Lowering extracellular glutamate via inhibiting the SHH-SMO-GLT-1 pathway, thus reducing infarct volume and ameliorating neurological functions following ischemia | [177] | Mice/Cynomolgus monkeys |
| Baicalin | Inhibition of glutamate–glutamine cycle | Suppressing ROS production and protecting GS protein stability via inactivating SDH, promoting the disposal of the glutamate in astrocytes and rat IS models | [183] | Rat MCAO |
| hrGOT | Scavenging of Glutamate | Attenuating infarct volume via displacing glutamate homeostasis between different pools | [210] | Rat MCAO |
| 2’-methoxy-6-methylflavone | Inhibition of GABAA δ | Reducing infarct volume and improving functional recovery via downregulating IL1b, TNFa, and IFg and dampening the IS-induced increase in circulating cytokines | [194] | Mice focal ischemia |
| S44819 | Inhibition of GABAA α5 | Improving stroke recovery and increasing peri-infarct cortical excitability | [49] | Phase II clinical |
| Edaravone Dexborneol injection | Selective block of PSD95-nNOS and GABA receptors | Exerting good neuroprotective functional outcomes via synergistic effects of antioxidant and anti-inflammatory | [211] | Phase III clinical |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Xie, X.; Xing, X.; Sun, X. Excitatory Synaptic Transmission in Ischemic Stroke: A New Outlet for Classical Neuroprotective Strategies. Int. J. Mol. Sci. 2022, 23, 9381. https://doi.org/10.3390/ijms23169381
Wang F, Xie X, Xing X, Sun X. Excitatory Synaptic Transmission in Ischemic Stroke: A New Outlet for Classical Neuroprotective Strategies. International Journal of Molecular Sciences. 2022; 23(16):9381. https://doi.org/10.3390/ijms23169381
Chicago/Turabian StyleWang, Fan, Xueheng Xie, Xiaoyan Xing, and Xiaobo Sun. 2022. "Excitatory Synaptic Transmission in Ischemic Stroke: A New Outlet for Classical Neuroprotective Strategies" International Journal of Molecular Sciences 23, no. 16: 9381. https://doi.org/10.3390/ijms23169381
APA StyleWang, F., Xie, X., Xing, X., & Sun, X. (2022). Excitatory Synaptic Transmission in Ischemic Stroke: A New Outlet for Classical Neuroprotective Strategies. International Journal of Molecular Sciences, 23(16), 9381. https://doi.org/10.3390/ijms23169381