
Structural Characterization of Human Heat Shock Protein 90 N-Terminal Domain and Its Variants K112R and K112A in Complex with a Potent 1,2,3-Triazole-Based Inhibitor

 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Synthesis and Characterization of JMC31
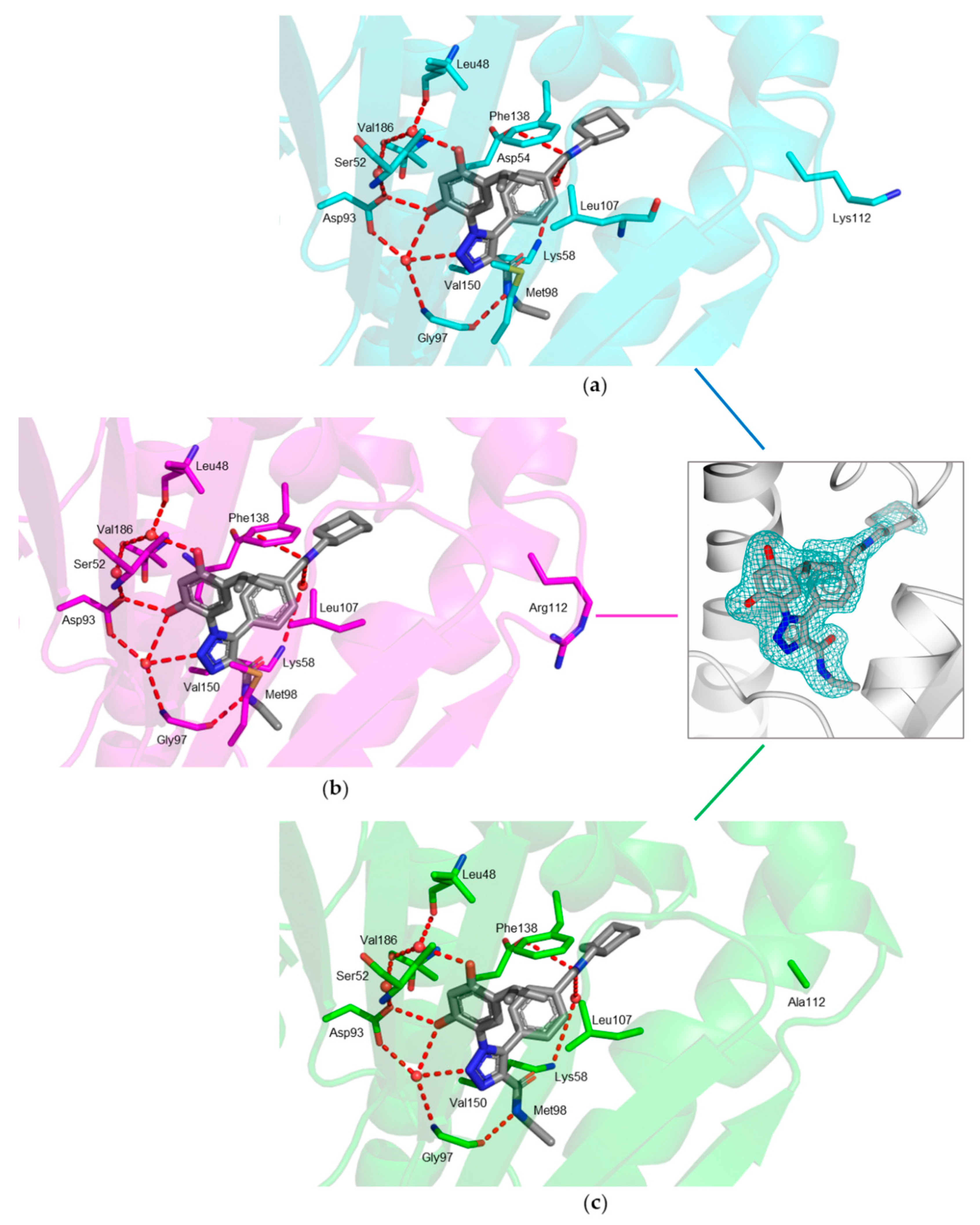
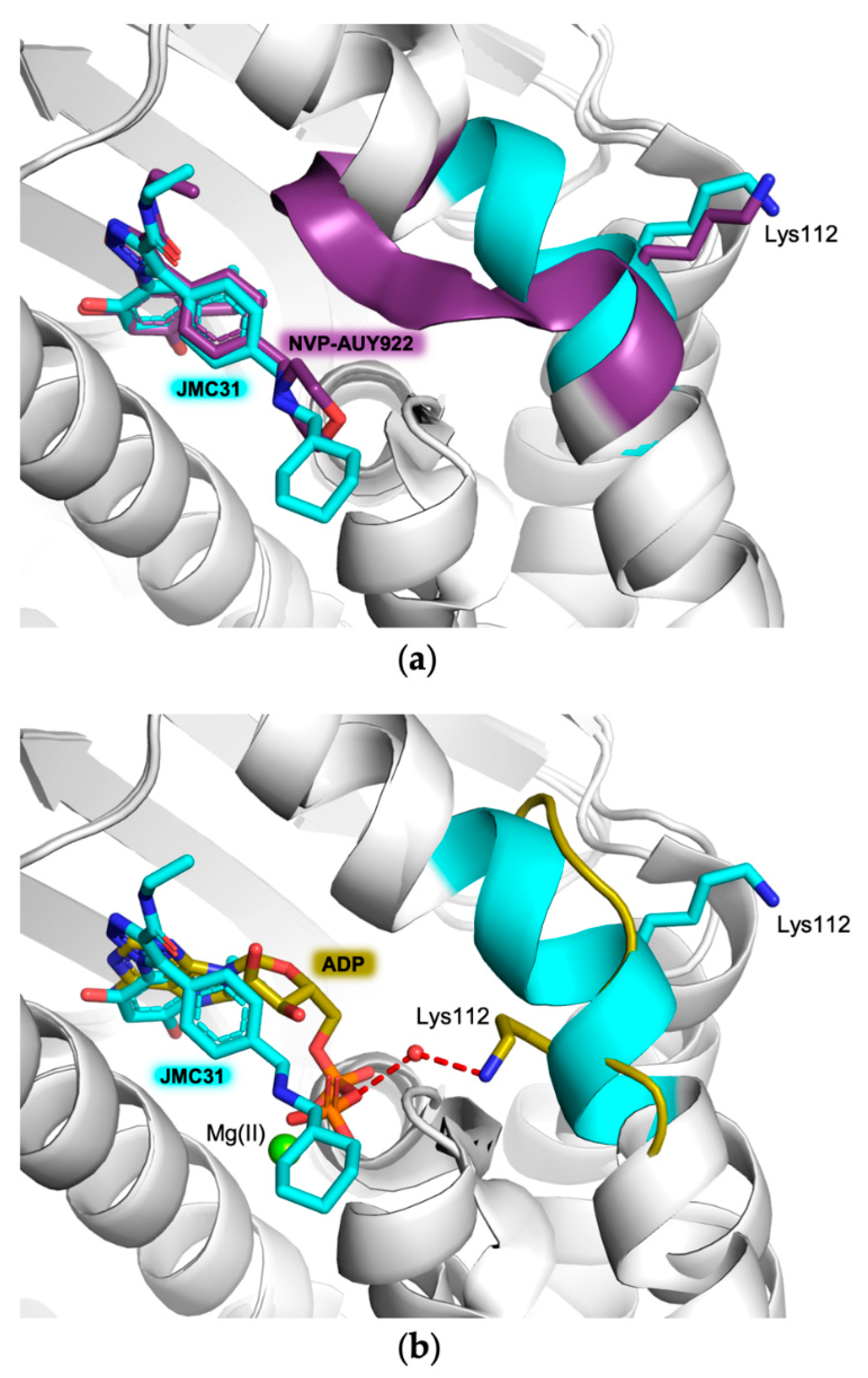
2.2. Crystal Structures of hHsp90-NTD and Variants K122R and K112A in Complex with JMC31
3. Discussion
4. Materials and Methods
4.1. Synthesis of JMC31
4.2. Proteins Expression, Purification, and Crystallization
4.3. Data Collection, Structure Solution and Refinement
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Z.; Srivastava, P. Heat-Shock Proteins. Curr. Protoc. Immunol. 2003, 58, A.1T.1–A.1T.6. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the Chaperoning of Cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H. Review: The HSP90 Molecular Chaperone—An Enigmatic ATPase. Biopolymers 2016, 105, 594–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, N.; Li, A.; Li, S.; Zhang, H. Heat Shock Protein 90 and Role of Its Chemical Inhibitors in Treatment of Hematologic Malignancies. Pharmaceuticals 2012, 5, 779–801. [Google Scholar] [CrossRef]
- Bhat, R.; Tummalapalli, S.R.; Rotella, D.P. Progress in the Discovery and Development of Heat Shock Protein 90 (Hsp90) Inhibitors. J. Med. Chem. 2014, 57, 8718–8728. [Google Scholar] [CrossRef]
- Mayer, M.P.; Le Breton, L. Hsp90: Breaking the Symmetry. Mol. Cell 2015, 58, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting HSP90 for Cancer Therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar] [CrossRef] [Green Version]
- Kryeziu, K.; Bruun, J.; Guren, T.K.; Sveen, A.; Lothe, R.A. Combination Therapies with HSP90 Inhibitors against Colorectal Cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2019, 1871, 240–247. [Google Scholar] [CrossRef]
- Bassanini, I.; Parapini, S.; Ferrandi, E.E.; Gabriele, E.; Basilico, N.; Taramelli, D.; Sparatore, A. Design, Synthesis and In Vitro Investigation of Novel Basic Celastrol Carboxamides as Bio-Inspired Leishmanicidal Agents Endowed with Inhibitory Activity against Leishmania Hsp90. Biomolecules 2021, 11, 56. [Google Scholar] [CrossRef]
- Wei, Q.; Ning, J.-Y.; Dai, X.; Gao, Y.-D.; Su, L.; Zhao, B.-X.; Miao, J.-Y. Discovery of Novel HSP90 Inhibitors that Induced Apoptosis and Impaired Autophagic Flux in A549 Lung Cancer Cells. Eur. J. Med. Chem. 2018, 145, 551–558. [Google Scholar] [CrossRef]
- Shonhai, A.; Maier, A.G.; Przyborski, J.M.; Blatch, G.L. Intracellular Protozoan Parasites of Humans: The Role of Molecular Chaperones in Development and Pathogenesis. Protein Pept. Lett. 2011, 18, 143–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, C.; Tassone, G.; Mangani, S. Chapter Five-X-Ray Crystallography Contributions to Drug Discovery against Parasite. Annual Reports in Medicinal Chemistry; Botta, M., Ed.; Neglected Diseases: Extensive Space for Modern Drug Discovery; Academic Press: Cambridge, MA, USA, 2018; Volume 51, pp. 175–230. [Google Scholar]
- Stofberg, M.L.; Caillet, C.; de Villiers, M.; Zininga, T. Inhibitors of the Plasmodium Falciparum Hsp90 towards Selective Antimalarial Drug Design: The Past, Present and Future. Cells 2021, 10, 2849. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Clos, J. No Stress—Hsp90 and Signal Transduction in Leishmania. Parasitology 2014, 141, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Batista, F.A.H.; Ramos, S.L.; Tassone, G.; Leitão, A.; Montanari, C.A.; Botta, M.; Mori, M.; Borges, J.C. Discovery of Small Molecule Inhibitors of Leishmania Braziliensis Hsp90 Chaperone. J. Enzym. Inhib. Med. Chem. 2020, 35, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graefe, S.E.B.; Wiesgigl, M.; Gaworski, I.; Macdonald, A.; Clos, J. Inhibition of HSP90 in Trypanosoma Cruzi Induces a Stress Response but no Stage Differentiation. Eukaryot. Cell 2002, 1, 936–943. [Google Scholar] [CrossRef] [Green Version]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 Chaperone Machinery. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar] [CrossRef] [Green Version]
- Sahasrabudhe, P.; Rohrberg, J.; Biebl, M.M.; Rutz, D.A.; Buchner, J. The Plasticity of the Hsp90 Co-Chaperone System. Mol. Cell 2017, 67, 947–961.e5. [Google Scholar] [CrossRef] [Green Version]
- Faya, N.; Penkler, D.L.; Tastan Bishop, Ö. Human, Vector and Parasite Hsp90 Proteins: A Comparative Bioinformatics Analysis. FEBS Open Bio 2015, 5, 916–927. [Google Scholar] [CrossRef] [Green Version]
- Tassone, G.; Mangani, S.; Botta, M.; Pozzi, C. Probing the Role of Arg97 in Heat Shock Protein 90 N-Terminal Domain from the Parasite Leishmania Braziliensis through Site-Directed Mutagenesis on the Human Counterpart. Biochim. Biophys. Acta Proteins Proteom 2018, 1866, 1190–1198. [Google Scholar] [CrossRef]
- Taddei, M.; Ferrini, S.; Giannotti, L.; Corsi, M.; Manetti, F.; Giannini, G.; Vesci, L.; Milazzo, F.M.; Alloatti, D.; Guglielmi, M.B.; et al. Synthesis and Evaluation of New Hsp90 Inhibitors Based on a 1,4,5-Trisubstituted 1,2,3-Triazole Scaffold. J. Med. Chem. 2014, 57, 2258–2274. [Google Scholar] [CrossRef]
- Giannini, G.; Cabri, W.; Vesci, L.; Cervoni, M.L.; Pisano, C.; Taddei, M.; Ferrini, S. Aryl Triazole Compounds with Antitumoural Activity. Patent WO2012/084602A1, 28 June 2012. Application No. PCT/EP2011/072558. [Google Scholar]
- Wright, L.; Barril, X.; Dymock, B.; Sheridan, L.; Surgenor, A.; Beswick, M.; Drysdale, M.; Collier, A.; Massey, A.; Davies, N.; et al. Structure-Activity Relationships in Purine-Based Inhibitor Binding to HSP90 Isoforms. Chem. Biol. 2004, 11, 775–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dymock, B.W.; Barril, X.; Brough, P.A.; Cansfield, J.E.; Massey, A.; McDonald, E.; Hubbard, R.E.; Surgenor, A.; Roughley, S.D.; Webb, P.; et al. Novel, Potent Small-Molecule Inhibitors of the Molecular Chaperone Hsp90 Discovered through Structure-Based Design. J. Med. Chem. 2005, 48, 4212–4215. [Google Scholar] [CrossRef] [PubMed]
- Kreusch, A.; Han, S.; Brinker, A.; Zhou, V.; Choi, H.; He, Y.; Lesley, S.A.; Caldwell, J.; Gu, X. Crystal Structures of Human HSP90α-Complexed with Dihydroxyphenylpyrazoles. Bioorganic Med. Chem. Lett. 2005, 15, 1475–1478. [Google Scholar] [CrossRef] [PubMed]
- Shahinas, D.; Folefoc, A.; Taldone, T.; Chiosis, G.; Crandall, I.; Pillai, D.R. A Purine Analog Synergizes with Chloroquine (CQ) by Targeting Plasmodium Falciparum Hsp90 (PfHsp90). PLoS ONE 2013, 8, e75446. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Bisson, W.H.; Mäser, P.; Scapozza, L.; Picard, D. Differences in Conformational Dynamics between Plasmodium Falciparum and Human Hsp90 Orthologues Enable the Structure-Based Discovery of Pathogen-Selective Inhibitors. J. Med. Chem. 2014, 57, 2524–2535. [Google Scholar] [CrossRef] [Green Version]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef]
- Soga, S.; Shiotsu, Y.; Akinaga, S.; Sharma, S.V. Development of Radicicol Analogues. Curr. Cancer Drug Targets 2003, 3, 359–369. [Google Scholar] [CrossRef]
- Corbett, K.D.; Berger, J.M. Structure of the ATP-Binding Domain of Plasmodium Falciparum Hsp90. Proteins 2010, 78, 2738–2744. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, J.C.; Hills, T.; Senisterra, G.; Wernimont, A.K.; Mackenzie, C.; Norcross, N.R.; Ferguson, M.A.J.; Wyatt, P.G.; Gilbert, I.H.; Hui, R. Exploring the Trypanosoma Brucei Hsp83 Potential as a Target for Structure Guided Drug Design. PLoS Negl. Trop. Dis. 2013, 7, e2492. [Google Scholar] [CrossRef] [Green Version]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.-M.J.; Collins, I.; Davies, N.G.M.; Drysdale, M.J.; et al. 4,5-Diarylisoxazole Hsp90 Chaperone Inhibitors: Potential Therapeutic Agents for the Treatment of Cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef]
- Benvenuti, M.; Mangani, S. Crystallization of Soluble Proteins in Vapor Diffusion for X-Ray Crystallography. Nat. Protoc. 2007, 2, 1633–1651. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P. Scaling and Assessment of Data Quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Section D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Vagin, A.; Teplyakov, A. Molecular Replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Potterton, L.; McNicholas, S.; Krissinel, E.; Gruber, J.; Cowtan, K.; Emsley, P.; Murshudov, G.N.; Cohen, S.; Perrakis, A.; Noble, M. Developments in the CCP4 Molecular-Graphics Project. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2288–2294. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.0.4; Schrödinger LLC: New York, NY, USA, 2018.


{kind=link}
{kind=link}
{kind=link}
| hHsp90-NTD JMC31 | hHsp90-NTD K112R JMC31 | hHsp90-NTD K112A JMC31 | |
|---|---|---|---|
| PDB ID codes | 8AGI | 8AGL | 8AGJ |
| Data Collection Statistics | |||
| Diffraction source | I04 (DLS) | I04 (DLS) | I04 (DLS) |
| Wavelength (Å) | 0.9795 | 0.9795 | 0.9795 |
| Temperature (K) | 100 | 100 | 100 |
| Detector | Eiger2 XE 16M | Eiger2 XE 16M | Eiger2 XE 16M |
| Crystal–detector distance (mm) | 258.4 | 294.9 | 258.4 |
| Exposure time per image (s) | 0.2 | 0.2 | 0.2 |
| Space group | P43212 | P43212 | P43212 |
| No. of subunits in ASU | 2 | 2 | 2 |
| a = b, c (Å) | 72.90, 212.19 | 72.86, 210.53 | 73.03, 209.29 |
| Resolution range (Å) | 212.19–2.10 (2.21–2.10) | 210.53–2.20 (2.32–2.20) | 209.29–2.32 (2.45–2.32) |
| Total no. of reflections | 397,371 (53116) | 394,512 (57473) | 333,207 (50672) |
| No. of unique reflections | 30,642 (4410) | 29,864 (4245) | 25,323 (3654) |
| Completeness (%) | 88.6 (89.7) | 100.0 (100.0) | 99.1 (100.0) |
| Redundancy | 13.0 (12.0) | 13.2 (13.5) | 13.2 (13.9) |
| 〈I/σ(I)〉 | 24.5 (2.8) | 25.7 (2.7) | 8.5 (2.5) |
| Rmeas | 0.057 (0.788) | 0.051 (0.973) | 0.170 (1.356) |
| Overall B factor from Wilson plot (Å2) | 40.8 | 55.2 | 42.9 |
| Refinements Statistics | |||
| Resolution range (Å) | 68.94–2.10 (2.15–2.10) | 68.86–2.20 (2.56–2.20) | 68.95–2.32 (2.38–2.32) |
| Completeness (%) | 88.7 (100.0) | 100.0 (100.0) | 98.6 (98.5) |
| No. of reflections, working set | 29,011 (2383) | 28,381 (2048) | 23,842 (1727) |
| No. of reflections, test set | 1547 (124) | 1393 (91) | 1289 (99) |
| Final Rcryst | 0.2099 (0.312) | 0.2295 (0.350) | 0.2219 (0.301) |
| Final Rfree | 0.2764 (0.393) | 0.2974 (0.374) | 0.2841 (0.344) |
| No. of non-H atoms | |||
| Protein | 3202 | 3195 | 3178 |
| JMC31 | 72 | 72 | 72 |
| Water | 161 | 125 | 156 |
| Total | 3435 | 3392 | 3406 |
| R.m.s. deviations Bonds (Å) | 0.009 | 0.007 | 0.009 |
| Angles (°) | 1.657 | 1.566 | 1.632 |
| Average B factors (Å2) | 45.4 | 61.5 | 51.7 |
| Estimate error on coordinates based on R value (Å) | 0.221 | 0.246 | 0.304 |
| Ramachandran plot | |||
| Most favored (%) | 98.6% | 94% | 91.6% |
| Allowed (%) | 1.4% | 6% | 8.4% |
| RSCC JMC31 chain A, B | 0.91, 0.88 | 0.97, 0.89 | 0.95, 0.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tassone, G.; Mazzorana, M.; Mangani, S.; Petricci, E.; Cini, E.; Giannini, G.; Pozzi, C.; Maramai, S. Structural Characterization of Human Heat Shock Protein 90 N-Terminal Domain and Its Variants K112R and K112A in Complex with a Potent 1,2,3-Triazole-Based Inhibitor. Int. J. Mol. Sci. 2022, 23, 9458. https://doi.org/10.3390/ijms23169458
Tassone G, Mazzorana M, Mangani S, Petricci E, Cini E, Giannini G, Pozzi C, Maramai S. Structural Characterization of Human Heat Shock Protein 90 N-Terminal Domain and Its Variants K112R and K112A in Complex with a Potent 1,2,3-Triazole-Based Inhibitor. International Journal of Molecular Sciences. 2022; 23(16):9458. https://doi.org/10.3390/ijms23169458
Chicago/Turabian StyleTassone, Giusy, Marco Mazzorana, Stefano Mangani, Elena Petricci, Elena Cini, Giuseppe Giannini, Cecilia Pozzi, and Samuele Maramai. 2022. "Structural Characterization of Human Heat Shock Protein 90 N-Terminal Domain and Its Variants K112R and K112A in Complex with a Potent 1,2,3-Triazole-Based Inhibitor" International Journal of Molecular Sciences 23, no. 16: 9458. https://doi.org/10.3390/ijms23169458