
Interactions between the Polysialylated Neural Cell Adhesion Molecule and the Transient Receptor Potential Canonical Channels 1, 4, and 5 Induce Entry of Ca2+ into Neurons



Abstract
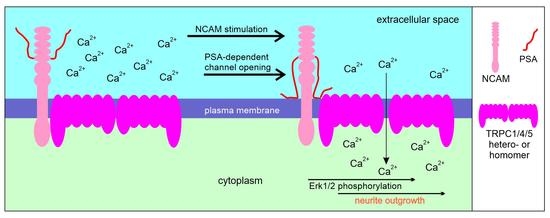
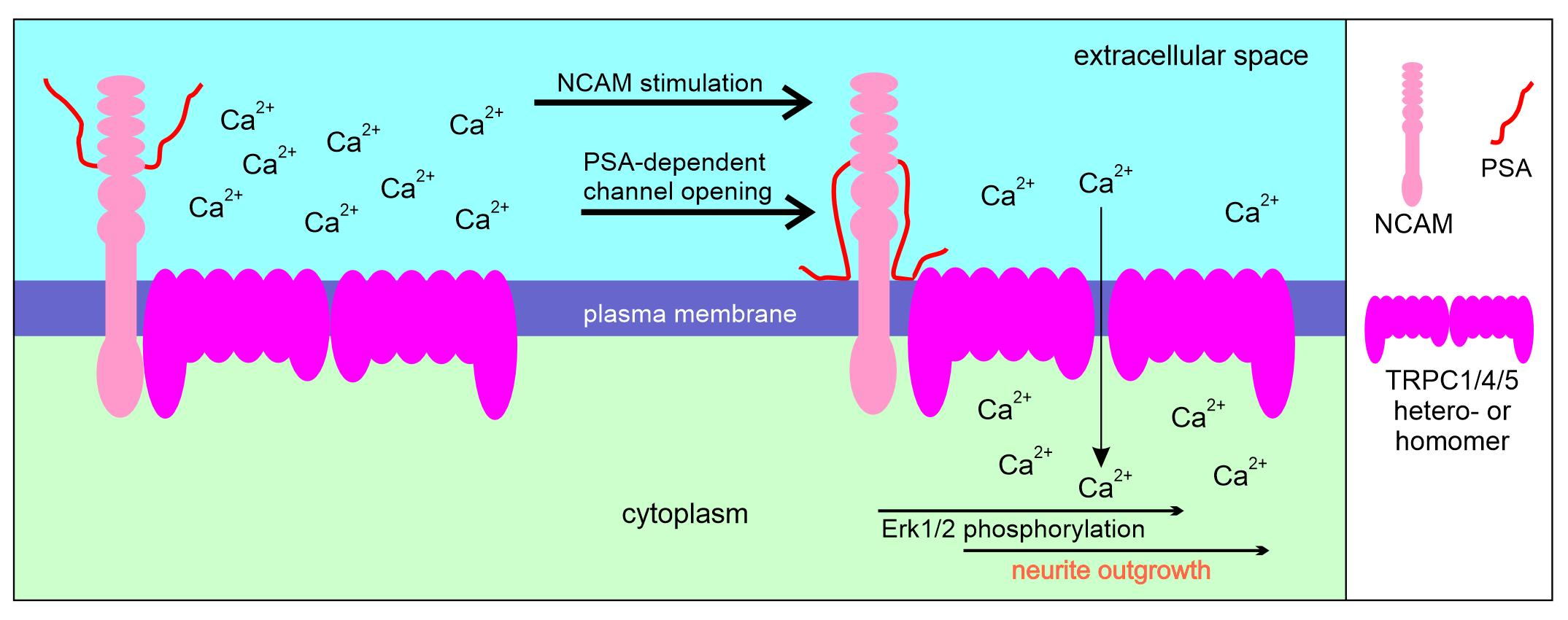{kind=link}
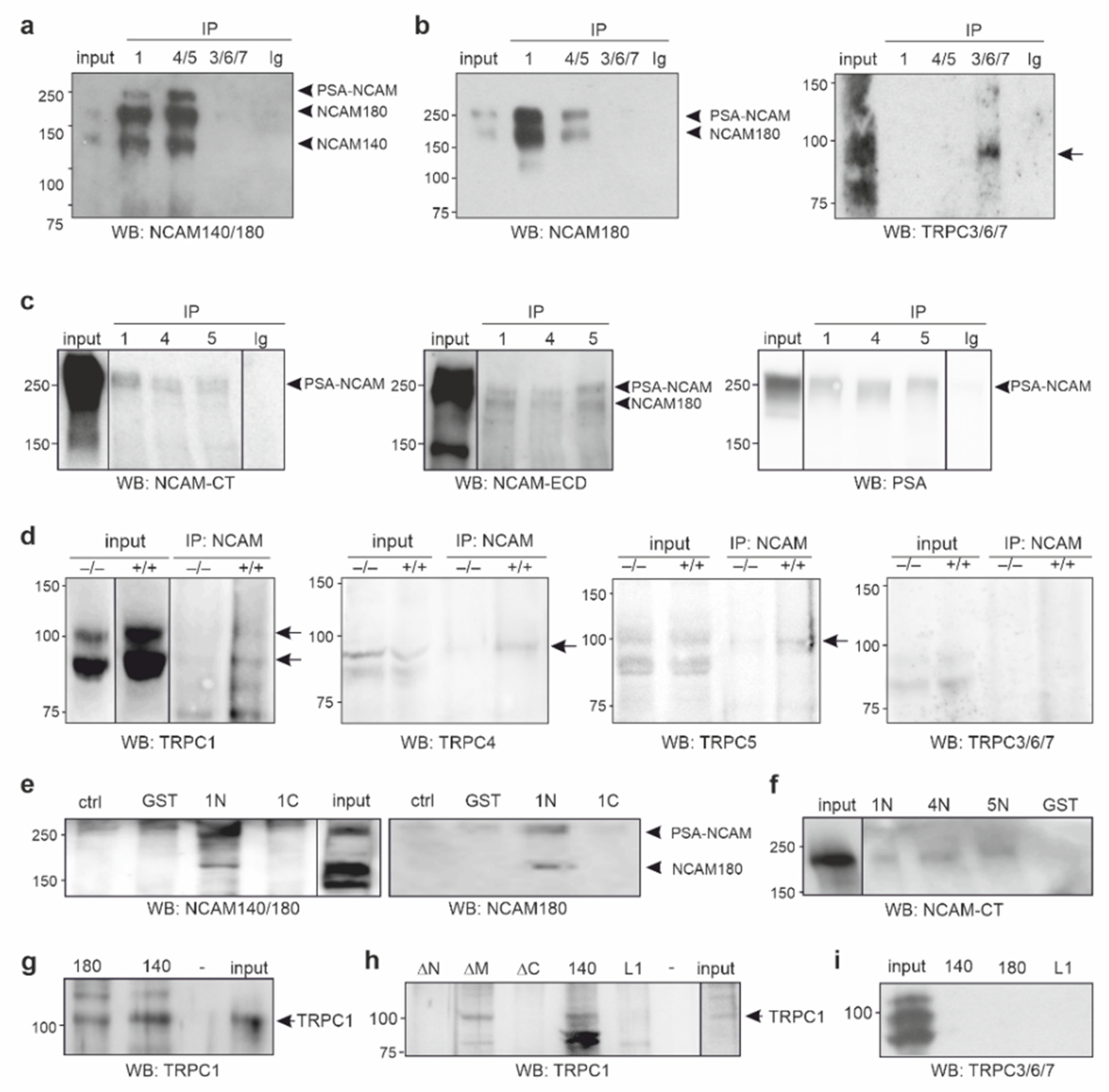{kind=link}
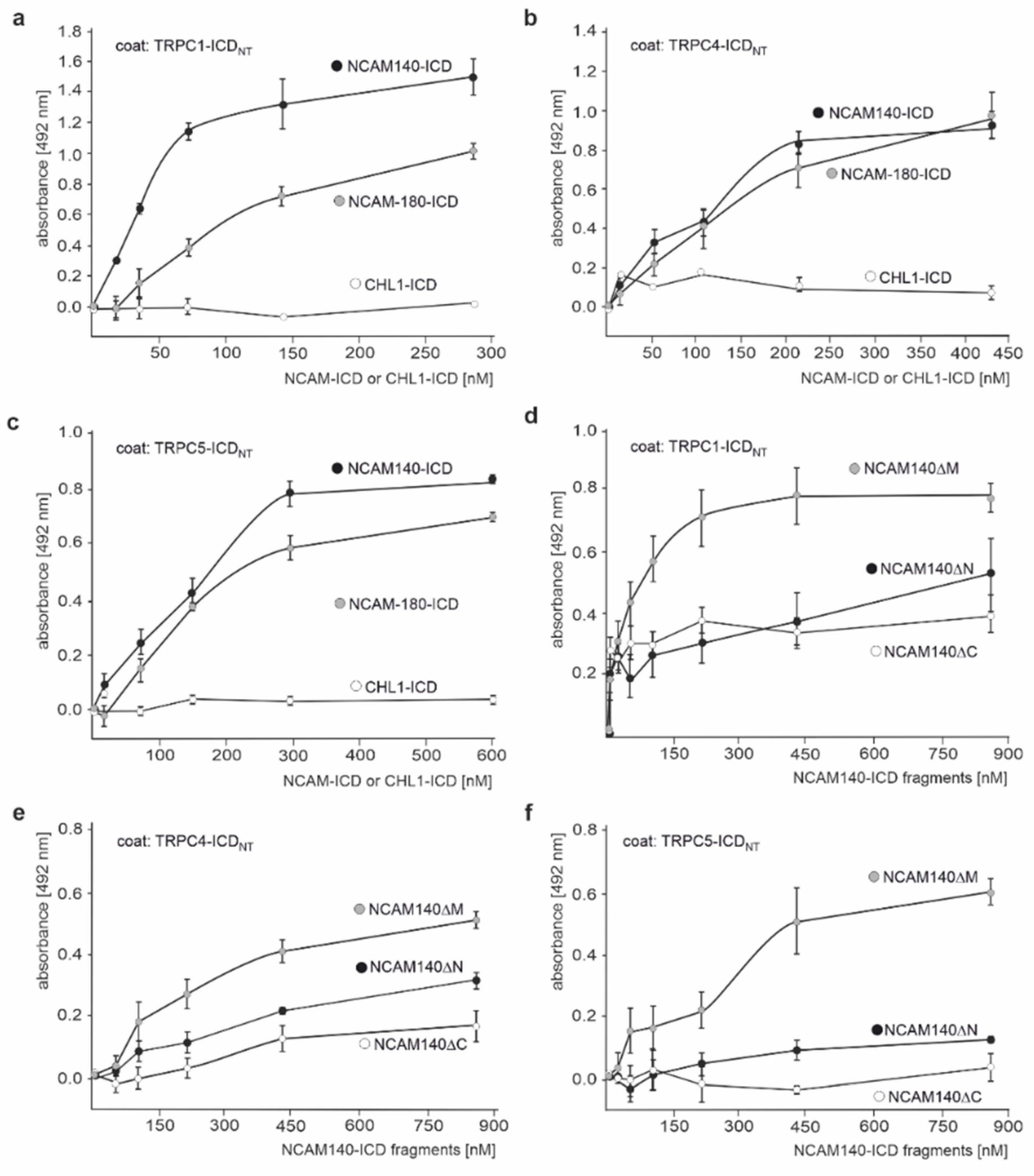{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. NCAM140 and NCAM180 Are Associated with TRPC1, −4, and −5 in the Mouse Brain
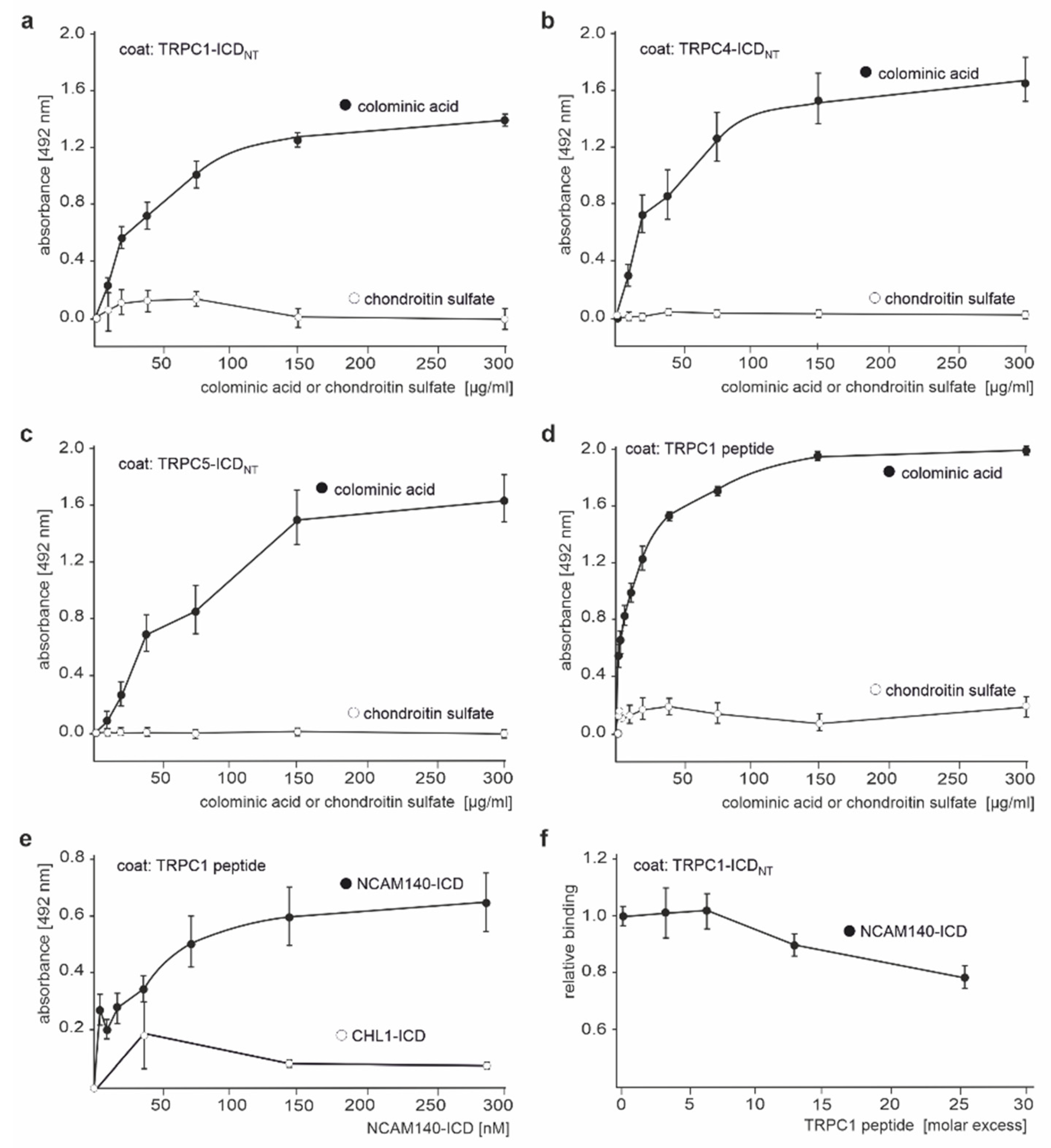
2.2. The ICDs of NCAM140 and NCAM180 Interact with the N-Terminal ICDs of TRPC1, −4, and −5
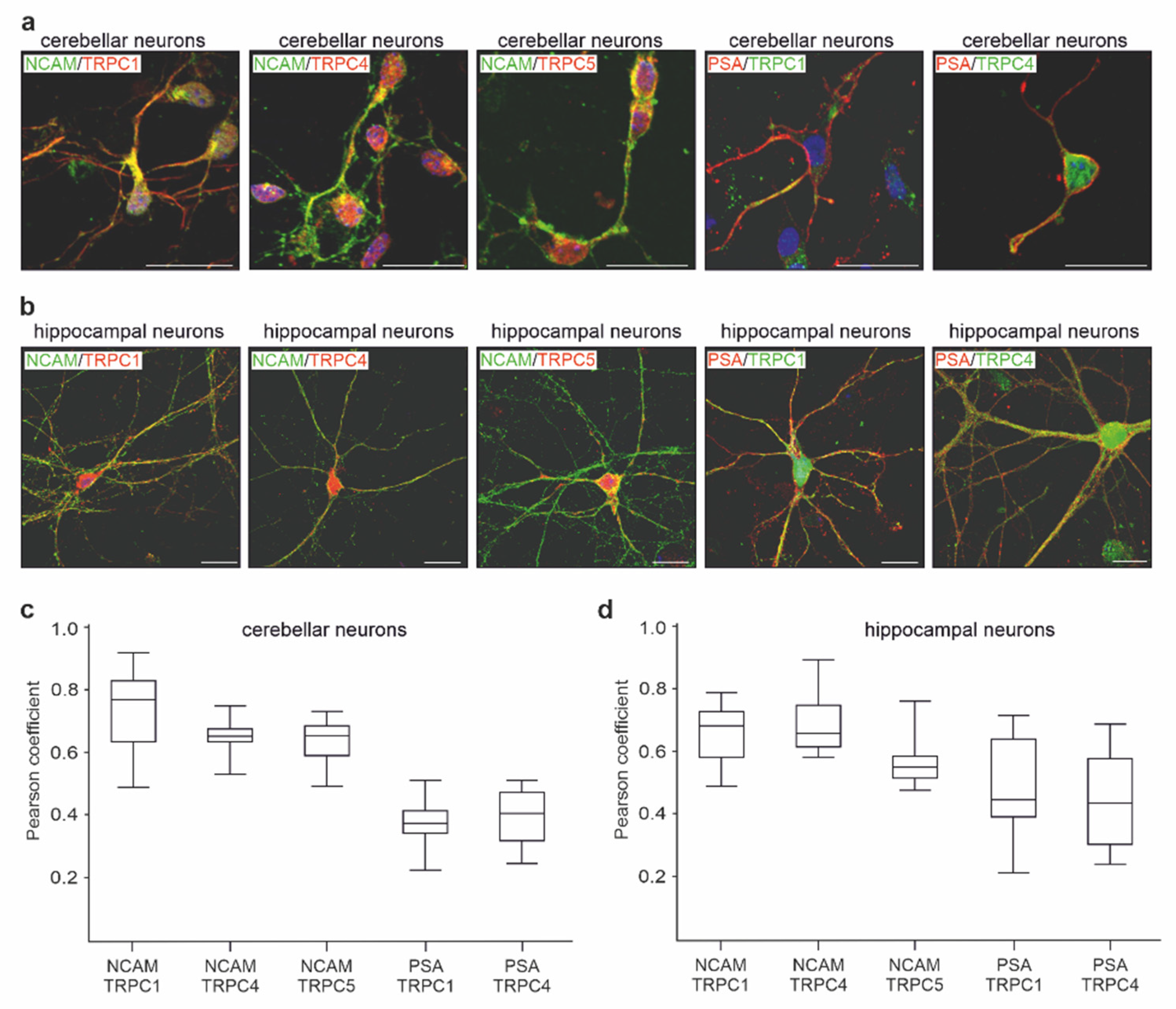
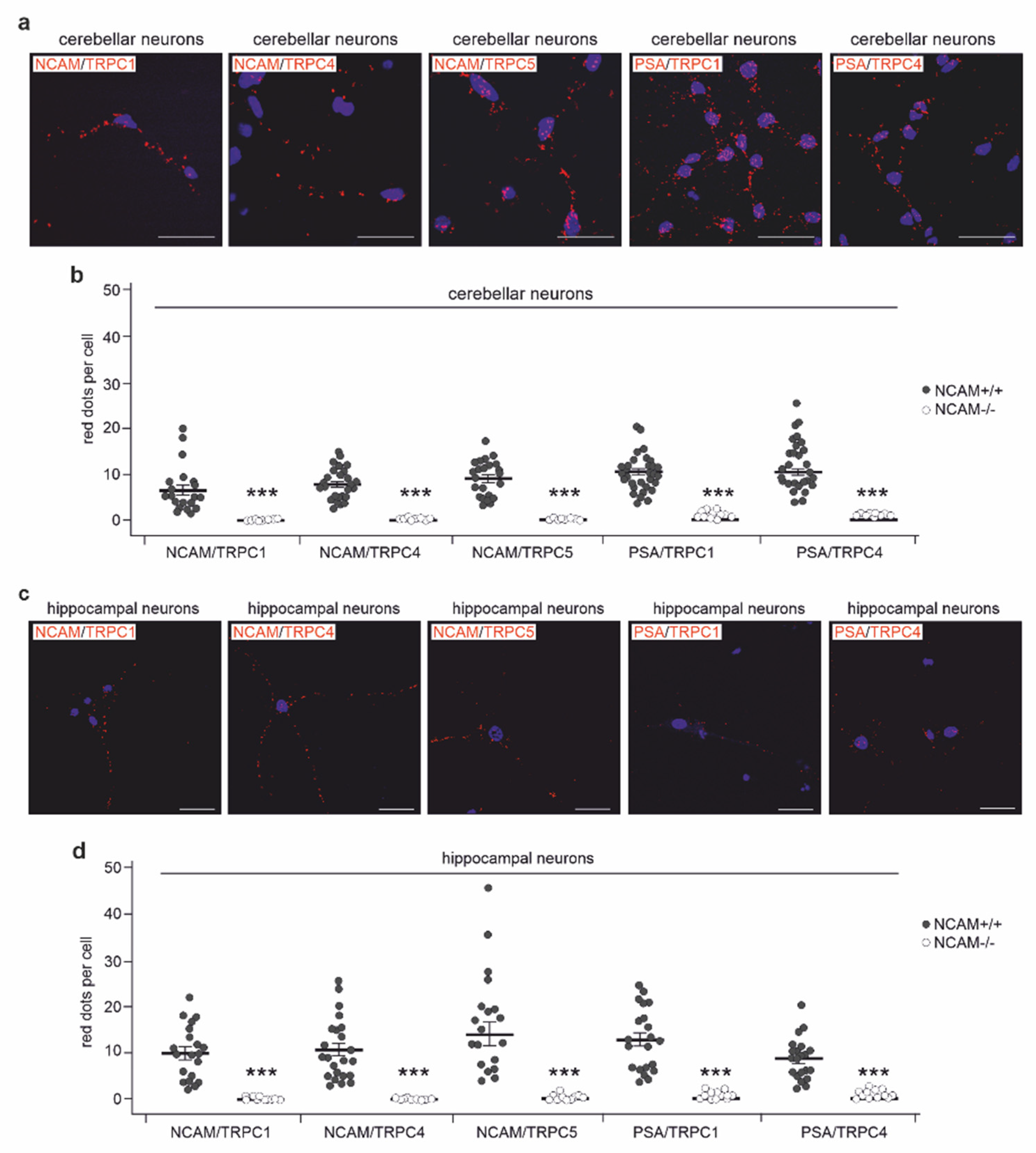
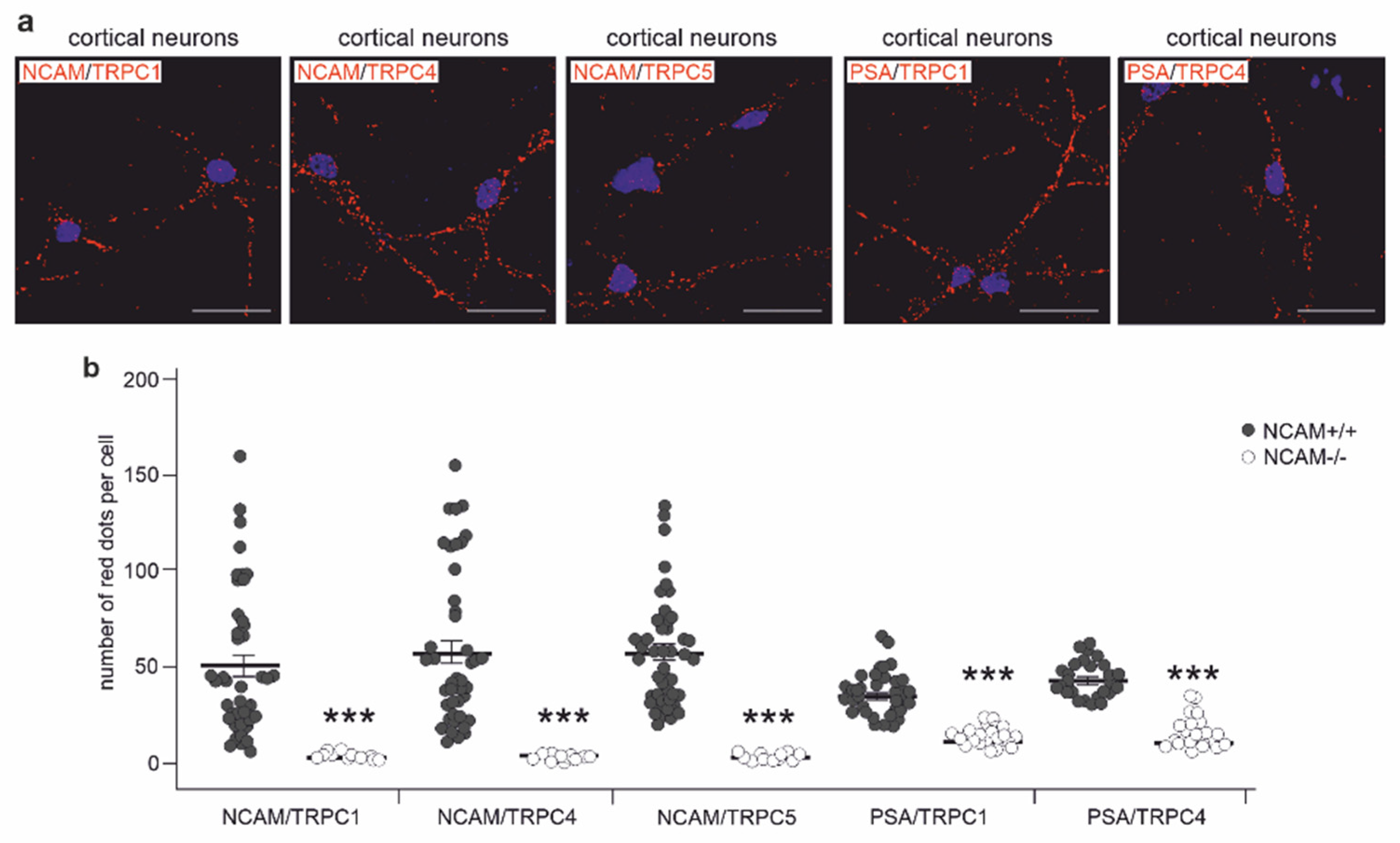
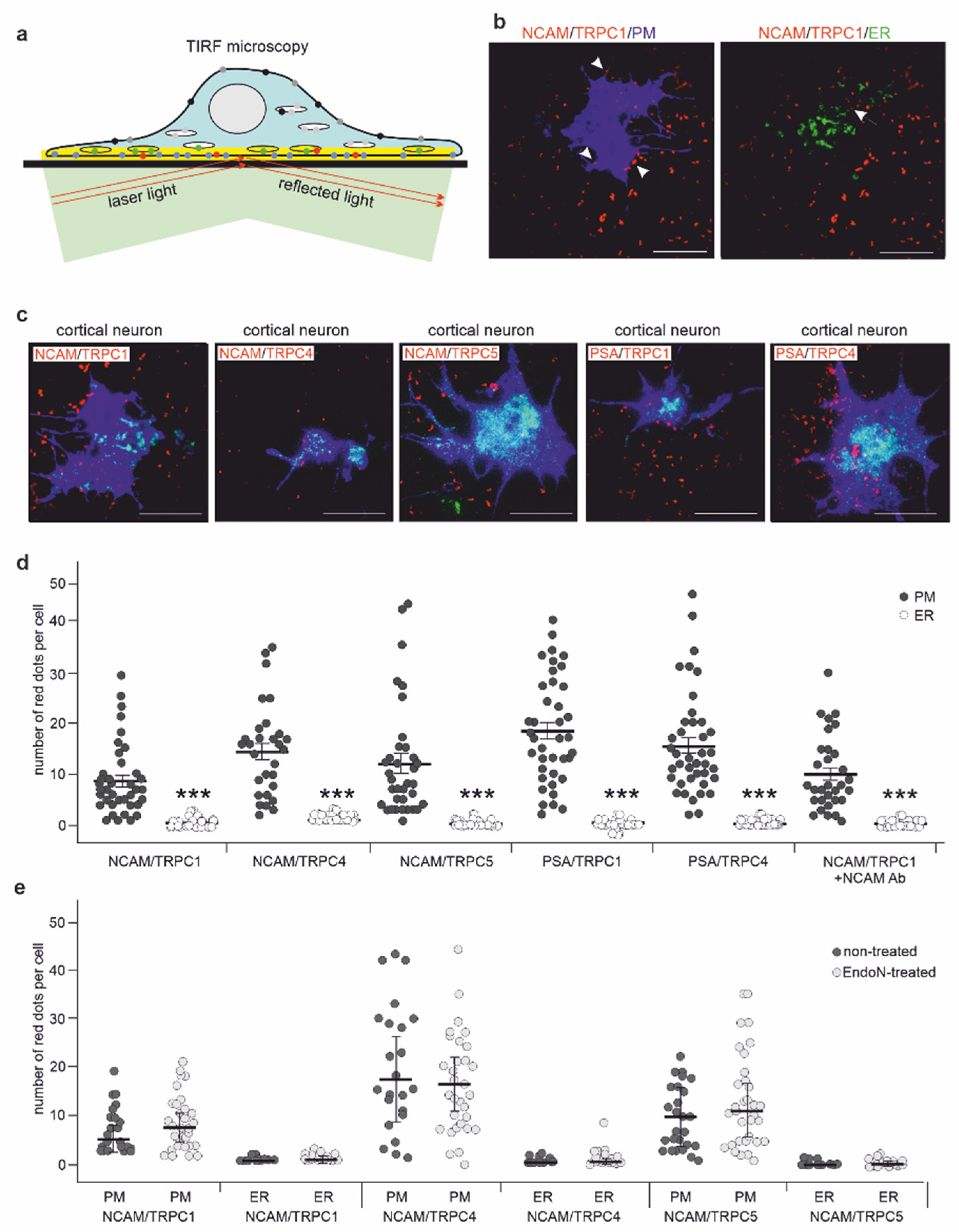
2.3. NCAM Colocalizes with TRPC1, −4, and −5 at the Neuronal Plasma Membrane
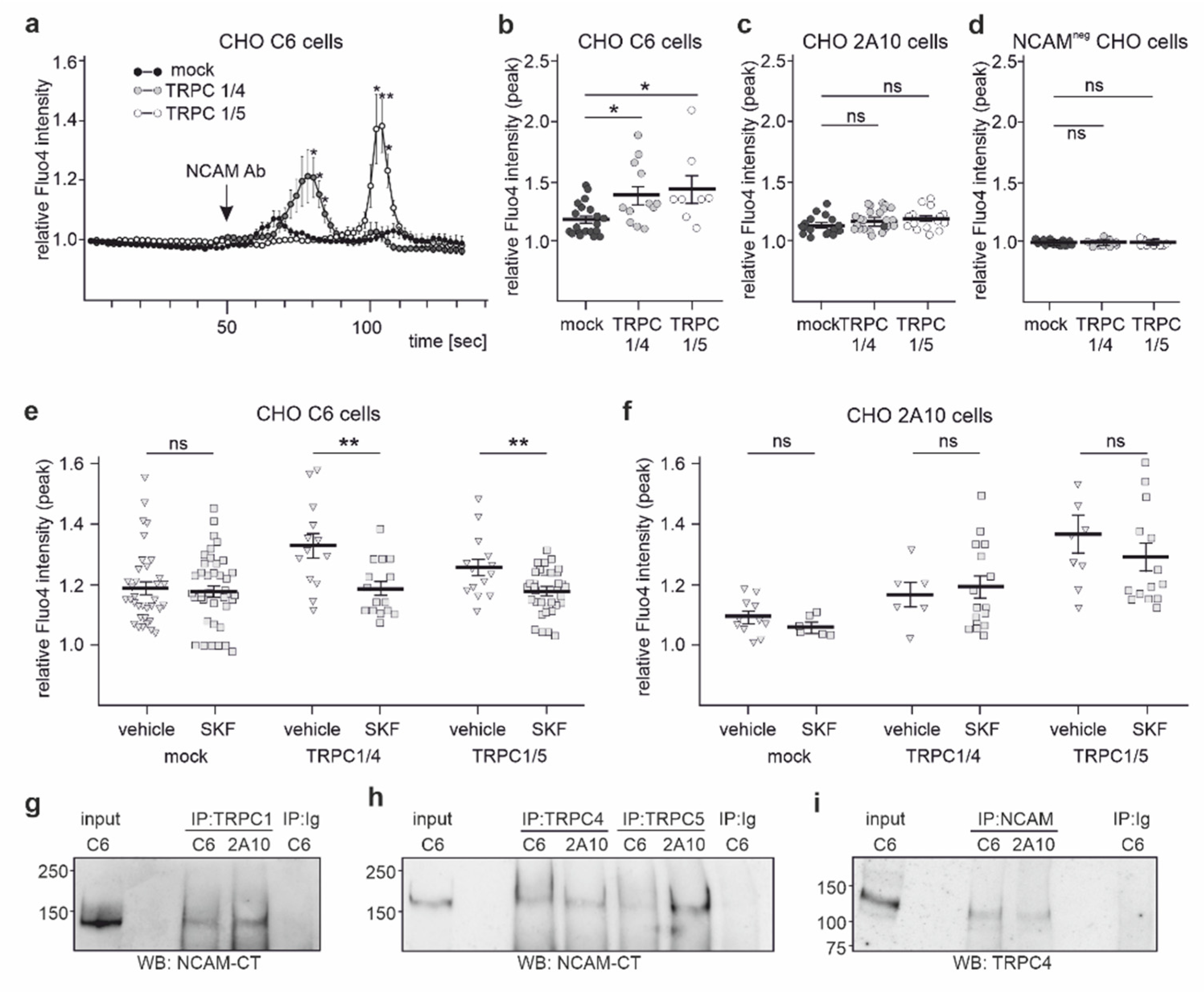
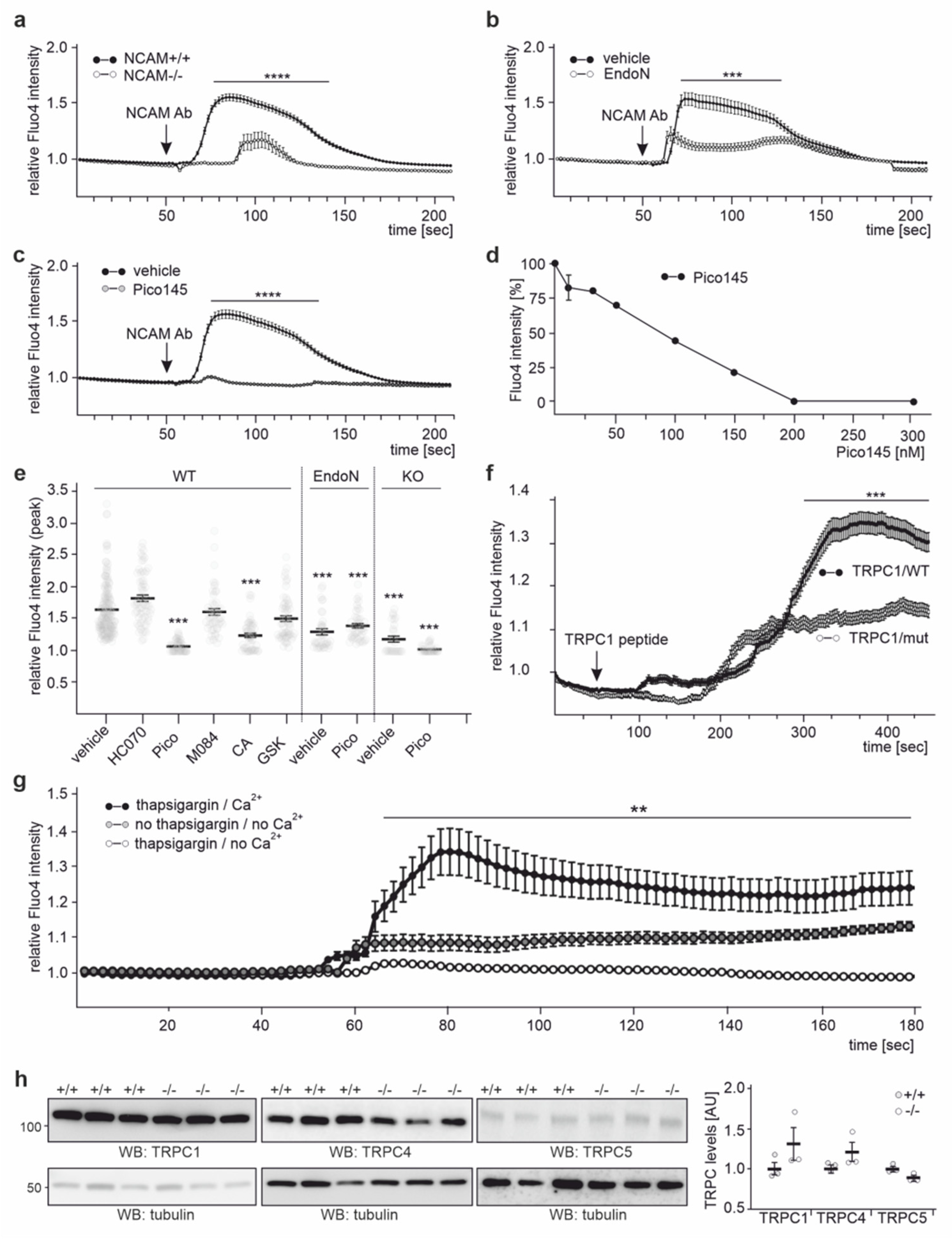
2.4. NCAM Regulates the Influx of Ca2+ via TRPC1, −4, and −5
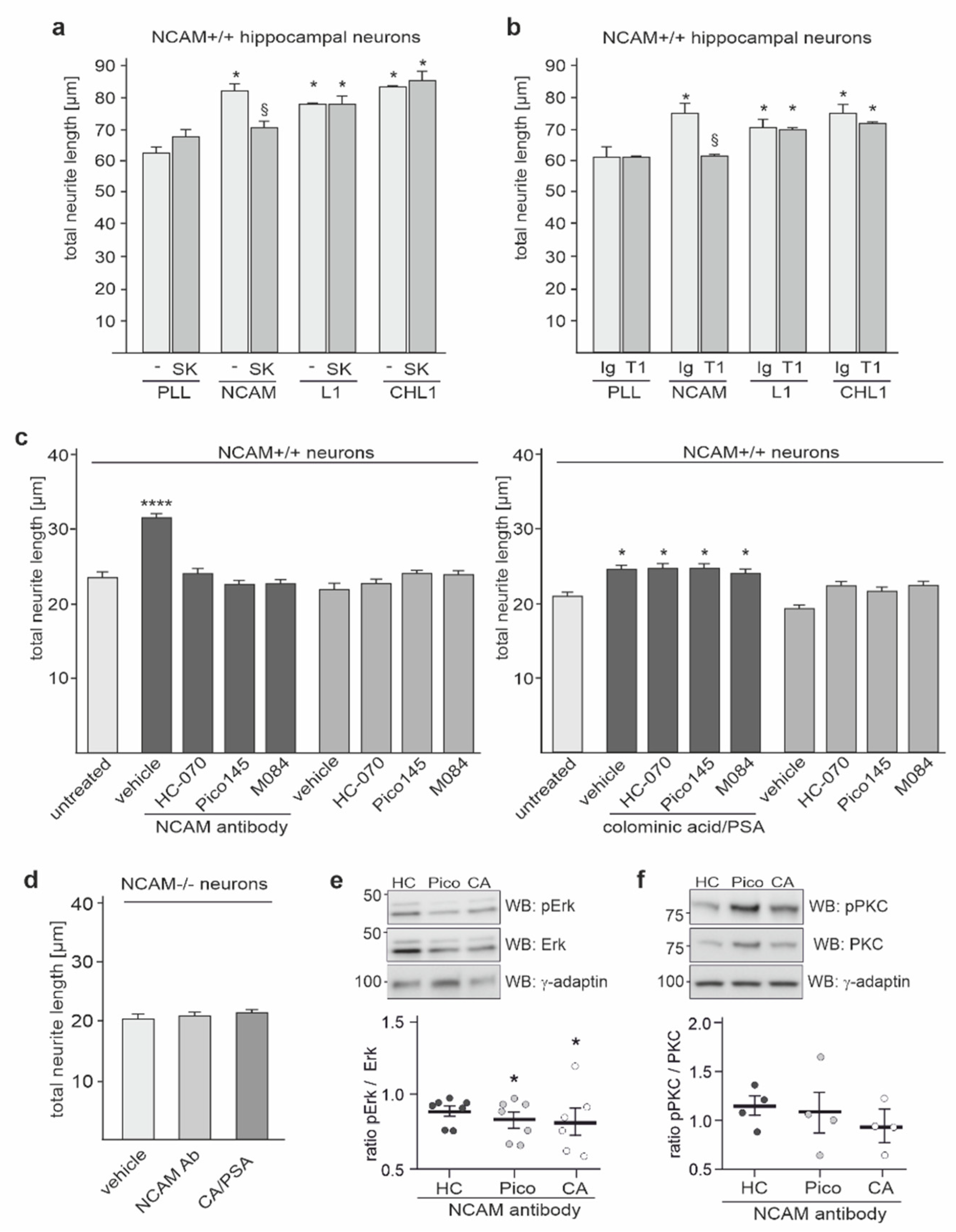
2.5. NCAM-Promoted Signal Transduction and Neurite Outgrowth Depend on TRPC1, −4, and −5
3. Discussion
4. Materials and Methods
4.1. Animals and Cell Lines
4.2. Antibodies and Reagents
4.3. Cloning of TRPC1/4 and TRPC1/5 Heteromers
4.4. Cloning and Expression of the N- and C-Terminal ICDs of TRPC1, 4, and 5
4.5. Cloning and Expression of His-Tagged NCAM140-ICD with Deletions of the N-Terminal, Middle, or C-Terminal Part
4.6. Immunoprecipitation, Pull-Down, and Western Blot Analysis
4.7. ELISA
4.8. Primary Culture of Mouse Cerebellar, Cortical, and Hippocampal Neurons
4.9. Culture of CHO Cells
4.10. Transfection of Cortical Cells and CHO Cells
4.11. Immunocytochemistry and Analysis of Colocalization
4.12. Proximity Ligation Assay
4.13. Total Internal Reflection Fluorescence (TIRF) Microscopy
4.14. Analysis of Neurite Outgrowth
4.15. Ca2+ Imaging of Cortical Neurons and CHO Cells
4.16. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schuch, U.; Lohse, M.J.; Schachner, M. Neural cell adhesion molecules influence second messenger systems. Neuron 1989, 3, 13–20. [Google Scholar] [CrossRef]
- Ditlevsen, D.K.; Kolkova, K. Signaling pathways involved in NCAM-induced neurite outgrowth. Adv. Exp. Med. Biol. 2010, 663, 151–168. [Google Scholar] [PubMed]
- Varbanov, H.; Dityatev, A. Regulation of extrasynaptic signaling by polysialylated NCAM: Impact for synaptic plasticity and cognitive functions. Mol. Cell. Neurosci. 2017, 81, 12–21. [Google Scholar] [CrossRef]
- Wedzony, K.; Chocyk, A.; Mackowiak, M. Potential roles of NCAM/PSA-NCAM proteins in depression and the mechanism of action of antidepressant drugs. Pharmacol. Rep. 2013, 65, 1471–1478. [Google Scholar] [CrossRef]
- Lu, B.; Liu, X.H.; Liao, S.M.; Lu, Z.L.; Chen, D.; Troy Ii, F.A.; Huang, R.B.; Zhou, G.P. A possible modulation mechanism of intramolecular and intermolecular interactions for NCAM polysialylation and cell migration. Curr. Top. Med. Chem. 2019, 19, 2271–2282. [Google Scholar] [CrossRef]
- Brennaman, L.H.; Maness, P.F. NCAM in neuropsychiatric and neurodegenerative disorders. Adv. Exp. Med. Biol. 2010, 663, 299–317. [Google Scholar] [PubMed]
- Aonurm-Helm, A.; Jaako, K.; Jurgenson, M.; Zharkovsky, A. Pharmacological approach for targeting dysfunctional brain plasticity: Focus on neural cell adhesion molecule (NCAM). Pharmacol. Res. 2016, 113 Pt B, 731–738. [Google Scholar] [CrossRef]
- Loers, G.; Schachner, M. Recognition molecules and neural repair. J. Neurochem. 2007, 101, 865–882. [Google Scholar] [CrossRef]
- Maness, P.F.; Schachner, M. Neural recognition molecules of the immunoglobulin superfamily: Signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007, 10, 19–26. [Google Scholar] [CrossRef]
- Hartz, B.P.; Ronn, L.C. NCAM in long-term potentiation and learning. Adv. Exp. Med. Biol. 2010, 663, 257–270. [Google Scholar]
- Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Neural cell adhesion molecules of the immunoglobulin superfamily regulate synapse formation, maintenance, and function. Trends Neurosci. 2017, 40, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Podesta, M.F.; Yam, P.; Codagnone, M.G.; Uccelli, N.A.; Colman, D.; Reines, A. Distinctive PSA-NCAM and NCAM hallmarks in glutamate-induced dendritic atrophy and synaptic disassembly. PLoS ONE 2014, 9, e108921. [Google Scholar]
- Niethammer, P.; Delling, M.; Sytnyk, V.; Dityatev, A.; Fukami, K.; Schachner, M. Cosignaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J. Cell Biol. 2002, 157, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, V.V.; Skladchikova, G.; Hinsby, A.M.; Jensen, P.H.; Kulahin, N.; Soroka, V.; Pedersen, N.; Tsetlin, V.; Poulsen, F.M.; Berezin, V.; et al. Structural basis for a direct interaction between FGFR1 and NCAM and evidence for a regulatory role of ATP. Structure 2003, 11, 691–701. [Google Scholar] [CrossRef]
- Kolkova, K.; Stensman, H.; Berezin, V.; Bock, E.; Larsson, C. Distinct roles of PKC isoforms in NCAM-mediated neurite outgrowth. J. Neurochem. 2005, 92, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Kleene, R.; Mzoughi, M.; Joshi, G.; Kalus, I.; Bormann, U.; Schulze, C.; Xiao, M.F.; Dityatev, A.; Schachner, M. NCAM-induced neurite outgrowth depends on binding of calmodulin to NCAM and on nuclear import of NCAM and fak fragments. J. Neurosci. 2010, 30, 10784–10798. [Google Scholar] [CrossRef]
- Doherty, P.; Ashton, S.V.; Moore, S.E.; Walsh, F.S. Morphoregulatory activities of NCAM and N-cadherin can be accounted for by G protein-dependent activation of L- and N-type neuronal Ca2+ channels. Cell 1991, 67, 21–33. [Google Scholar] [CrossRef]
- Kiryushko, D.; Korshunova, I.; Berezin, V.; Bock, E. Neural cell adhesion molecule induces intracellular signaling via multiple mechanisms of Ca2+ homeostasis. Mol. Biol. Cell 2006, 17, 2278–2286. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Singh, B.B. TRPC channels and their implication in neurological diseases. CNS Neurol. Disord. Drug Targets 2010, 9, 94–104. [Google Scholar] [CrossRef]
- Salido, G.M.; Sage, S.O.; Rosado, J.A. TRPC channels and store-operated Ca2+ entry. Biochim. Biophys. Acta 2009, 1793, 223–230. [Google Scholar] [CrossRef]
- Vazquez, G.; Wedel, B.J.; Aziz, O.; Trebak, M.; Putney, J.W., Jr. The mammalian TRPC cation channels. Biochim. Biophys. Acta 2004, 1742, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Beech, D.J. TRPC1: Store-operated channel and more. Pflugers Arch. 2005, 451, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Greka, A.; Navarro, B.; Oancea, E.; Duggan, A.; Clapham, D.E. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat. Neurosci. 2003, 6, 837–845. [Google Scholar] [CrossRef]
- Plant, T.D.; Schaefer, M. Receptor-operated cation channels formed by TRPC4 and TRPC5. Naunyn-Schmiedebergs Arch. Pharmacol. 2005, 371, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Plant, T.D.; Schaefer, M. TRPC4 and TRPC5: Receptor-operated Ca2+-permeable nonselective cation channels. Cell Calcium 2003, 33, 441–450. [Google Scholar] [CrossRef]
- Freichel, M.; Philipp, S.; Cavalie, A.; Flockerzi, V. TRPC4 and TRPC4-deficient mice. Novartis Found. Symp. 2004, 258, 189–199; discussion 199–203, 263–266. [Google Scholar]
- Freichel, M.; Tsvilovskyy, V.; Camacho-Londono, J.E. TRPC4- and TRPC4-containing channels. Handb. Exp. Pharmacol. 2014, 222, 85–128. [Google Scholar]
- Ambudkar, I.S. TRPC1: Getting physical in space. Cell Cycle 2013, 12, 3355–3356. [Google Scholar] [CrossRef][Green Version]
- Ambudkar, I.S. TRPC1: A core component of store-operated calcium channels. Biochem. Soc. Trans. 2007, 35 Pt 1, 96–100. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; Ong, H.L.; Liu, X.; Bandyopadhyay, B.C.; Cheng, K.T. TRPC1: The link between functionally distinct store-operated calcium channels. Cell Calcium 2007, 42, 213–223. [Google Scholar] [CrossRef]
- Rychkov, G.; Barritt, G.J. TRPC1 Ca2+-permeable channels in animal cells. Handb. Exp. Pharmacol. 2007, 179, 23–52. [Google Scholar]
- Nesin, V.; Tsiokas, L. Trpc1. Handb. Exp. Pharmacol. 2014, 222, 15–51. [Google Scholar] [PubMed]
- Sharma, S.; Hopkins, C.R. Review of transient receptor potential canonical (TRPC5) channel modulators and diseases. J. Med. Chem. 2019, 62, 7589–7602. [Google Scholar] [CrossRef]
- Zholos, A.V. Trpc5. Handb. Exp. Pharmacol. 2014, 222, 129–156. [Google Scholar] [PubMed]
- Kim, H.; Kim, J.; Jeon, J.P.; Myeong, J.; Wie, J.; Hong, C.; Kim, H.J.; Jeon, J.H.; So, I. The roles of G proteins in the activation of TRPC4 and TRPC5 transient receptor potential channels. Channels 2012, 6, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ko, J.; Myeong, J.; Kwak, M.; Hong, C.; So, I. TRPC1 as a negative regulator for TRPC4 and TRPC5 channels. Pflugers Arch. 2019, 471, 1045–1053. [Google Scholar] [CrossRef]
- Dietrich, A.; Fahlbusch, M.; Gudermann, T. Classical transient receptor potential 1 (TRPC1): Channel or channel regulator? Cells 2014, 3, 939–962. [Google Scholar] [CrossRef]
- Cavalie, A. Ionic channels formed by TRPC4. Handb. Exp. Pharmacol. 2007, 179, 93–108. [Google Scholar]
- Ong, H.L.; Ambudkar, I.S. The dynamic complexity of the TRPC1 channelosome. Channels 2011, 5, 424–431. [Google Scholar] [CrossRef]
- Wu, D.; Huang, W.; Richardson, P.M.; Priestley, J.V.; Liu, M. TRPC4 in rat dorsal root ganglion neurons is increased after nerve injury and is necessary for neurite outgrowth. J. Biol. Chem. 2008, 283, 416–426. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Obukhov, A.G.; Nowycky, M.C.; Ledeen, R.W. Induction of calcium influx through TRPC5 channels by cross-linking of GM1 ganglioside associated with α5β1 integrin initiates neurite outgrowth. J. Neurosci. 2007, 27, 7447–7458. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Yamamoto, H.; Matsumoto, H.; Ishizaki, Y.; Shibasaki, K. TRPC5 regulates axonal outgrowth in developing retinal ganglion cells. Lab. Investig. 2020, 100, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Beech, D.J.; Xu, S.Z.; McHugh, D.; Flemming, R. TRPC1 store-operated cationic channel subunit. Cell Calcium 2003, 33, 433–440. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, X.; Tian, J.; Xiao, Y.; Tian, T.; Xu, F.; Hong, X.; Zhu, M.X. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 2020, 209, 107497. [Google Scholar] [CrossRef]
- Heo, D.K.; Chung, W.Y.; Park, H.W.; Yuan, J.P.; Lee, M.G.; Kim, J.Y. Opposite regulatory effects of TRPC1 and TRPC5 on neurite outgrowth in PC12 cells. Cell. Signal. 2012, 24, 899–906. [Google Scholar] [CrossRef]
- Hui, H.; McHugh, D.; Hannan, M.; Zeng, F.; Xu, S.Z.; Khan, S.U.; Levenson, R.; Beech, D.J.; Weiss, J.L. Calcium-sensing mechanism in TRPC5 channels contributing to retardation of neurite outgrowth. J. Physiol. 2006, 572 Pt 1, 165–172. [Google Scholar] [CrossRef]
- Kumar, S.; Chakraborty, S.; Barbosa, C.; Brustovetsky, T.; Brustovetsky, N.; Obukhov, A.G. Mechanisms controlling neurite outgrowth in a pheochromocytoma cell line: The role of TRPC channels. J. Cell. Physiol. 2012, 227, 1408–1419. [Google Scholar] [CrossRef]
- Li, H.S.; Xu, X.Z.; Montell, C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron 1999, 24, 261–273. [Google Scholar] [CrossRef]
- Fiorio Pla, A.; Maric, D.; Brazer, S.C.; Giacobini, P.; Liu, X.; Chang, Y.H.; Ambudkar, I.S.; Barker, J.L. Canonical transient receptor potential 1 plays a role in basic fibroblast growth factor (bFGF)/FGF receptor-1-induced Ca2+ entry and embryonic rat neural stem cell proliferation. J. Neurosci. 2005, 25, 2687–2701. [Google Scholar]
- Hannan, M.A.; Kabbani, N.; Paspalas, C.D.; Levenson, R. Interaction with dopamine D2 receptor enhances expression of transient receptor potential channel 1 at the cell surface. Biochim. Biophys. Acta 2008, 1778, 974–982. [Google Scholar] [CrossRef][Green Version]
- Xiao, M.F.; Xu, J.C.; Tereshchenko, Y.; Novak, D.; Schachner, M.; Kleene, R. Neural cell adhesion molecule modulates dopaminergic signaling and behavior by regulating dopamine D2 receptor internalization. J. Neurosci. 2009, 29, 14752–14763. [Google Scholar] [CrossRef] [PubMed]
- Cassens, C.; Kleene, R.; Xiao, M.F.; Friedrich, C.; Dityateva, G.; Schafer-Nielsen, C.; Schachner, M. Binding of the receptor tyrosine kinase TrkB to the neural cell adhesion molecule (NCAM) regulates phosphorylation of NCAM and NCAM-dependent neurite outgrowth. J. Biol. Chem. 2010, 285, 28959–28967. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.; Lauridsen, J.B.; Berezin, V.; Bock, E.; Kiselyov, V.V. The neural cell adhesion molecule binds to fibroblast growth factor receptor 2. FEBS Lett. 2006, 580, 3386–3390. [Google Scholar] [CrossRef] [PubMed]
- Theis, T.; Mishra, B.; von der Ohe, M.; Loers, G.; Prondzynski, M.; Pless, O.; Blackshear, P.J.; Schachner, M.; Kleene, R. Functional role of the interaction between polysialic acid and myristoylated alanine-rich C kinase substrate at the plasma membrane. J. Biol. Chem. 2013, 288, 6726–6742. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Myristoylated alanine-rich C kinase substrate coordinates native TRPC1 channel activation by phosphatidylinositol 4,5-bisphosphate and protein kinase C in vascular smooth muscle. FASEB J. 2014, 28, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Jahan, K.S.; Shi, J.; Greenberg, H.Z.E.; Khavandi, S.; Baudel, M.M.; Barrese, V.; Greenwood, I.A.; Albert, A.P. MARCKS mediates vascular contractility through regulating interactions between voltage-gated Ca2+ channels and PIP2. Vasc. Pharmacol. 2020, 132, 106776. [Google Scholar] [CrossRef] [PubMed]
- Gerardy-Schahn, R.; Bethe, A.; Brennecke, T.; Muhlenhoff, M.; Eckhardt, M.; Ziesing, S.; Lottspeich, F.; Frosch, M. Molecular cloning and functional expression of bacteriophage PK1E-encoded endoneuraminidase Endo NE. Mol. Microbiol. 1995, 16, 441–450. [Google Scholar] [CrossRef]
- Neuner, S.M.; Wilmott, L.A.; Hope, K.A.; Hoffmann, B.; Chong, J.A.; Abramowitz, J.; Birnbaumer, L.; O’Connell, K.M.; Tryba, A.K.; Greene, A.S.; et al. TRPC3 channels critically regulate hippocampal excitability and contextual fear memory. Behav. Brain Res. 2015, 281, 69–77. [Google Scholar] [CrossRef]
- Liu, X.; Singh, B.B.; Ambudkar, I.S. TRPC1 is required for functional store-operated Ca2+ channels. Role of acidic amino acid residues in the S5-S6 region. J. Biol. Chem. 2003, 278, 11337–11343. [Google Scholar] [CrossRef]
- Francavilla, C.; Cattaneo, P.; Berezin, V.; Bock, E.; Ami, D.; de Marco, A.; Christofori, G.; Cavallaro, U. The binding of NCAM to FGFR1 induces a specific cellular response mediated by receptor trafficking. J. Cell Biol. 2009, 187, 1101–1116. [Google Scholar] [CrossRef]
- Rubaiy, H.N.; Ludlow, M.J.; Bon, R.S.; Beech, D.J. Pico145—Powerful new tool for TRPC1/4/5 channels. Channels 2017, 11, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Rubaiy, H.N.; Ludlow, M.J.; Henrot, M.; Gaunt, H.J.; Miteva, K.; Cheung, S.Y.; Tanahashi, Y.; Hamzah, N.; Musialowski, K.E.; Blythe, N.M.; et al. Picomolar, selective, and subtype-specific small-molecule inhibition of TRPC1/4/5 channels. J. Biol. Chem. 2017, 292, 8158–8173. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.Y.; Tian, W.; Zhang, H.N.; Sun, Y.; Meng, J.R.; Cao, W.; Li, X.Q. Canonical transient receptor potential channels and their modulators: Biology, pharmacology and therapeutic potentials. Arch. Pharm. Res. 2021, 44, 354–377. [Google Scholar] [CrossRef]
- Wright, D.J.; Simmons, K.J.; Johnson, R.M.; Beech, D.J.; Muench, S.P.; Bon, R.S. Human TRPC5 structures reveal interaction of a xanthine-based TRPC1/4/5 inhibitor with a conserved lipid binding site. Commun. Biol. 2020, 3, 704. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Wei, M.; Guo, W.; Quan, L.; Kang, Y.; Wu, J.X.; Chen, L. Structural basis for human TRPC5 channel inhibition by two distinct inhibitors. eLife 2021, 10, e63429. [Google Scholar] [CrossRef]
- Westphal, N.; Theis, T.; Loers, G.; Schachner, M.; Kleene, R. Nuclear fragments of the neural cell adhesion molecule NCAM with or without polysialic acid differentially regulate gene expression. Sci. Rep. 2017, 7, 13631. [Google Scholar] [CrossRef]
- Westphal, N.; Kleene, R.; Lutz, D.; Theis, T.; Schachner, M. Polysialic acid enters the cell nucleus attached to a fragment of the neural cell adhesion molecule NCAM to regulate the circadian rhythm in mouse brain. Mol. Cell. Neurosci. 2016, 74, 114–127. [Google Scholar] [CrossRef]
- Westphal, N.; Loers, G.; Lutz, D.; Theis, T.; Kleene, R.; Schachner, M. Generation and intracellular trafficking of a polysialic acid-carrying fragment of the neural cell adhesion molecule NCAM to the cell nucleus. Sci. Rep. 2017, 7, 8622. [Google Scholar] [CrossRef]
- Stork, O.; Welzl, H.; Wolfer, D.; Schuster, T.; Mantei, N.; Stork, S.; Hoyer, D.; Lipp, H.; Obata, K.; Schachner, M. Recovery of emotional behaviour in neural cell adhesion molecule (NCAM) null mutant mice through transgenic expression of NCAM180. Eur. J. Neurosci. 2000, 12, 3291–3306. [Google Scholar] [CrossRef]
- Stork, O.; Welzl, H.; Wotjak, C.T.; Hoyer, D.; Delling, M.; Cremer, H.; Schachner, M. Anxiety and increased 5-HT1A receptor response in NCAM null mutant mice. J. Neurobiol. 1999, 40, 343–355. [Google Scholar] [CrossRef]
- Brandewiede, J.; Stork, O.; Schachner, M. NCAM deficiency in the mouse forebrain impairs innate and learned avoidance behaviours. Genes Brain Behav. 2014, 13, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Angata, K.; Long, J.M.; Bukalo, O.; Lee, W.; Dityatev, A.; Wynshaw-Boris, A.; Schachner, M.; Fukuda, M.; Marth, J.D. Sialyltransferase ST8Sia-II assembles a subset of polysialic acid that directs hippocampal axonal targeting and promotes fear behavior. J. Biol. Chem. 2004, 279, 32603–32613. [Google Scholar] [CrossRef]
- Pillai-Nair, N.; Panicker, A.K.; Rodriguiz, R.M.; Gilmore, K.L.; Demyanenko, G.P.; Huang, J.Z.; Wetsel, W.C.; Maness, P.F. Neural cell adhesion molecule-secreting transgenic mice display abnormalities in GABAergic interneurons and alterations in behavior. J. Neurosci. 2005, 25, 4659–4671. [Google Scholar] [CrossRef]
- Bisaz, R.; Conboy, L.; Sandi, C. Learning under stress: A role for the neural cell adhesion molecule NCAM. Neurobiol. Learn. Mem. 2009, 91, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Bisaz, R.; Sandi, C. The role of NCAM in auditory fear conditioning and its modulation by stress: A focus on the amygdala. Genes Brain Behav. 2010, 9, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fernandez, M.A.; Montaron, M.F.; Varea, E.; Rougon, G.; Venero, C.; Abrous, D.N.; Sandi, C. Upregulation of polysialylated neural cell adhesion molecule in the dorsal hippocampus after contextual fear conditioning is involved in long-term memory formation. J. Neurosci. 2007, 27, 4552–4561. [Google Scholar] [CrossRef]
- Markram, K.; Lopez Fernandez, M.A.; Abrous, D.N.; Sandi, C. Amygdala upregulation of NCAM polysialylation induced by auditory fear conditioning is not required for memory formation, but plays a role in fear extinction. Neurobiol. Learn. Mem. 2007, 87, 573–582. [Google Scholar] [CrossRef]
- Senkov, O.; Sun, M.; Weinhold, B.; Gerardy-Schahn, R.; Schachner, M.; Dityatev, A. Polysialylated neural cell adhesion molecule is involved in induction of long-term potentiation and memory acquisition and consolidation in a fear-conditioning paradigm. J. Neurosci. 2006, 26, 10888–109898. [Google Scholar] [CrossRef]
- Riccio, A.; Li, Y.; Tsvetkov, E.; Gapon, S.; Yao, G.L.; Smith, K.S.; Engin, E.; Rudolph, U.; Bolshakov, V.Y.; Clapham, D.E. Decreased anxiety-like behavior and Galphaq/11-dependent responses in the amygdala of mice lacking TRPC4 channels. J. Neurosci. 2014, 34, 3653–3667. [Google Scholar] [CrossRef]
- Just, S.; Chenard, B.L.; Ceci, A.; Strassmaier, T.; Chong, J.A.; Blair, N.T.; Gallaschun, R.J.; Del Camino, D.; Cantin, S.; D’Amours, M.; et al. Treatment with HC-070, a potent inhibitor of TRPC4 and TRPC5, leads to anxiolytic and antidepressant effects in mice. PLoS ONE 2018, 13, e0191225. [Google Scholar] [CrossRef]
- de Oliveira, A.R.; Reimer, A.E.; Brandao, M.L. Dopamine D2 receptor mechanisms in the expression of conditioned fear. Pharmacol. Biochem. Behav. 2006, 84, 102–111. [Google Scholar] [CrossRef] [PubMed]
- de Vita, V.M.; Zapparoli, H.R.; Reimer, A.E.; Brandao, M.L.; de Oliveira, A.R. Dopamine D2 receptors in the expression and extinction of contextual and cued conditioned fear in rats. Exp. Brain Res. 2021, 239, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.D.; Neve, K.A.; Lattal, K.M. Dopamine and extinction: A convergence of theory with fear and reward circuitry. Neurobiol. Learn. Mem. 2014, 108, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Cremer, H.; Lange, R.; Christoph, A.; Plomann, M.; Vopper, G.; Roes, J.; Brown, R.; Baldwin, S.; Kraemer, P.; Scheff, S.; et al. Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature 1994, 367, 455–459. [Google Scholar] [CrossRef]
- Kwan, H.Y.; Shen, B.; Ma, X.; Kwok, Y.C.; Huang, Y.; Man, Y.B.; Yu, S.; Yao, X. TRPC1 associates with BK(Ca) channel to form a signal complex in vascular smooth muscle cells. Circ. Res. 2009, 104, 670–678. [Google Scholar] [CrossRef]
- Kleene, R.; Loers, G.; Castillo, G.; Schachner, M. Cell adhesion molecule L1 interacts with the chromo shadow domain of heterochromatin protein 1 isoforms alpha, beta, and via its intracellular domain. FASEB J. 2022, 36, e22074. [Google Scholar] [CrossRef]
- Adler, J.; Parmryd, I. Quantifying colocalization by correlation: The Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytom. A 2010, 77, 733–742. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224 Pt 3, 213–232. [Google Scholar] [CrossRef]
- Goulden, B.D.; Pacheco, J.; Dull, A.; Zewe, J.P.; Deiters, A.; Hammond, G.R.V. A high-avidity biosensor reveals plasma membrane PI(3,4)P2 is predominantly a class I PI3K signaling product. J. Cell Biol. 2019, 218, 1066–1079. [Google Scholar] [CrossRef]
- Holbro, N.; Grunditz, A.; Oertner, T.G. Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc. Natl. Acad. Sci. USA 2009, 106, 15055–15060. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amores-Bonet, L.; Kleene, R.; Theis, T.; Schachner, M. Interactions between the Polysialylated Neural Cell Adhesion Molecule and the Transient Receptor Potential Canonical Channels 1, 4, and 5 Induce Entry of Ca2+ into Neurons. Int. J. Mol. Sci. 2022, 23, 10027. https://doi.org/10.3390/ijms231710027
Amores-Bonet L, Kleene R, Theis T, Schachner M. Interactions between the Polysialylated Neural Cell Adhesion Molecule and the Transient Receptor Potential Canonical Channels 1, 4, and 5 Induce Entry of Ca2+ into Neurons. International Journal of Molecular Sciences. 2022; 23(17):10027. https://doi.org/10.3390/ijms231710027
Chicago/Turabian StyleAmores-Bonet, Laura, Ralf Kleene, Thomas Theis, and Melitta Schachner. 2022. "Interactions between the Polysialylated Neural Cell Adhesion Molecule and the Transient Receptor Potential Canonical Channels 1, 4, and 5 Induce Entry of Ca2+ into Neurons" International Journal of Molecular Sciences 23, no. 17: 10027. https://doi.org/10.3390/ijms231710027
APA StyleAmores-Bonet, L., Kleene, R., Theis, T., & Schachner, M. (2022). Interactions between the Polysialylated Neural Cell Adhesion Molecule and the Transient Receptor Potential Canonical Channels 1, 4, and 5 Induce Entry of Ca2+ into Neurons. International Journal of Molecular Sciences, 23(17), 10027. https://doi.org/10.3390/ijms231710027