
PRMT1, a Key Modulator of Unliganded Progesterone Receptor Signaling in Breast Cancer


 and
and
Abstract
:1. Introduction
2. Results
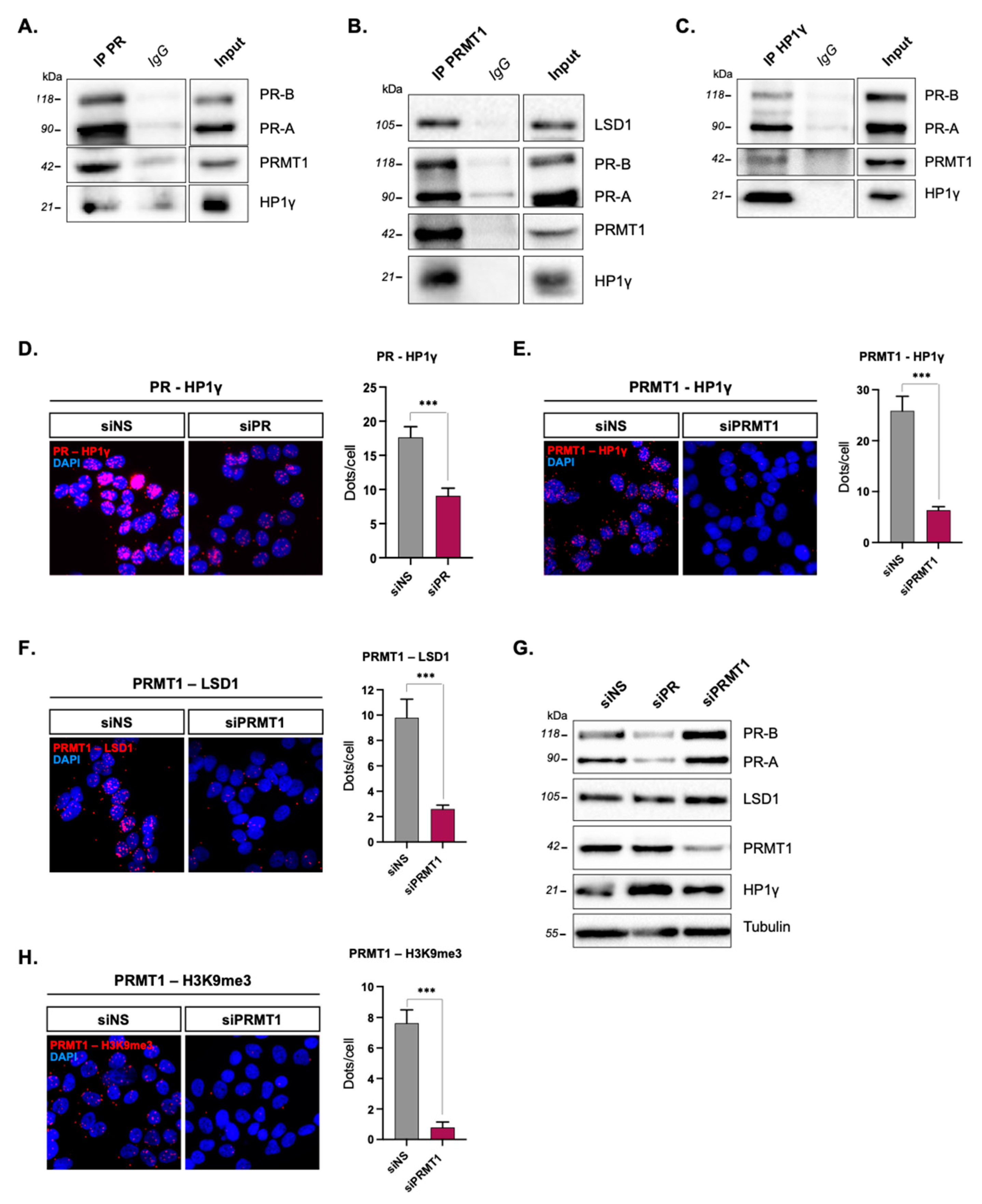
2.1. PRMT1 Interacts with uPR and Its Partners Prior to Hormonal Treatment
2.2. PRMT1 Targets Progesterone-Inducible Promoters in Unstimulated T47D Cells
2.3. PRMT1 Represses the Basal Expression of an R5020-Activated Set of Genes
2.4. PRMT1 Is an Interesting Candidate in BRCA1-Dependent uPR Recycling
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. SiRNA and Plasmid Transfection
4.3. Immunoprecipitation (IP), Immunoblot (IB), and Antibodies
4.4. Proximity Ligation Assays (PLA), Image Acquisition, and Analysis
4.5. RNA Extraction and Real-Time qPCR Analysis
4.6. Chromatin Immunoprecipitation (ChIP)
4.7. RNA Sequencing and RNA Seq Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brisken, C.; Scabia, V. 90 YEARS OF PROGESTERONE: Progesterone receptor signaling in the normal breast and its implications for cancer. J. Mol. Endocrinol. 2020, 65, T81–T94. [Google Scholar] [CrossRef]
- DeMayo, F.J.; Lydon, J. New insights into progesterone receptor signaling in the endometrium required for embryo implantation. J. Mol. Endocrinol. 2020, 65, T1–T14. [Google Scholar] [CrossRef]
- Catenaccio, E.; Mu, W.; Lipton, M.L. Estrogen- and progesterone-mediated structural neuroplasticity in women: Evidence from neuroimaging. Brain Struct. Funct. 2016, 221, 3845–3867. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Afrin, S.; Jones, S.I.; Segars, J. Selective Progesterone Receptor Modulators—Mechanisms and Therapeutic Utility. Endocr. Rev. 2020, 41, bnaa012. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, K.B.; Sartorius, C.A. 90 YEARS OF PROGESTERONE: Progesterone and progesterone receptors in breast cancer: Past, present, future. J. Mol. Endocrinol. 2020, 65, T49–T63. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.H.; Lange, C.A. Deciphering Steroid Receptor Crosstalk in Hormone-Driven Cancers. Endocrinology 2018, 159, 3897–3907. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hafiz, H.A.; Horwitz, K.B. Post-translational modifications of the progesterone receptors. J. Steroid Biochem. Mol. Biol. 2014, 140, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malbeteau, L.; Poulard, C.; Languilaire, C.; Mikaelian, I.; Flamant, F.; Le Romancer, M.; Corbo, L. PRMT1 Is Critical for the Transcriptional Activity and the Stability of the Progesterone Receptor. iScience 2020, 23, 101236. [Google Scholar] [CrossRef]
- Dwyer, A.R.; Truong, T.H.; Ostrander, J.H.; Lange, C.A. 90 YEARS OF PROGESTERONE: Steroid receptors as MAPK signaling sensors in breast cancer: Let the fates decide. J. Mol. Endocrinol. 2020, 65, T35–T48. [Google Scholar] [CrossRef]
- Treviño, L.S.; Weigel, N.L. Phosphorylation: A fundamental regulator of steroid receptor action. Trends Endocrinol. Metab. 2013, 24, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Daniel, A.R.; Lange, C.A. Protein kinases mediate ligand-independent derepression of sumoylated progesterone receptors in breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14287–14292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giguère, V. Orphan Nuclear Receptors: From Gene to Function. Endocr. Rev. 1999, 20, 689–725. [Google Scholar] [CrossRef] [PubMed]
- Vicent, G.P.; Nacht, A.S.; Zaurin, R.; Font-Mateu, J.; Soronellas, D.; Le Dily, F.; Reyes, D.; Beato, M. Unliganded progesterone receptor-mediated targeting of an RNA-containing repressive complex silences a subset of hormone-inducible genes. Genes Dev. 2013, 27, 1179–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicent, G.P.; Nacht, A.S.; Ballaré, C.; Zaurin, R.; Soronellas, D.; Beato, M. Progesterone Receptor Interaction with Chromatin. In Steroid Receptors; Humana: New York, NY, USA, 2014; Volume 1204, pp. 1–14. [Google Scholar] [CrossRef]
- Söderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstråle, K.; Leuchowius, K.-J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.-G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef]
- Poulard, C.; Rambaud, J.; Le Romancer, M.; Corbo, L. Proximity Ligation Assay to Detect and Localize the Interactions of ERα with PI3-K and Src in Breast Cancer Cells and Tumor Samples. Methods Mol. Biol. 2014, 1204, 135–143. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Nakayama, J.-I.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I.S. Role of Histone H3 Lysine 9 Methylation in Epigenetic Control of Heterochromatin Assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef] [Green Version]
- Litt, M.; Qiu, Y.; Huang, S. Histone arginine methylations: Their roles in chromatin dynamics and transcriptional regulation. Biosci. Rep. 2009, 29, 131–141. [Google Scholar] [CrossRef]
- Stallcup, M.R. Role of protein methylation in chromatin remodeling and transcriptional regulation. Oncogene 2001, 20, 3014–3020. [Google Scholar] [CrossRef] [Green Version]
- Malbeteau, L.; Pham, H.T.; Eve, L.; Stallcup, M.R.; Poulard, C.; Le Romancer, M. How Protein Methylation Regulates Steroid Receptor Function. Endocr. Rev. 2022, 43, 160–197. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, C.; Eve, L.; Poulard, C.; Le Romancer, M. Structure, Activity, and Function of PRMT1. Life 2021, 11, 1147. [Google Scholar] [CrossRef] [PubMed]
- Eram, M.S.; Shen, Y.; Szewczyk, M.M.; Wu, H.; Senisterra, G.; Li, F.; Butler, K.V.; Kaniskan, H.; Speed, B.A.; Seña, C.D.; et al. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, J.Y.; Pignata, L.; Goy, P.-A.; Kawabata, K.C.; Lee, S.C.-W.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e9. [Google Scholar] [CrossRef]
- Poulard, C.; Bittencourt, D.; Wu, D.; Hu, Y.; Gerke, D.S.; Stallcup, M.R. A post-translational modification switch controls coactivator function of histone methyltransferases G9a and GLP. EMBO Rep. 2017, 18, 1442–1459. [Google Scholar] [CrossRef]
- Koh, S.S.; Chen, D.; Lee, Y.-H.; Stallcup, M.R. Synergistic Enhancement of Nuclear Receptor Function by p160 Coactivators and Two Coactivators with Protein Methyltransferase Activities. J. Biol. Chem. 2001, 276, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Briggs, S.D.; Brame, C.J.; Caldwell, J.A.; Koh, S.S.; Ma, H.; Cook, R.G.; Shabanowitz, J.; Hunt, D.F.; Stallcup, M.R.; et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol. 2001, 11, 996–1000. [Google Scholar] [CrossRef] [Green Version]
- Stendahl, M.; Kronblad, Å.; Rydén, L.; Emdin, S.; Bengtsson, N.O.; Landberg, G. Cyclin D1 overexpression is a negative predictive factor for tamoxifen response in postmenopausal breast cancer patients. Br. J. Cancer 2004, 90, 1942–1948. [Google Scholar] [CrossRef] [Green Version]
- Bucan, V.; Mandel, K.; Bertram, C.; Lazaridis, A.; Reimers, K.; Park-Simon, T.-W.; Vogt, P.M.; Hass, R. LEF-1 regulates proliferation and MMP-7 transcription in breast cancer cells. Genes Cells 2012, 17, 559–567. [Google Scholar] [CrossRef]
- Bai, X.-Y.; Li, S.; Wang, M.; Li, X.; Yang, Y.; Xu, Z.; Li, B.; Li, Y.; Xia, K.; Chen, H.; et al. Krüppel-like factor 9 down-regulates matrix metalloproteinase 9 transcription and suppresses human breast cancer invasion. Cancer Lett. 2018, 412, 224–235. [Google Scholar] [CrossRef]
- Haines, C.N.; Braunreiter, K.M.; Mo, X.M.; Burd, C.J. GREB1 isoforms regulate proliferation independent of ERα co-regulator activities in breast cancer. Endocr. Relat. Cancer 2018, 25, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Ge, N.; Wang, X.; Sun, W.; Mao, R.; Bu, W.; Creighton, C.J.; Zheng, P.; Vasudevan, S.; An, L.; et al. Amplification and over-expression of MAP3K3 gene in human breast cancer promotes formation and survival of breast cancer cells. J. Pathol. 2014, 232, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, C.A.; Shen, T.; Horwitz, K.B. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc. Natl. Acad. Sci. USA 2000, 97, 1032–1037. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; Horwitz, K.B.; Lange, C.A. Transcriptional Hyperactivity of Human Progesterone Receptors Is Coupled to Their Ligand-Dependent Down-Regulation by Mitogen-Activated Protein Kinase-Dependent Phosphorylation of Serine. Mol. Cell. Biol. 2001, 21, 6122–6131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonard, D.M.; Nawaz, Z.; Smith, C.L.; O’Malley, B.W. The 26S Proteasome Is Required for Estrogen Receptor-α and Coactivator Turnover and for Efficient Estrogen Receptor-α Transactivation. Mol. Cell 2000, 5, 939–948. [Google Scholar] [CrossRef]
- Calvo, V.; Beato, M. BRCA1 Counteracts Progesterone Action by Ubiquitination Leading to Progesterone Receptor Degradation and Epigenetic Silencing of Target Promoters. Cancer Res. 2011, 71, 3422–3431. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.M.; Fan, S.; Pestell, R.G.; Goldberg, I.D. BRCA1 in hormone-responsive cancers. Trends Endocrinol. Metab. 2003, 14, 378–385. [Google Scholar] [CrossRef]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Katiyar, P.; Jones, L.P.; Fan, S.; Zhang, Y.; Furth, P.A.; Rosen, E.M. The Breast Cancer Susceptibility Gene BRCA1 Regulates Progesterone Receptor Signaling in Mammary Epithelial Cells. Mol. Endocrinol. 2006, 20, 14–34. [Google Scholar] [CrossRef]
- Guendel, I.; Carpio, L.; Pedati, C.; Schwartz, A.; Teal, C.; Kashanchi, F.; Kehn-Hall, K. Methylation of the Tumor Suppressor Protein, BRCA1, Influences Its Transcriptional Cofactor Function. PLoS ONE 2010, 5, e11379. [Google Scholar] [CrossRef] [Green Version]
- Montenegro, M.F.; González-Guerrero, R.; Sánchez-Del-Campo, L.; Piñero-Madrona, A.; Cabezas-Herrera, J.; Rodríguez-López, J.N. PRMT1-dependent methylation of BRCA1 contributes to the epigenetic defense of breast cancer cells against ionizing radiation. Sci. Rep. 2020, 10, 13275. [Google Scholar] [CrossRef]
- Knutson, T.P.; Lange, C.A. Tracking progesterone receptor-mediated actions in breast cancer. Pharmacol. Ther. 2014, 142, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Beato, M.; Vicent, G.P. Impact of chromatin structure and dynamics on PR signaling. The initial steps in hormonal gene regulation. Mol. Cell. Endocrinol. 2012, 357, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Hübner, M.R.; Métivier, R.; Brand, H.; Denger, S.; Manu, D.; Beaudouin, J.; Ellenberg, J.; Gannon, F. Cyclic, Proteasome-Mediated Turnover of Unliganded and Liganded ERα on Responsive Promoters Is an Integral Feature of Estrogen Signaling. Mol. Cell 2003, 11, 695–707. [Google Scholar] [CrossRef]
- Zheng, L.; Annab, L.A.; Afshari, C.A.; Lee, W.-H.; Boyer, T.G. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 9587–9592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verde, G.; De Llobet, L.I.; Wright, R.H.; Quilez, J.; Peiró, S.; Le Dily, F.; Beato, M. Unliganded Progesterone Receptor Governs Estrogen Receptor Gene Expression by Regulating DNA Methylation in Breast Cancer Cells. Cancers 2018, 10, 371. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, K.B.; Koseki, Y.; McGuire, W.L. Estrogen Control of Progesterone Receptor in Human Breast Cancer: Role of Estradiol and Antiestrogen. Endocrinology 1978, 103, 1742–1751. [Google Scholar] [CrossRef]
- Kastner, P.; Krust, A.; Turcotte, B.; Stropp, U.; Tora, L.; Gronemeyer, H.; Chambon, P. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990, 9, 1603–1614. [Google Scholar] [CrossRef]
- Rosen, J.M. On Hormone Action in the Mammary Gland. Cold Spring Harb. Perspect. Biol. 2010, 12, a013086. [Google Scholar] [CrossRef] [Green Version]
- Miller, T.W.; Balko, J.M.; Fox, E.M.; Ghazoui, Z.; Dunbier, A.; Anderson, H.; Dowsett, M.; Jiang, A.; Smith, R.A.; Maira, S.-M.; et al. ERα-Dependent E2F Transcription Can Mediate Resistance to Estrogen Deprivation in Human Breast Cancer. Cancer Discov. 2011, 1, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Caizzi, L.; Ferrero, G.; Cutrupi, S.; Cordero, F.; Ballaré, C.; Miano, V.; Reineri, S.; Ricci, L.; Friard, O.; Testori, A.; et al. Genome-wide activity of unliganded estrogen receptor-α in breast cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, 4892–4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, S.; Huard, S.; Brisson, A.; Némati, F.; Dakroub, R.; Poulard, C.; Ye, M.; Martel, E.; Reyes, C.; Silvestre, D.C.; et al. PRMT1 Regulates EGFR and Wnt Signaling Pathways and Is a Promising Target for Combinatorial Treatment of Breast Cancer. Cancers 2022, 14, 306. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-M.; Sun, W.-Z.; Fan, X.-Z.; Xu, Y.-L.; Cheng, M.-B.; Zhang, Y. Methylation of C/EBPα by PRMT1 Inhibits Its Tumor-Suppressive Function in Breast Cancer. Cancer Res. 2019, 79, 2865–2877. [Google Scholar] [CrossRef] [Green Version]
- Kao, J.; Salari, K.; Bocanegra, M.; Choi, Y.-L.; Girard, L.; Gandhi, J.; Kwei, K.A.; Hernandez-Boussard, T.; Wang, P.; Gazdar, A.F.; et al. Molecular Profiling of Breast Cancer Cell Lines Defines Relevant Tumor Models and Provides a Resource for Cancer Gene Discovery. PLoS ONE 2009, 4, e6146. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Kim, T.; Yoo, K.H.; Kang, K. The T47D cell line is an ideal experimental model to elucidate the progesterone-specific effects of a luminal A subtype of breast cancer. Biochem. Biophys. Res. Commun. 2017, 486, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Robin-Lespinasse, Y.; Sentis, S.; Kolytcheff, C.; Rostan, M.-C.; Corbo, L.; Le Romancer, M. hCAF1, a new regulator of PRMT1-dependent arginine methylation. J. Cell Sci. 2007, 120, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulard, C.; Jacquemetton, J.; Pham, T.H.; Le Romancer, M. Using proximity ligation assay to detect protein arginine methylation. Methods 2020, 175, 66–71. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Ref. (Company) | Species | Experiments |
|---|---|---|---|
| Progesterone Receptor (PR) | sc-7208 (SCBT) | Rb | IP/ChIP/IB |
| PR | MA1-12626 (ThermoF) | Ms | PLA/IF |
| PRMT1 | P1620 (Millipore) | Ms | IB/PLA |
| PRMT1 | #07-404 (Millipore) | Rb | IB/PLA |
| PRMT1 | #A300-722A (Bethyl) | Rb | IP/ChIP |
| HP1 gamma (HP1γ) | ab10480 (Abcam) | Rb | IP |
| HP1γ | ab66617 (Abcam) | Rb | IB/PLA |
| LSD1 | sc-53875 (SCBT) | Ms | IB/PLA |
| H3K9me3 | #39161 (ActiveMotif) | Rb | PLA |
| BRCA1 | #OP92 (Millipore) | Ms | PLA |
| Tubulin | T6074 (Sigma) | Ms | IB |
| Target Gene | Experiment | Forward Sequence | Reverse Sequence |
|---|---|---|---|
| (5′–3′) | (5′–3′) | ||
| PRMT1 | qPCR | CGCCTCTTGAAGAAGTGTCCT | GATGCCAAAGTGTGCGTAGG |
| EGFR | GACAGGCCACCTCGTCG | CCGGCTCTCCCGATCAATAC | |
| STAT5A | AAGCCCCACTGGAATGATGG | GGAGTCAAACTTCCAGGCGA | |
| BCL-X | CCATCCACTCTACCCTCCCA | GTGTGGGGGTCTCACAGAA | |
| CCND1 | AAGCTCAAGTGGAACCT | AGGAAGTTGTTGGGGC | |
| GREB1 | CAAAGAATAACCTGTTGGCCCTGC | GACATGCCTGCGCTCTCATACTTA | |
| PDK4 | CATACTCCACTGCACCAACG | AGAAATTGGCAAGCCGTAAC | |
| BRCA1 | GTTGTTATGAAAACAGATGCTGAGTTTGTG | CTGGGTCACCCAGAAATAGCTAAC | |
| 28S | CGATCCATCATCCGCAATG | AGCCAAGCTCAGCGCAAC | |
| EGFR | ChIP | [13] | |
| BCL-X | |||
| STAT5A | |||
| hChr1 (NG) | CGGGGGTCTTTTTGGACCTT | GAAACACGGCTGCCAGAAAC | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malbeteau, L.; Jacquemetton, J.; Languilaire, C.; Corbo, L.; Le Romancer, M.; Poulard, C. PRMT1, a Key Modulator of Unliganded Progesterone Receptor Signaling in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 9509. https://doi.org/10.3390/ijms23179509
Malbeteau L, Jacquemetton J, Languilaire C, Corbo L, Le Romancer M, Poulard C. PRMT1, a Key Modulator of Unliganded Progesterone Receptor Signaling in Breast Cancer. International Journal of Molecular Sciences. 2022; 23(17):9509. https://doi.org/10.3390/ijms23179509
Chicago/Turabian StyleMalbeteau, Lucie, Julien Jacquemetton, Cécile Languilaire, Laura Corbo, Muriel Le Romancer, and Coralie Poulard. 2022. "PRMT1, a Key Modulator of Unliganded Progesterone Receptor Signaling in Breast Cancer" International Journal of Molecular Sciences 23, no. 17: 9509. https://doi.org/10.3390/ijms23179509
APA StyleMalbeteau, L., Jacquemetton, J., Languilaire, C., Corbo, L., Le Romancer, M., & Poulard, C. (2022). PRMT1, a Key Modulator of Unliganded Progesterone Receptor Signaling in Breast Cancer. International Journal of Molecular Sciences, 23(17), 9509. https://doi.org/10.3390/ijms23179509