
Histone Deacetylases Cooperate with NF-κB to Support the Immediate Migratory Response after Zebrafish Pronephros Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
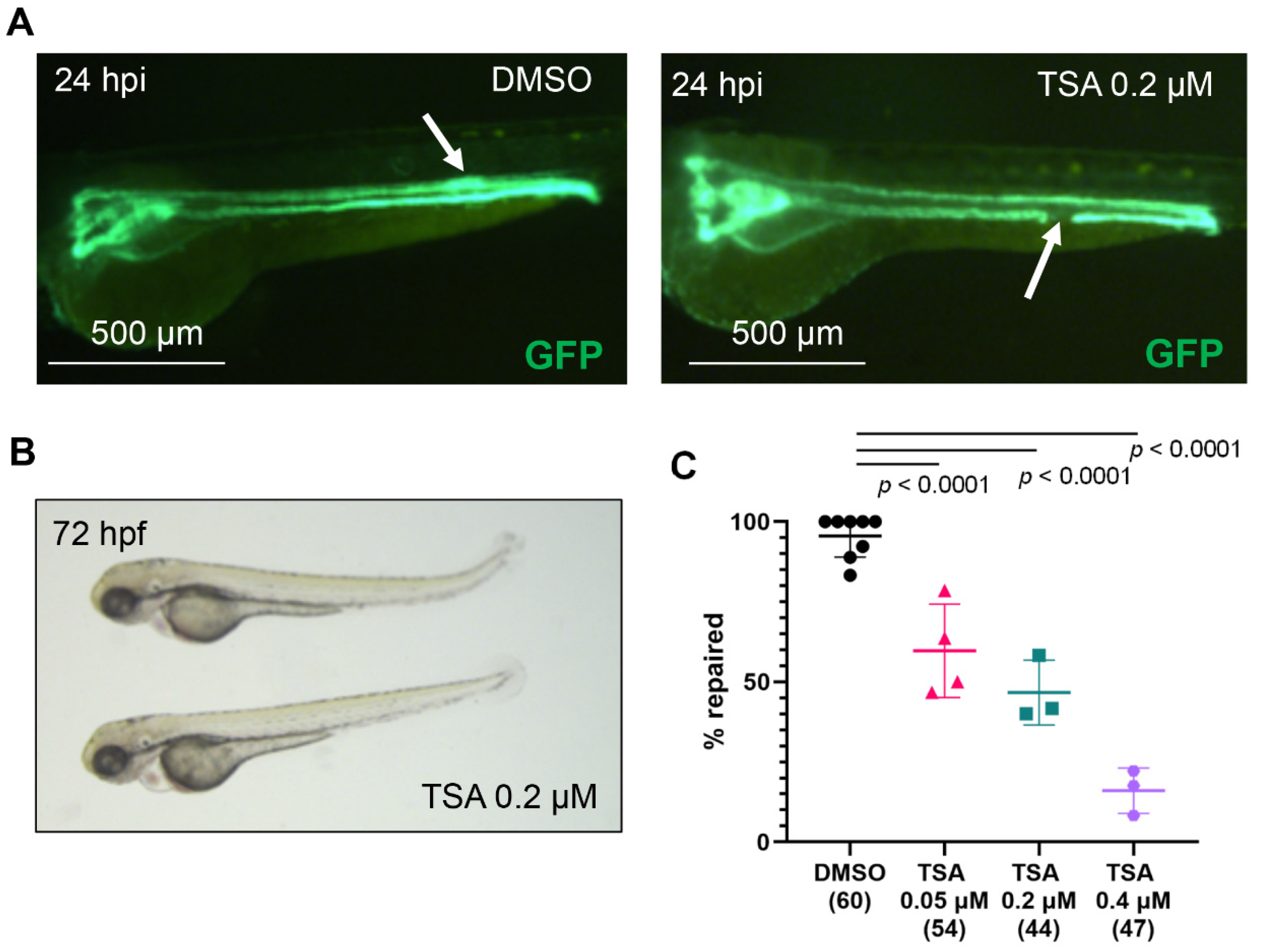
2.1. Histone Deacetylase (Hdac) Activity Is Necessary for Pronephros Repair after Laser-Induced Injury
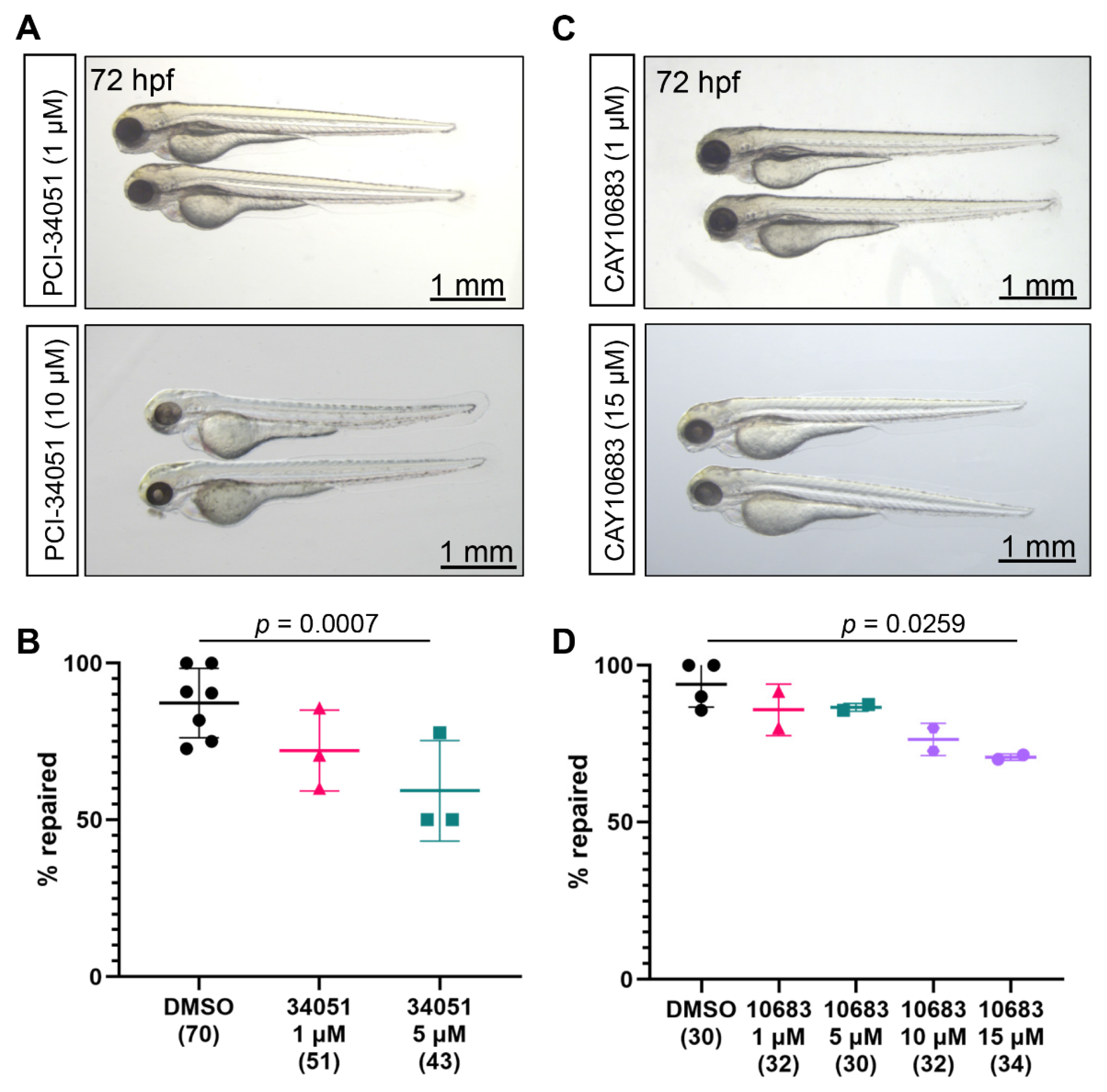
2.2. Role of Class I Hdac in the Zebrafish Pronephros Repair
2.3. Role of Class IIa and Class IIb Hdac in the Zebrafish Pronephros Repair
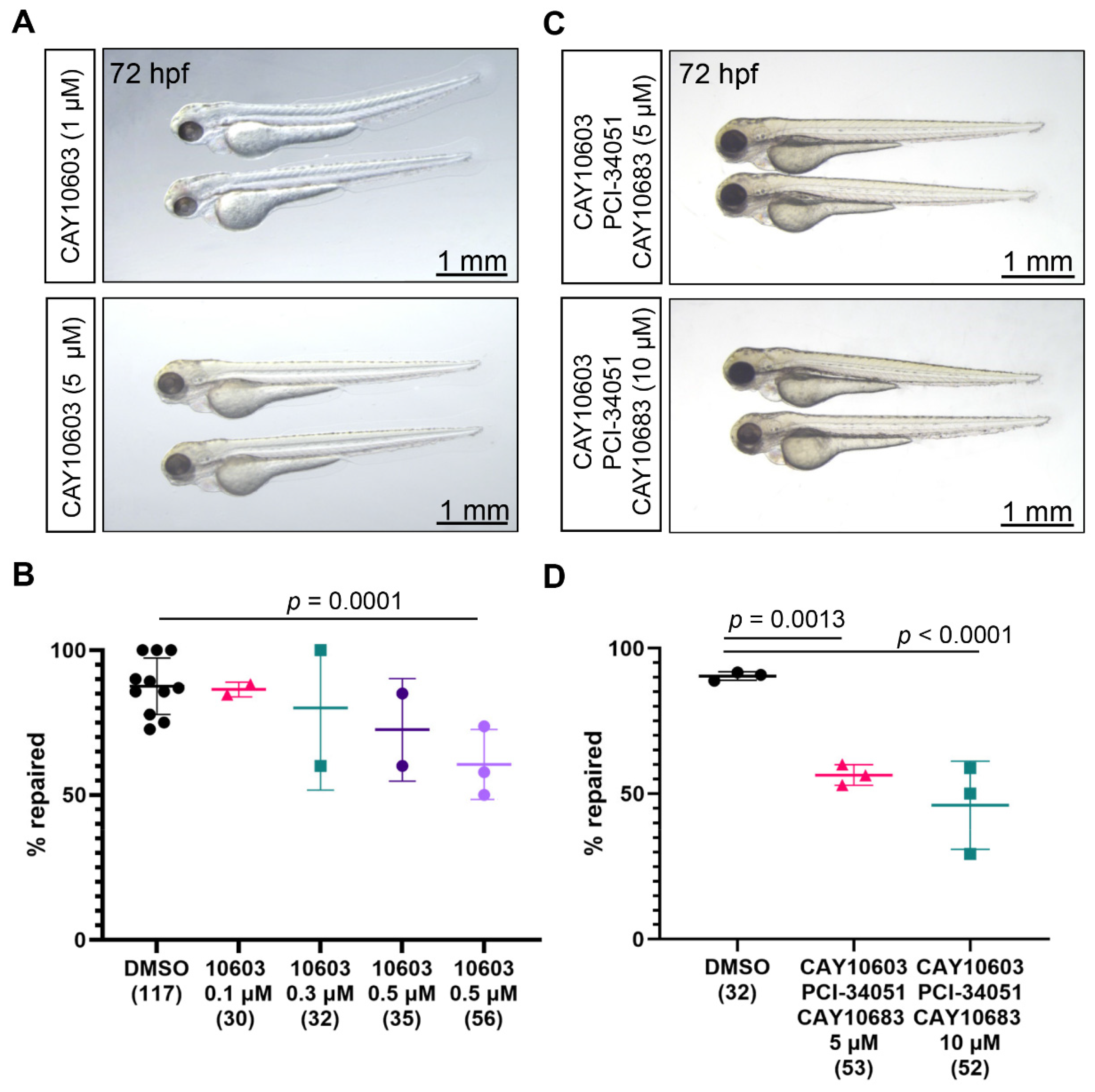
2.4. Combined Inhibition of Hdac2, Hdac6, and Hdac8
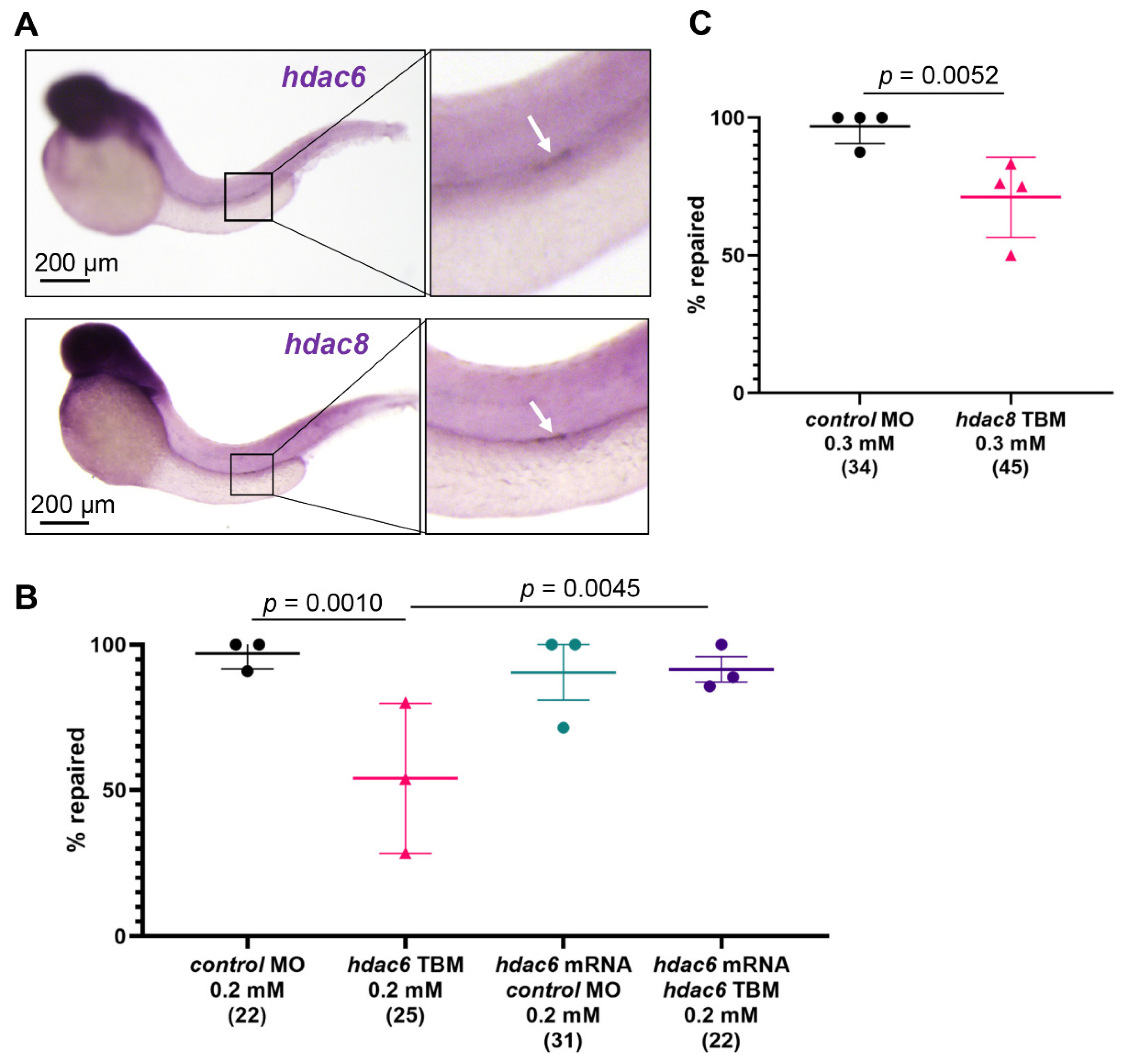
2.5. Hdac6 and Hdac8 Are Necessary for the Pronephros Repair
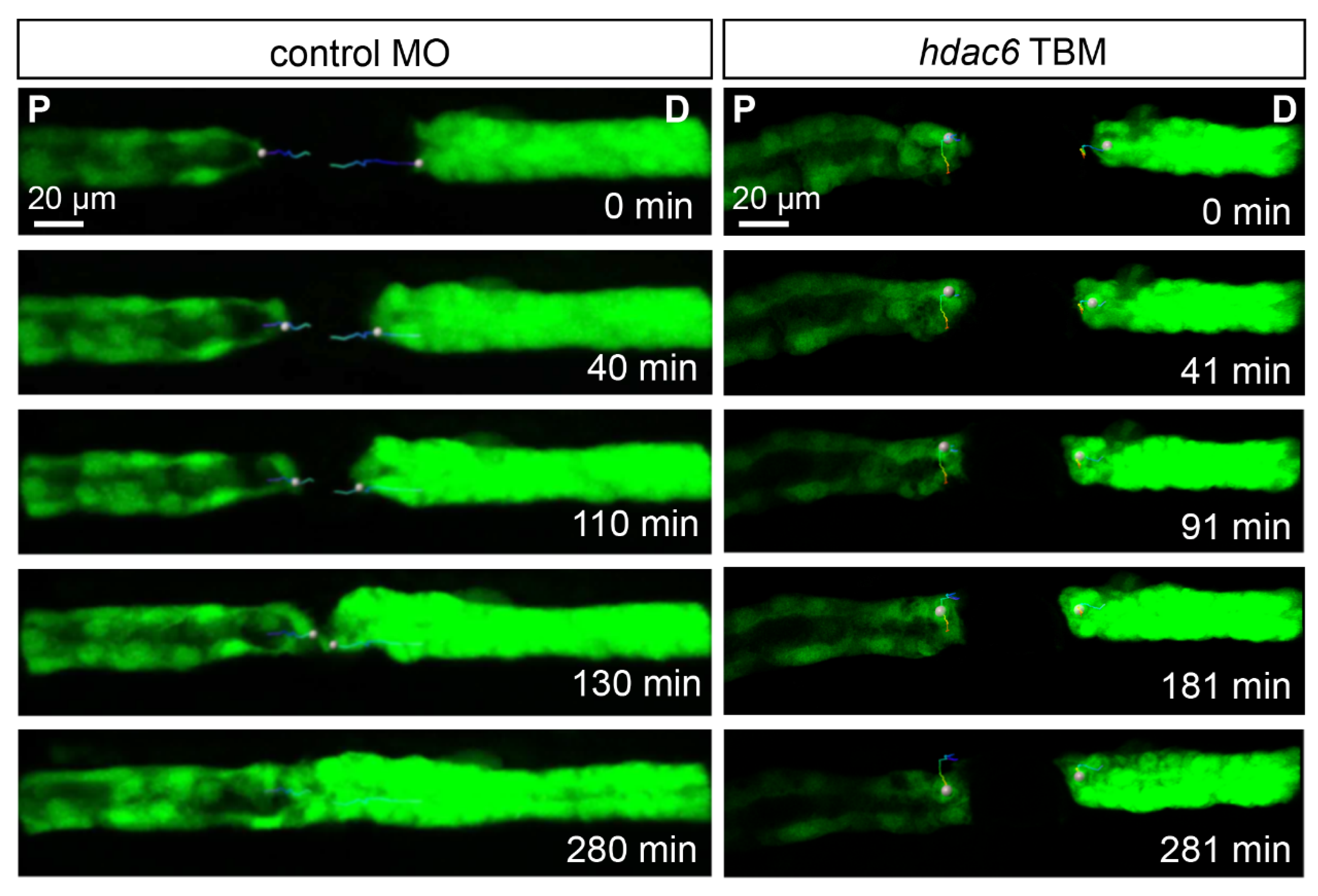
2.6. Depletion of Hdac6 Suppresses the Migratory Response
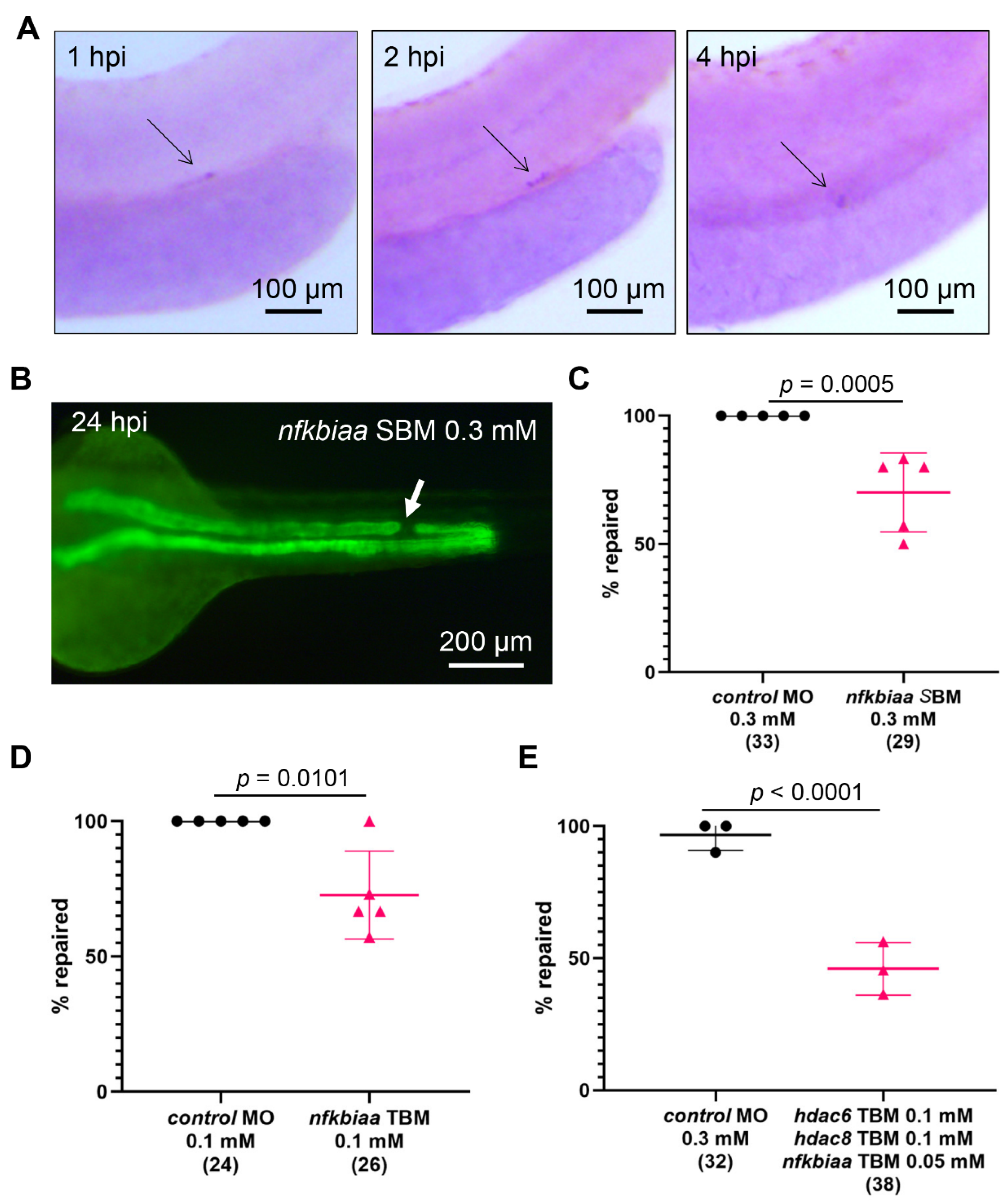
2.7. Cooperation between NF-κB Signaling and Histone Deacetylase Activity Promotes the Repair Process
3. Discussion
4. Materials and Methods
4.1. Zebrafish Lines Maintenance and Pharmacological Treatment
4.2. Photon Laser-Induced Injury, Iepair Quantification, and Image Acquisition
4.3. Whole-Mount In Situ Hybridization (WISH)
| 5′-GACAATATGCGAGCCTTGGG–3′ |
| 5′–GATCCAGGTTCTGCAGGTCT–3′ |
| 5′-TCTGCCCATTCAATTTCACA–3′ |
| 5′-GAAGAAGCGCCACATGTTTT–3′ |
| 5′–TTCCCAAACTCAGAGGATGC–3′ |
| 5′–TGGTCTAGAGAAGGCGGAGA–3′ |
4.4. Morpholino Oligonucleotides (MO)-Mediated Gene Depletion and RT-PCR
| 5′-CTTTGGTATCTGGAACCGCATCCAT-3′ [41]; |
| 5′-ATTACTGTCGCTTTTTTCACTCATT-3′ [42]; |
| 5′-TGCGGCTCTGTGTAAATCCATGTTC-3′ [48]; |
| 5′-CTTTCAGATGTGACTGAACTCACCG-3′ (this study); |
| 5′-CCTTGCCATCATTCACGAGG–3′ |
| 5′-CTTTGCGTCTACATCTGCCC–3′ |
| 5′-ATCTACAAATGCGGTGGAAT–3′ |
| 5′-ATACCAGCCTCAAACTCACC–3′ |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lewington, A.J.P.; Cerdá, J.; Mehta, R.L. Raising Awareness of Acute Kidney Injury: A Global Perspective of a Silent Killer. Kidney Int. 2013, 84, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Cerdá, J.; Burdmann, E.A.; Tonelli, M.; García-García, G.; Jha, V.; Susantitaphong, P.; Rocco, M.; Vanholder, R.; Sever, M.S.; et al. International Society of Nephrology’s 0by25 Initiative for Acute Kidney Injury (Zero Preventable Deaths by 2025): A Human Rights Case for Nephrology. Lancet 2015, 385, 2616–2643. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Czerniak, S.; DiRocco, D.P.; Hasnain, W.; Cheema, R.; Bonventre, J.V. Repair of Injured Proximal Tubule Does Not Involve Specialized Progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9226–9231. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic Epithelial Cells Repair the Kidney after Injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Chang-Panesso, M.; Humphreys, B.D. Cellular Plasticity in Kidney Injury and Repair. Nat. Rev. Nephrol. 2017, 13, 39–46. [Google Scholar] [CrossRef]
- Palmyre, A.; Lee, J.; Ryklin, G.; Camarata, T.; Selig, M.K.; Duchemin, A.-L.; Nowak, P.; Arnaout, M.A.; Drummond, I.A.; Vasilyev, A. Collective Epithelial Migration Drives Kidney Repair after Acute Injury. PLoS ONE 2014, 9, e101304. [Google Scholar] [CrossRef]
- Yakulov, T.A.; Todkar, A.P.; Slanchev, K.; Wiegel, J.; Bona, A.; Groß, M.; Scholz, A.; Hess, I.; Wurditsch, A.; Grahammer, F.; et al. CXCL12 and MYC Control Energy Metabolism to Support Adaptive Responses after Kidney Injury. Nat. Commun. 2018, 9, 3660. [Google Scholar] [CrossRef]
- Gessler, S.; Guthmann, C.; Schuler, V.; Lilienkamp, M.; Walz, G.; Yakulov, T.A. Control of Directed Cell Migration after Tubular Cell Injury by Nucleotide Signaling. Int. J. Mol. Sci. 2022, 23, 7870. [Google Scholar] [CrossRef]
- Tang, J.; Zhuang, S. Epigenetics in Acute Kidney Injury. Curr. Opin. Nephrol. Hypertens. 2015, 24, 351–358. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Sanchez-Niño, M.D.; Valiño-Rivas, L.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting Epigenetic DNA and Histone Modifications to Treat Kidney Disease. Nephrol. Dial. Transplant. 2018, 33, 1875–1886. [Google Scholar] [CrossRef]
- Allison, S.J. Trans-Epigenetic Modulation of Target Genes in Acute Kidney Injury. Nat. Rev. Nephrol. 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S. Epigenetic Targeting for Acute Kidney Injury. Nephrology 2018, 23, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zang, X.; Ponnusamy, M.; Masucci, M.V.; Tolbert, E.; Gong, R.; Zhao, T.C.; Liu, N.; Bayliss, G.; Dworkin, L.D.; et al. Enhancer of Zeste Homolog 2 Inhibition Attenuates Renal Fibrosis by Maintaining Smad7 and Phosphatase and Tensin Homolog Expression. J. Am. Soc. Nephrol. 2016, 27, 2092–2108. [Google Scholar] [CrossRef]
- Wen, L.; Tao, S.-H.; Guo, F.; Li, L.-Z.; Yang, H.-L.; Liang, Y.; Zhang, L.-D.; Ma, L.; Fu, P. Selective EZH2 Inhibitor Zld1039 Alleviates Inflammation in Cisplatin-Induced Acute Kidney Injury Partially by Enhancing RKIP and Suppressing NF-ΚB P65 Pathway. Acta Pharmacol. Sin. 2021, 43, 2067–2080. [Google Scholar] [CrossRef]
- Gao, X.; Peng, Y.; Fang, Z.; Li, L.; Ming, S.; Dong, H.; Li, R.; Zhu, Y.; Zhang, W.; Zhu, B.; et al. Inhibition of EZH2 Ameliorates Hyperoxaluria-Induced Kidney Injury through the JNK/FoxO3a Pathway. Life Sci. 2022, 291, 120258. [Google Scholar] [CrossRef]
- Tang, J.; Yan, Y.; Zhao, T.C.; Bayliss, G.; Yan, H.; Zhuang, S. Class I Histone Deacetylase Activity Is Required for Proliferation of Renal Epithelial Cells. Am. J. Physiol. Renal. Physiol. 2013, 305, F244–F254. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; He, S.; Ma, L.; Ponnusamy, M.; Tang, J.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Yan, H.; Zhuang, S. Blocking the Class I Histone Deacetylase Ameliorates Renal Fibrosis and Inhibits Renal Fibroblast Activation via Modulating TGF-Beta and EGFR Signaling. PLoS ONE 2013, 8, e54001. [Google Scholar] [CrossRef]
- Tang, J.; Shi, Y.; Liu, N.; Xu, L.; Zang, X.; Li, P.; Zhang, J.; Zheng, X.; Qiu, A.; Zhuang, S. Blockade of Histone Deacetylase 6 Protects against Cisplatin-Induced Acute Kidney Injury. Clin. Sci. 2018, 132, 339–359. [Google Scholar] [CrossRef]
- Long, K.; Vaughn, Z.; McDaniels, M.D.; Joyasawal, S.; Przepiorski, A.; Parasky, E.; Sander, V.; Close, D.; Johnston, P.A.; Davidson, A.J.; et al. Validation of HDAC8 Inhibitors as Drug Discovery Starting Points to Treat Acute Kidney Injury. ACS Pharmacol. Transl. Sci. 2022, 5, 207–215. [Google Scholar] [CrossRef]
- Shi, Y.; Xu, L.; Tang, J.; Fang, L.; Ma, S.; Ma, X.; Nie, J.; Pi, X.; Qiu, A.; Zhuang, S.; et al. Inhibition of HDAC6 Protects against Rhabdomyolysis-Induced Acute Kidney Injury. Am. J. Physiol. Renal. Physiol. 2017, 312, F502–F515. [Google Scholar] [CrossRef]
- Liu, J.; Cui, X.; Guo, F.; Li, X.; Li, L.; Pan, J.; Tao, S.; Huang, R.; Feng, Y.; Ma, L.; et al. 2-Methylquinazoline Derivative F7 as a Potent and Selective HDAC6 Inhibitor Protected against Rhabdomyolysis-Induced Acute Kidney Injury. PLoS ONE 2019, 14, e0224158. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, H.; Shi, Y.; Ma, X.; Zhuang, S.; Liu, N. The Role and Mechanism of Histone Deacetylases in Acute Kidney Injury. Front. Pharmacol. 2021, 12, 1449. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Novitskaya, T.; McDermott, L.; Zhang, K.X.; Chiba, T.; Paueksakon, P.; Hukriede, N.A.; de Caestecker, M.P. A PTBA Small Molecule Enhances Recovery and Reduces Postinjury Fibrosis after Aristolochic Acid-Induced Kidney Injury. Am. J. Physiol. Renal. Physiol. 2014, 306, F496–F504. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Huang, R.; Guo, F.; Liang, Y.; Xiang, J.; Lei, S.; Shi, M.; Li, L.; Liu, J.; Feng, Y.; et al. Selective Histone Deacetylase 6 Inhibitor 23BB Alleviated Rhabdomyolysis-Induced Acute Kidney Injury by Regulating Endoplasmic Reticulum Stress and Apoptosis. Front. Pharmacol. 2018, 9, 274. [Google Scholar] [CrossRef]
- Hao, Y.; Guo, F.; Huang, Z.; Feng, Y.; Xia, Z.; Liu, J.; Li, L.; Huang, R.; Lin, L.; Ma, L.; et al. 2-Methylquinazoline Derivative 23BB as a Highly Selective Histone Deacetylase 6 Inhibitor Alleviated Cisplatin-Induced Acute Kidney Injury. Biosci. Rep. 2020, 40, BSR20191538. [Google Scholar] [CrossRef]
- Marumo, T.; Hishikawa, K.; Yoshikawa, M.; Fujita, T. Epigenetic Regulation of BMP7 in the Regenerative Response to Ischemia. J. Am. Soc. Nephrol. 2008, 19, 1311–1320. [Google Scholar] [CrossRef]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 Counteracts TGF-Beta1-Induced Epithelial-to-Mesenchymal Transition and Reverses Chronic Renal Injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Hsing, C.-H.; Lin, C.-F.; So, E.; Sun, D.-P.; Chen, T.-C.; Li, C.-F.; Yeh, C.-H. A2-Adrenoceptor Agonist Dexmedetomidine Protects Septic Acute Kidney Injury through Increasing BMP-7 and Inhibiting HDAC2 and HDAC5. Am. J. Physiol. Renal. Physiol. 2012, 303, F1443–F1453. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. The Functions of E(Z)/EZH2-Mediated Methylation of Lysine 27 in Histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 Are Histone H3K27 Demethylases Involved in HOX Gene Regulation and Development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Cho, Y.-W.; Yu, L.-R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC Domain-Containing UTX and JMJD3 as Histone H3 Lysine 27 Demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef] [PubMed]
- Frederiks, F.; Tzouros, M.; Oudgenoeg, G.; van Welsem, T.; Fornerod, M.; Krijgsveld, J.; van Leeuwen, F. Nonprocessive Methylation by Dot1 Leads to Functional Redundancy of Histone H3K79 Methylation States. Nat. Struct. Mol. Biol. 2008, 15, 550–557. [Google Scholar] [CrossRef]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef]
- Guenther, M.G.; Jenner, R.G.; Chevalier, B.; Nakamura, T.; Croce, C.M.; Canaani, E.; Young, R.A. Global and Hox-Specific Roles for the MLL1 Methyltransferase. Proc. Natl. Acad. Sci. USA 2005, 102, 8603–8608. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Mattevi, A.; Battaglioli, E. Histone Demethylation Catalysed by LSD1 Is a Flavin-Dependent Oxidative Process. FEBS Lett. 2005, 579, 2203–2207. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Kim, J.-S. A Short Guide to Histone Deacetylases Including Recent Progress on Class II Enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Jiao, F.; Li, X.; Zhang, H.; Wang, L.; Gong, Z. The Nephroprotective Effect of MS-275 on Lipopolysaccharide (LPS)-Induced Acute Kidney Injury by Inhibiting Reactive Oxygen Species (ROS)-Oxidative Stress and Endoplasmic Reticulum Stress. Med. Sci. Monit. 2018, 24, 2620–2630. [Google Scholar] [CrossRef]
- Ha, S.-D.; Solomon, O.; Akbari, M.; Sener, A.; Kim, S.O. Histone Deacetylase 8 Protects Human Proximal Tubular Epithelial Cells from Hypoxia-Mimetic Cobalt- and Hypoxia/Reoxygenation-Induced Mitochondrial Fission and Cytotoxicity. Sci. Rep. 2018, 8, 11332. [Google Scholar] [CrossRef]
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.-P.; Boon, R.; Hergenreider, E.; Tjwa, M.; Rössig, L.; Seto, E.; Augustin, H.; et al. Class IIb HDAC6 Regulates Endothelial Cell Migration and Angiogenesis by Deacetylation of Cortactin. EMBO J. 2011, 30, 4142–4156. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-T.; Kathrein, K.L.; Barton, A.; Gitlin, Z.; Huang, Y.-H.; Ward, T.P.; Hofmann, O.; Dibiase, A.; Song, A.; Tyekucheva, S.; et al. A Network of Epigenetic Regulators Guides Developmental Haematopoiesis in Vivo. Nat. Cell. Biol. 2013, 15, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Atalaya, J.P.; Ito, S.; Valor, L.M.; Benito, E.; Barco, A. Genomic Targets, and Histone Acetylation and Gene Expression Profiling of Neural HDAC Inhibition. Nucleic Acids Res. 2013, 41, 8072–8084. [Google Scholar] [CrossRef]
- Furumai, R.; Ito, A.; Ogawa, K.; Maeda, S.; Saito, A.; Nishino, N.; Horinouchi, S.; Yoshida, M. Histone Deacetylase Inhibitors Block Nuclear Factor-ΚB-Dependent Transcription by Interfering with RNA Polymerase II Recruitment. Cancer Sci. 2011, 102, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Imre, G.; Gekeler, V.; Leja, A.; Beckers, T.; Boehm, M. Histone Deacetylase Inhibitors Suppress the Inducibility of Nuclear Factor-KappaB by Tumor Necrosis Factor-Alpha Receptor-1 down-Regulation. Cancer Res. 2006, 66, 5409–5418. [Google Scholar] [CrossRef]
- Es-haghi, M.; Soltanian, S.; Dehghani, H. Perspective: Cooperation of Nanog, NF-ΚΒ, and CXCR4 in a Regulatory Network for Directed Migration of Cancer Stem Cells. Tumor Biol. 2016, 37, 1559–1565. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. TNF-Alpha/NF-KappaB/Snail Pathway in Cancer Cell Migration and Invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef]
- He, Q.; Zhang, C.; Wang, L.; Zhang, P.; Ma, D.; Lv, J.; Liu, F. Inflammatory Signaling Regulates Hematopoietic Stem and Progenitor Cell Emergence in Vertebrates. Blood 2015, 125, 1098–1106. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Chapter Six—Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. In Advances in Cancer Research; Tew, K.D., Fisher, P.B., Eds.; Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2018; Volume 138, pp. 183–211. [Google Scholar]
- Hesham, H.M.; Lasheen, D.S.; Abouzid, K.A.M. Chimeric HDAC Inhibitors: Comprehensive Review on the HDAC-Based Strategies Developed to Combat Cancer. Med. Res. Rev. 2018, 38, 2058–2109. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Li, Z.; Li, N. Epigenetic Modification Drives Acute Kidney Injury-to-Chronic Kidney Disease Progression. Nephron Exp. Nephrol. 2021, 145, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, A.; Kakuki, T.; Ohwada, K.; Kurose, M.; Kondoh, A.; Obata, K.; Nomura, K.; Miyata, R.; Kaneko, Y.; Konno, T.; et al. HDAC Inhibitors Suppress the Proliferation, Migration and Invasiveness of Human Head and Neck Squamous Cell Carcinoma Cells via P63-mediated Tight Junction Molecules and P21-mediated Growth Arrest. Oncol. Rep. 2021, 45, 46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, J.; Wang, H.; Wu, L.; Yuan, W.; Du, J.; Cai, S. Trichostatin A, a Histone Deacetylase Inhibitor, Reverses Epithelial–Mesenchymal Transition in Colorectal Cancer SW480 and Prostate Cancer PC3 Cells. Biochem. Biophys. Res. Commun. 2015, 456, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ali, A.; Khan, S.; Ibrahim, M.; Alshehri, M.A.; Thirupathi, A. Inhibition of HDACs Suppresses Cell Proliferation and Cell Migration of Gastric Cancer by Regulating E2F5 Targeting BCL2. Life 2021, 11, 1425. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-H.; Lee, J.-W.; Lee, J.; Moon, E.-Y. Dynamic Rearrangement of F-Actin Is Required to Maintain the Antitumor Effect of Trichostatin A. PLoS ONE 2014, 9, e97352. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute Kidney Injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Penzo, M.; Habiel, D.M.; Ramadass, M.; Kew, R.R.; Marcu, K.B. Cell Migration to CXCL12 Requires Simultaneous IKKα and IKKβ-Dependent NF-ΚB Signaling. Biochim. Biophys. Acta 2014, 1843, 1796–1804. [Google Scholar] [CrossRef]
- Kew, R.R.; Penzo, M.; Habiel, D.M.; Marcu, K.B. The IKKα-Dependent NF-ΚB P52/RelB Noncanonical Pathway Is Essential to Sustain a CXCL12 Autocrine Loop in Cells Migrating in Response to HMGB1. J. Immunol. 2012, 188, 2380–2386. [Google Scholar] [CrossRef]
- Marcet, B.; Horckmans, M.; Libert, F.; Hassid, S.; Boeynaems, J.-M.; Communi, D. Extracellular Nucleotides Regulate CCL20 Release from Human Primary Airway Epithelial Cells, Monocytes and Monocyte-Derived Dendritic Cells. J. Cell. Physiol. 2007, 211, 716–727. [Google Scholar] [CrossRef]
- Beg, A.A. Endogenous Ligands of Toll-like Receptors: Implications for Regulating Inflammatory and Immune Responses. Trends Immunol. 2002, 23, 509–512. [Google Scholar] [CrossRef]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 Activation Mediates Kidney Ischemia/Reperfusion Injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef] [PubMed]
- Bosch, X.; Poch, E.; Grau, J.M. Rhabdomyolysis and Acute Kidney Injury. N. Engl. J. Med. 2009, 361, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Bai, Y.; Jayne, L.A.; Cianciolo, R.E.; Bajwa, A.; Pabla, N.S. Involvement of the CDKL5-SOX9 Signaling Axis in Rhabdomyolysis-Associated Acute Kidney Injury. Am. J. Physiol. Renal. Physiol. 2020, 319, F920–F929. [Google Scholar] [CrossRef] [PubMed]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio); University of Oregon: Eugene, OR, USA, 2007. [Google Scholar]
- Schoels, M.; Zhuang, M.; Fahrner, A.; Küchlin, S.; Sagar; Franz, H.; Schmitt, A.; Walz, G.; Yakulov, T.A. Single-Cell MRNA Profiling Reveals Changes in Solute Carrier Expression and Suggests a Metabolic Switch during Zebrafish Pronephros Development. Am. J. Physiol. Renal. Physiol. 2021, 320, F826–F837. [Google Scholar] [CrossRef]
- Robu, M.E.; Larson, J.D.; Nasevicius, A.; Beiraghi, S.; Brenner, C.; Farber, S.A.; Ekker, S.C. P53 Activation by Knockdown Technologies. PLoS Genet. 2007, 3, e78. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuang, M.; Scholz, A.; Walz, G.; Yakulov, T.A. Histone Deacetylases Cooperate with NF-κB to Support the Immediate Migratory Response after Zebrafish Pronephros Injury. Int. J. Mol. Sci. 2022, 23, 9582. https://doi.org/10.3390/ijms23179582
Zhuang M, Scholz A, Walz G, Yakulov TA. Histone Deacetylases Cooperate with NF-κB to Support the Immediate Migratory Response after Zebrafish Pronephros Injury. International Journal of Molecular Sciences. 2022; 23(17):9582. https://doi.org/10.3390/ijms23179582
Chicago/Turabian StyleZhuang, Mingyue, Alexander Scholz, Gerd Walz, and Toma Antonov Yakulov. 2022. "Histone Deacetylases Cooperate with NF-κB to Support the Immediate Migratory Response after Zebrafish Pronephros Injury" International Journal of Molecular Sciences 23, no. 17: 9582. https://doi.org/10.3390/ijms23179582
APA StyleZhuang, M., Scholz, A., Walz, G., & Yakulov, T. A. (2022). Histone Deacetylases Cooperate with NF-κB to Support the Immediate Migratory Response after Zebrafish Pronephros Injury. International Journal of Molecular Sciences, 23(17), 9582. https://doi.org/10.3390/ijms23179582