
Expression of the C-Terminal Domain of Phospholipase Cβ3 Inhibits Signaling via Gαq-Coupled Receptors and Transient Receptor Potential Channels

Abstract
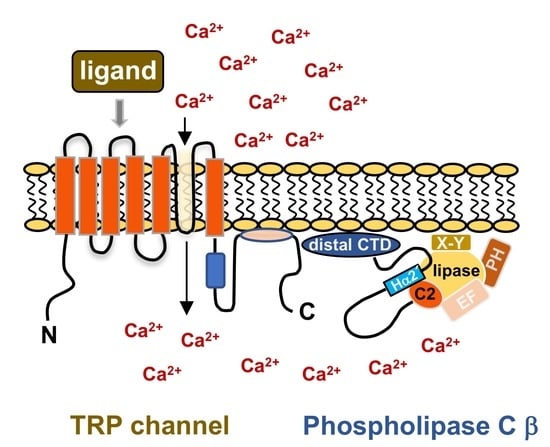
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Expression of the C-Terminal Domain of Phospholipase Cβ3 Blocks Signaling Induced by Stimulation of a Gαq-Coupled Designer Receptor
2.2. Expression of the C-Terminal Domain of Phospholipase Cβ3 Blocks Phosphorylation of Extracellular Signal-Regulated Protein Kinase (ERK1/2) after Stimulation of a Gαq-Coupled Designer Receptor
2.3. Expression of Membrane-Tagged Cyan Fluorescence Protein Containing the Helix-Turn-Helix Motif (Hα1—Hα2) of PLCβ3 Has No Effect on Downstream Gαq Signaling
2.4. Expression of the C-Terminal Domain of Phospholipase Cβ1 Blocks Signaling Induced by Stimulation of a Gαq-Coupled Designer Receptor
2.5. Expression of the C-Terminal Domain of Phospholipase Cβ3 Blocks Intracellular Signaling Triggered by Transient Receptor Potential (TRP) M8 Channel Stimulation
2.6. Expression of the C-Terminal Domain of Phospholipase Cβ3 Blocks Intracellular Signaling Induced by Stimulation of TRPM3 Channels
2.7. The Benzenesulfonamide m-3M3FBS Blocks TRPM3 and TRPM8 Signaling
2.8. The Compound m-3M3FBS Has No Effect on B-Raf-Induced Signal Transduction
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Lentiviral Gene Transfer
4.3. Reporter Gene Assay
4.4. Western Blots
4.5. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nilius, B.; Szallasi, A. A Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacol. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef] [PubMed]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.-C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Badheka, D.; Borbiro, I.; Rohacs, T. Transient receptor potential melastatin 3 is a phosphoinositide-dependent ion channel. J. Gen. Physiol. 2015, 146, 65–77. [Google Scholar] [CrossRef]
- Tóth, B.I.; Konrad, M.; Ghosh, D.; Mohr, F.; Halaszovich, C.R.; Leitner, M.G.; Vriens, J.; Oberwinkler, J.; Voets, T. Regulation of the transient receptor potential channel TRPM3 by phosphoinositides. J. Gen. Physiol. 2015, 146, 51–63. [Google Scholar] [CrossRef]
- Yudin, Y.; Lukacs, V.; Cao, C.; Rohács, T. Decrease in phosphatidylinositol 4,5-bisphosphate levels mediates desensitization of the cold sensor TRPM8 channels. J. Physiol. 2011, 589, 6007–6027. [Google Scholar] [CrossRef]
- Liu, B.; Qin, F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005, 25, 1674–1681. [Google Scholar] [CrossRef]
- Rohács, T.; Lopez, C.M.B.; Michailidis, I.; Logothetis, D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels though the TRP domain. Nat. Neurosci. 2005, 8, 626–634. [Google Scholar] [CrossRef]
- Wagner, T.F.J.; Loch, S.; Lambert, S.; Straub, I.; Mannebach, S.; Mathar, I.; Düfer, M.; Lis, A.; Flockerzi, V.; Philipp, S.E.; et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat. Cell Biol. 2008, 10, 1421–1430. [Google Scholar] [CrossRef]
- Lesch, A.; Rubil, S.; Thiel, G. Activation and inhibition of transient receptor potential TRPM3-induced gene transcription. Br. J. Pharmacol. 2014, 171, 2645–2658. [Google Scholar] [CrossRef]
- Thiel, G.; Rubil, S.; Lesch, A.; Guethlein, L.A.; Rössler, O.G. Transient receptor potential TRPM3 channels: Pharmacology, signaling, and biological functions. Pharmacol. Res. 2017, 124, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Vandewauw, I.; de Clercq, K.; Mulier, M.; Held, K.; Pinto, S.; van Ranst, N.; Segal, A.; Voet, T.; Vennekens, R.; Zimmermann, K.; et al. A TRP channel trio mediates acute noxious heat sensing. Nature 2018, 555, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Almaraz, L.; Manenschijn, J.-A.; de la Peña, E.; Viana, F. TRPM8. In Mammalian Transient Receptor Potential (TRP) Cation Channels; Handbook of Experimental Pharmacology; Nilius, B., Flockerzi, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 222, pp. 547–579. [Google Scholar]
- Colburn, R.W.; Lubin, M.L.; Stone, D.J., Jr.; Wang, Y.; Lawrence, D.; D´Andrea, M.R.; Brandt, M.R.; Liu, Y.; Flores, C.M.; Qin, N. Attenuated cold sensitivity in TRPM8 null mice. Neuron 2007, 54, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, A.; Murray, A.N.; Mathur, J.; Earley, T.J.; Petrus, M.J.A. Patapoutian, TRPM8 is required for cold sensation in mice. Neuron 2007, 54, 371–378. [Google Scholar] [CrossRef]
- Wills, R.C.; Goulden, B.D.; Hammond, G.R.V. Genetically encoded lipid biosensors. Mol. Biol. Cell 2018, 29, 1526–1532. [Google Scholar]
- Kang, J.K.; Kim, O.-H.; Hur, J.; Yu, S.H.; Lamichhane, S.; Lee, J.W.; Ojha, U.; Hong, J.H.; Lee, C.S.; Cha, J.-Y.; et al. Increased intracellular Ca2+ concentrations prevent membrane localization of PH domains through the formation of Ca2+-phosphoinositides. Proc. Natl. Acad. Sci. USA 2017, 114, 11926–11931. [Google Scholar] [CrossRef]
- Mogami, H.; Mills, C.L.; Gallacher, D.V. Phospholipase C inhibitor, U73122, releases intracellular Ca2+, potentiates Ins(1,4,5)P3-mediated Ca2+ release and directly activates ion channels in mouse pancreatic acinar cells. Biochem. J. 1997, 324, 645–651. [Google Scholar] [CrossRef]
- Berven, L.A.; Barritt, G.J. Evidence obtained using single hepatocytes for inhibition by the phospholipase C inhibitor U73122 of store-operated Ca2+ inflow. Biochem. Pharmacol. 1995, 49, 1373–1379. [Google Scholar] [CrossRef]
- Macmillan, D.; McCarron, J.G. The phospholipase C inhibitor U-73122 inhibits Ca2+ release from the intracellular sarcoplasmic reticulum Ca2+ store by inhibiting Ca2+ pumps in smooth muscle. Brit. J. Pharmacol. 2010, 160, 1295–1301. [Google Scholar] [CrossRef]
- Klose, A.; Huth, T.; Alzheimer, C. 1-[6-[[(17beta)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122) selectively inhibits Kir3 and BK channels in a phospholipase C-independent fashion. Mol. Pharmacol. 2008, 74, 1203–1214. [Google Scholar] [CrossRef]
- Leitner, M.G.; Michel, N.; Behrendt, M.; Dierich, M.; Dembla, S.; Wilke, B.U.; Konrad, M.; Lindner, M.; Oberwinkler, J.; Oliver, D. Direct modulation of TRPM4 and TRPM3 channels by the phospholipase C inhibitor U73122. Brit. J. Pharmacol. 2016, 173, 2555–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, B.N.; Hyun, S.-H.; Thompson, D.H.; Lyon, A.M. Phospholipase Cβ3 membrane adsorption and activation are regulated by its C-terminal domains and phosphatidylinositol 4,5-bisphosphate. Biochemistry 2017, 56, 5604–5614. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Keim, A.; Thiel, G. Regulation of immediate-early gene transcription following activation of Gαq-coupled designer receptors. J. Cell. Biochem. 2013, 114, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Kaufmann, A.; Rössler, O.G. G-protein-coupled designer receptors—New chemical-genetic tools for signal transduction research. Biol. Chem. 2013, 394, 1615–1622. [Google Scholar] [CrossRef]
- Urban, D.J.; Roth, B.L. DREADDs (designer receptors exclusively activated by designer drugs): Chemogenetic tools with therapeutic utility. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 399–417. [Google Scholar] [CrossRef]
- Thiel, G.; Backes, T.M.; Guethlein, L.A.; Rössler, O.G. Chromatin-embedded reporter genes: Quantification of stimulus-induced gene transcription. Gene 2021, 787, 145645. [Google Scholar] [CrossRef]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G protein. Nat. Commun. 2018, 9, 341. [Google Scholar] [CrossRef]
- Marinissen, M.J.; Gutkind, J.S. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Nakajima, K.; Jain, S.; de Azua, I.R.; McMillin, S.M.; Rossi, M.; Wess, J. Novel aspects of M3 muscarinic receptor signaling in pancreatic β-cells. Mol. Endocrinol. 2013, 27, 1208–1216. [Google Scholar] [CrossRef]
- Waldo, G.L.; Ricks, T.K.; Hicks, S.N.; Cheever, M.L.; Kawano, T.; Tsuboi, K.; Wang, X.; Montell, C.; Kozasa, T.; Sondek, J.; et al. Kinetic scaffolding mediated by a phospholipase C-β and Gq signaling complex. Science 2010, 330, 974–980. [Google Scholar] [CrossRef]
- Charpentier, T.H.; Waldo, G.L.; Lowery-Gionta, E.G.; Krajewski, K.; Strahl, B.D.; Kash, T.L.; Harden, T.H.; Sondek, J. Potent and selective peptide-based inhibition of the G protein Gq. J. Biol. Chem. 2016, 291, 25608–25616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adjobo-Hermans, M.J.W.; Crosby, K.C.; Putyrski, M.; Bhageloe, A.; van Weeren, L.; Schultz, C.; Goedhart, J.; Gadella, T.W.J., Jr. PLCβ isoforms differ in their subcellular location and their CT-domain dependent interaction with Gαq. Cell. Signal. 2013, 25, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Jhon, D.-Y.; Lee, H.-H.; Park, D.; Lee, C.-W.; Lee, K.-H.; Yoo, O.J.; Rhee, S.G. Cloning, sequencing, purification, and Gq-dependent activation of phospholipase C-β3. J. Biol. Chem. 1993, 268, 6654–6661. [Google Scholar] [CrossRef]
- Lyon, A.M.; Tesmer, J.J. Structural insights into phospholipase C-β function. Mol. Pharmacol. 2013, 84, 488–500. [Google Scholar] [CrossRef]
- Rohács, T. Regulation of transient receptor potential channels by the phospholipase C pathway. Adv. Biol. Reg. 2013, 53, 341–355. [Google Scholar] [CrossRef]
- Thiel, G.; Welck, J.; Wissenbach, U.; Rössler, O.G. Dihydrotestosterone activates AP-1 in LNCaP prostate cancer cells. Int. J. Biochem. Cell Biol. 2019, 110, 9–20. [Google Scholar] [CrossRef]
- Bae, Y.-S.; Lee, T.G.; Park, J.C.; Hur, J.H.; Kim, Y.; Heo, K.; Kwak, J.-Y.; Suh, P.-G.; Ryu, S.H. Identification of a compound that directly stimulates phospholipase C activity. Mol. Pharmacol. 2003, 63, 1043–1050. [Google Scholar] [CrossRef]
- Lyon, A.M.; Tesmer, V.M.; Dhamsania, V.D.; Thal, D.M.; Gutierrez, J.; Chowdhury, S.; Suddala, K.C.; Northup, J.K.; Tesmer, J.J.G. An autoinhibitory helix in the C-terminal region of phospholipase C-β mediates Gαq activation, Nature Struct. Mol. Biol. 2011, 18, 999–1005. [Google Scholar]
- Lyon, A.M.; Begley, J.A.; Manett, T.D.; Tesmer, J.J.G. Molecular mechanisms of phospholipase C β3 autoinhibition. Structure 2014, 22, 1844–1854. [Google Scholar] [CrossRef]
- Schnabel, P.; Camps, M.; Carozzi, A.; Parker, P.J.; Gierschik, P. Mutational analysis of phospholipase C-β2. Identification of regions required for membrane association and stimulation by guanine-nucleotide-binding protein βγ subunits. Eur. J. Biochem. 1993, 217, 1109–1115. [Google Scholar] [CrossRef]
- Jenco, J.M.; Becker, K.P.; Morris, A.J. Membrane-binding properties of phospholipase C-β1 and phospholipase C-β2: Role of the C-terminus and effects of polyphosphoinositides, G proteins and Ca2+. Biochem. J. 1997, 327, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Ilkaeva, O.; Kinch, L.N.; Paulssen, R.H.; Ross, E.M. Mutations in the carboxy-terminal domain of phospholipase Cβ-1 delineate the dimer interface and a potential Gαq interaction site. J. Biol. Chem. 2002, 277, 4294–4300. [Google Scholar] [CrossRef] [Green Version]
- Lyon, A.M.; Dutta, S.; Boguth, C.A.; Skiniotis, G.; Tesmer, J.J. Full-length Gαq-phospholipase C-β3 structure reveals interfaces of the C-terminal coiled-coil domain. Nat. Struct. Mol. Biol. 2013, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Melliti, K.; Meza, U.; Adams, B. Muscarinic stimulation of α1E Ca channels is selectively blocked by the effector antagonist function of RGS2 and phospholipase C-β1. J. Neurosci. 2000, 20, 7167–7173. [Google Scholar] [CrossRef]
- Varnai, P.; Thyagarajan, B.; Rohacs, T.; Balla, T. Rapid inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J. Cell Biol. 2006, 175, 377–382. [Google Scholar] [CrossRef]
- Daniels, R.L.; Takashima, Y.; McKemy, D.D. activity of the neuronal cold sensor TRPM8 is regulated by phospholipase C via the phospholipid phosphoinositol 4,5-bisphosphate. J. Biol. Chem. 2009, 284, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Fujita, F.; Uchida, K.; Takaishi, M.; Sokabe, T.; Tominaga, M. Ambient temperature affects the temperature threshold for TRPM8 activation through interaction of phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2013, 33, 6154–6159. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, R.M.; Magoski, N.S. A closely associated phospholipase C regulates cation channel function through phosphoinositide hydrolysis. J. Neurosci. 2018, 38, 7622–7634. [Google Scholar] [CrossRef]
- Krjukova, J.; Holmqvist, T.; Danis, A.S.; Åkerman, K.E.O.; Kukkonen, J.P. Phospholipase C activator m-3M3FBS affects Ca2+ homeostasis independently of phospholipase C activation. Brit. J. Pharmacol. 2004, 143, 3–7. [Google Scholar] [CrossRef]
- Bödding, M.; Wissenbach, U.; Flockerzi, V. Characterization of TRPM8 as a pharmacophore receptor. Cell Calcium 2007, 42, 618–628. [Google Scholar] [CrossRef]
- Al-Sarraj, A.; Day, R.M.; Thiel, G. Specificity of transcriptional regulation by the zinc finger transcription factor Sp1, Sp3, and Egr-1. J. Cell. Biochem. 2005, 94, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Rössler, O.G.; Thiel, G. Regulation of gene transcription following stimulation of Gαq-coupled designer receptors. In Designer Receptors Exclusively Activated by Designer Drugs; Thiel, G., Ed.; Springer: New York, NY, USA; Humana Press, Neuromethods: Totowa, NJ, USA, 2015; Volume 108, pp. 49–60. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thiel, G.; Rössler, O.G. Expression of the C-Terminal Domain of Phospholipase Cβ3 Inhibits Signaling via Gαq-Coupled Receptors and Transient Receptor Potential Channels. Int. J. Mol. Sci. 2022, 23, 9590. https://doi.org/10.3390/ijms23179590
Thiel G, Rössler OG. Expression of the C-Terminal Domain of Phospholipase Cβ3 Inhibits Signaling via Gαq-Coupled Receptors and Transient Receptor Potential Channels. International Journal of Molecular Sciences. 2022; 23(17):9590. https://doi.org/10.3390/ijms23179590
Chicago/Turabian StyleThiel, Gerald, and Oliver G. Rössler. 2022. "Expression of the C-Terminal Domain of Phospholipase Cβ3 Inhibits Signaling via Gαq-Coupled Receptors and Transient Receptor Potential Channels" International Journal of Molecular Sciences 23, no. 17: 9590. https://doi.org/10.3390/ijms23179590