
RNA Editing Enzyme ADAR1 Regulates METTL3 in an Editing Dependent Manner to Promote Breast Cancer Progression via METTL3/ARHGAP5/YTHDF1 Axis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
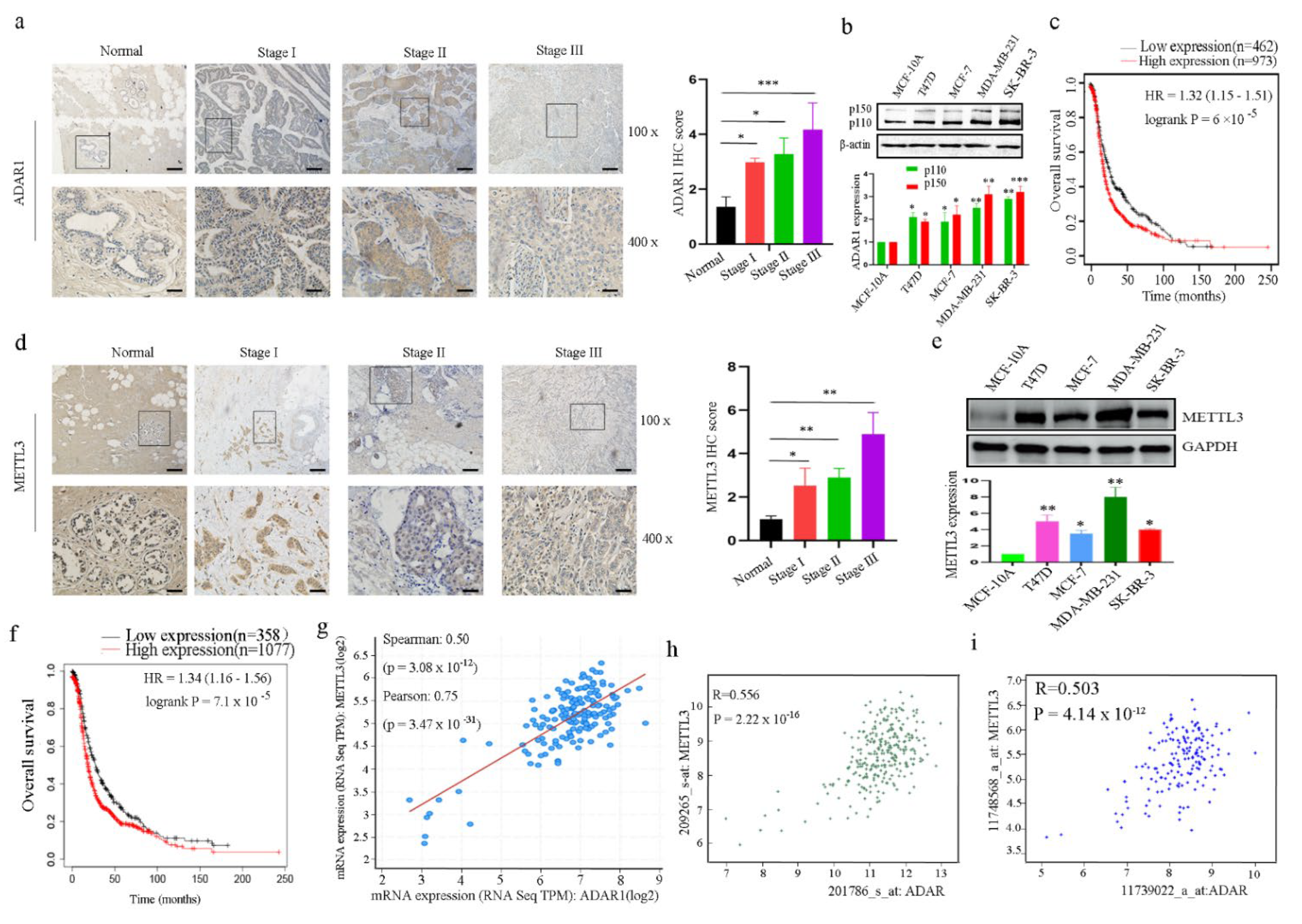
2.1. ADAR1 and METTL3 Proteins Are Upregulated in Both Breast Cancer Tissues and Cell Lines
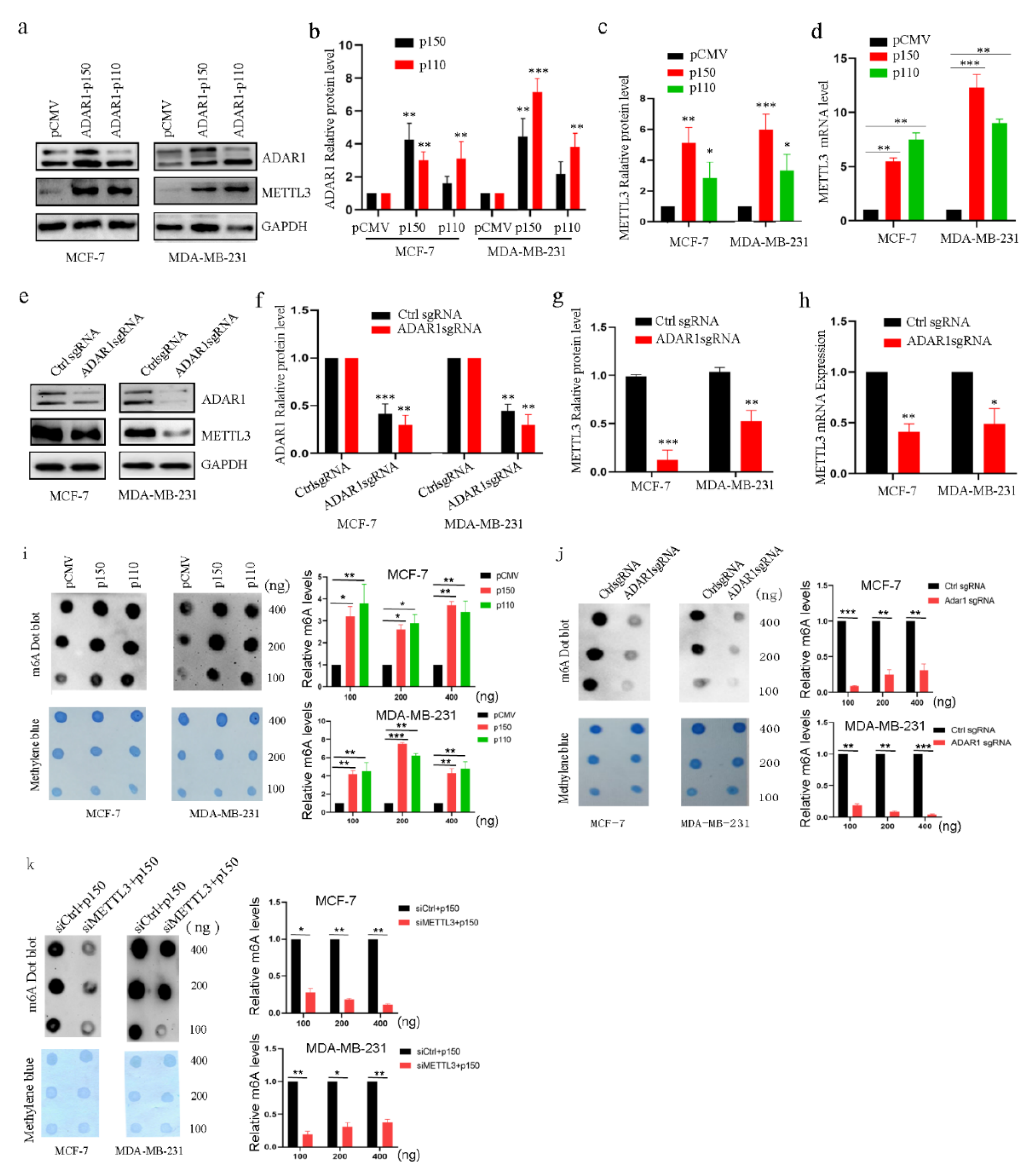
2.2. ADAR1 Upregulates METTL3 and Increases mRNA m6A Levels in Breast Cancer Cell Lines
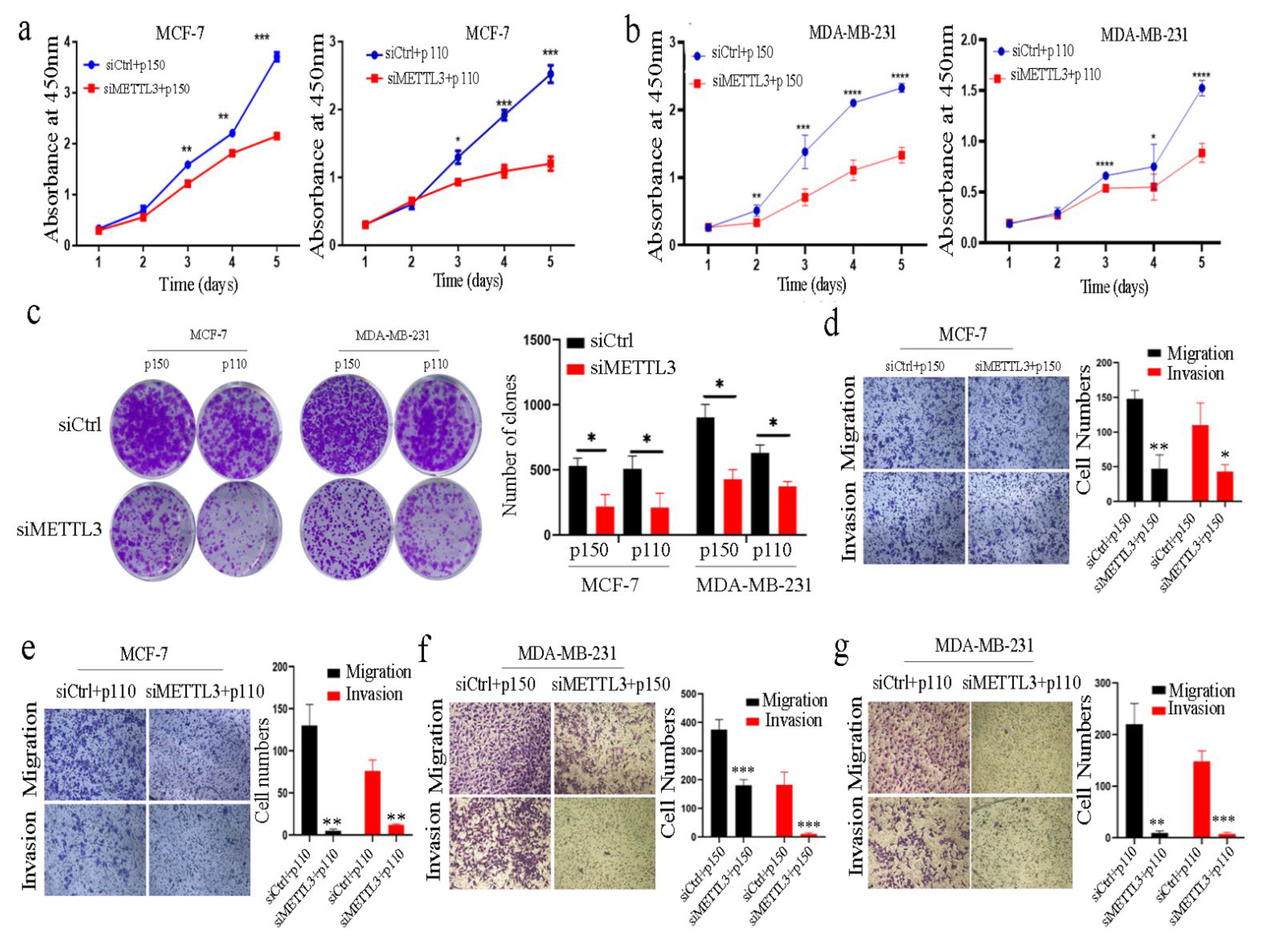
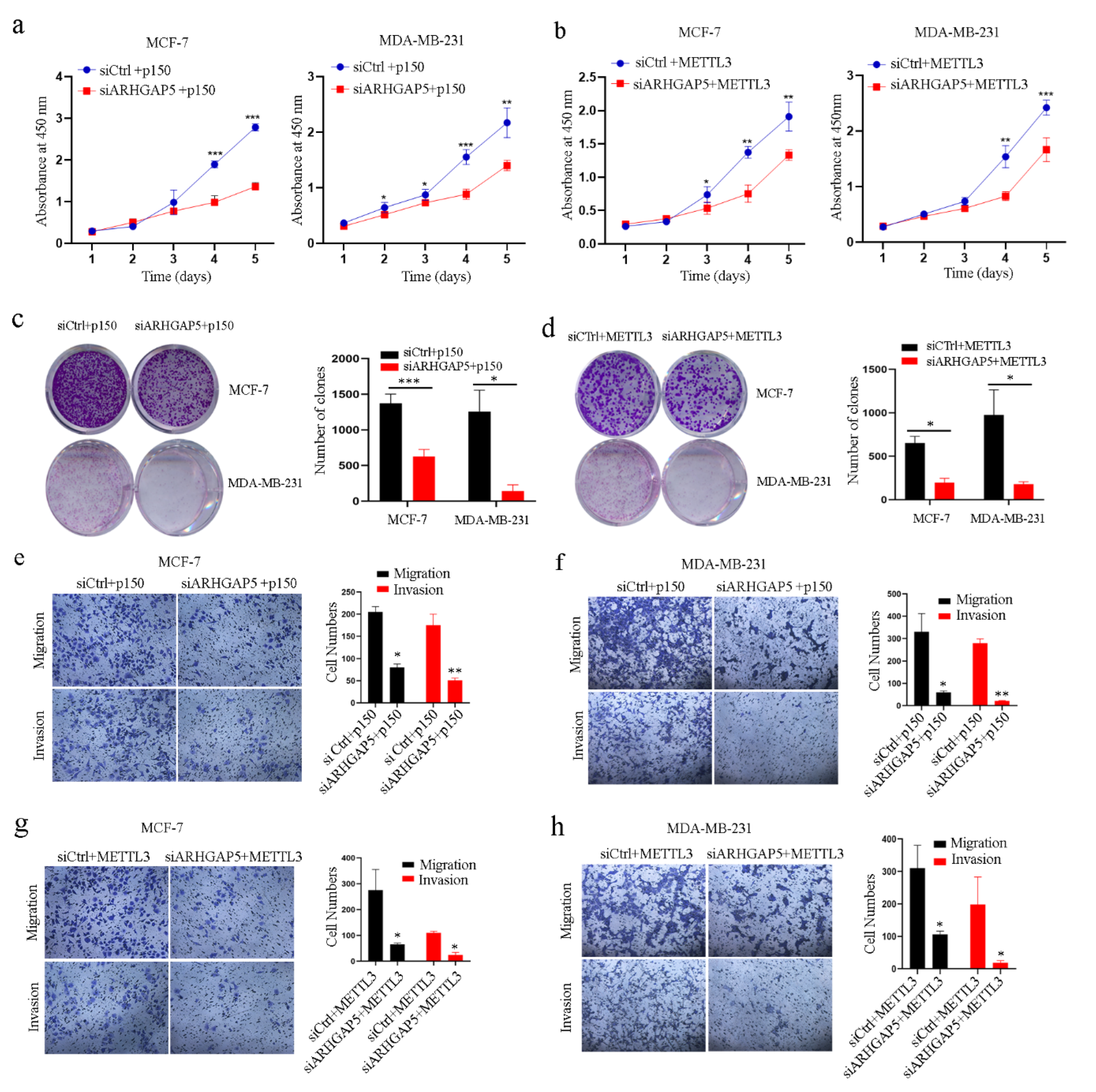
2.3. ADAR1 Promotes the Progression of Breast Cancer through METTL3
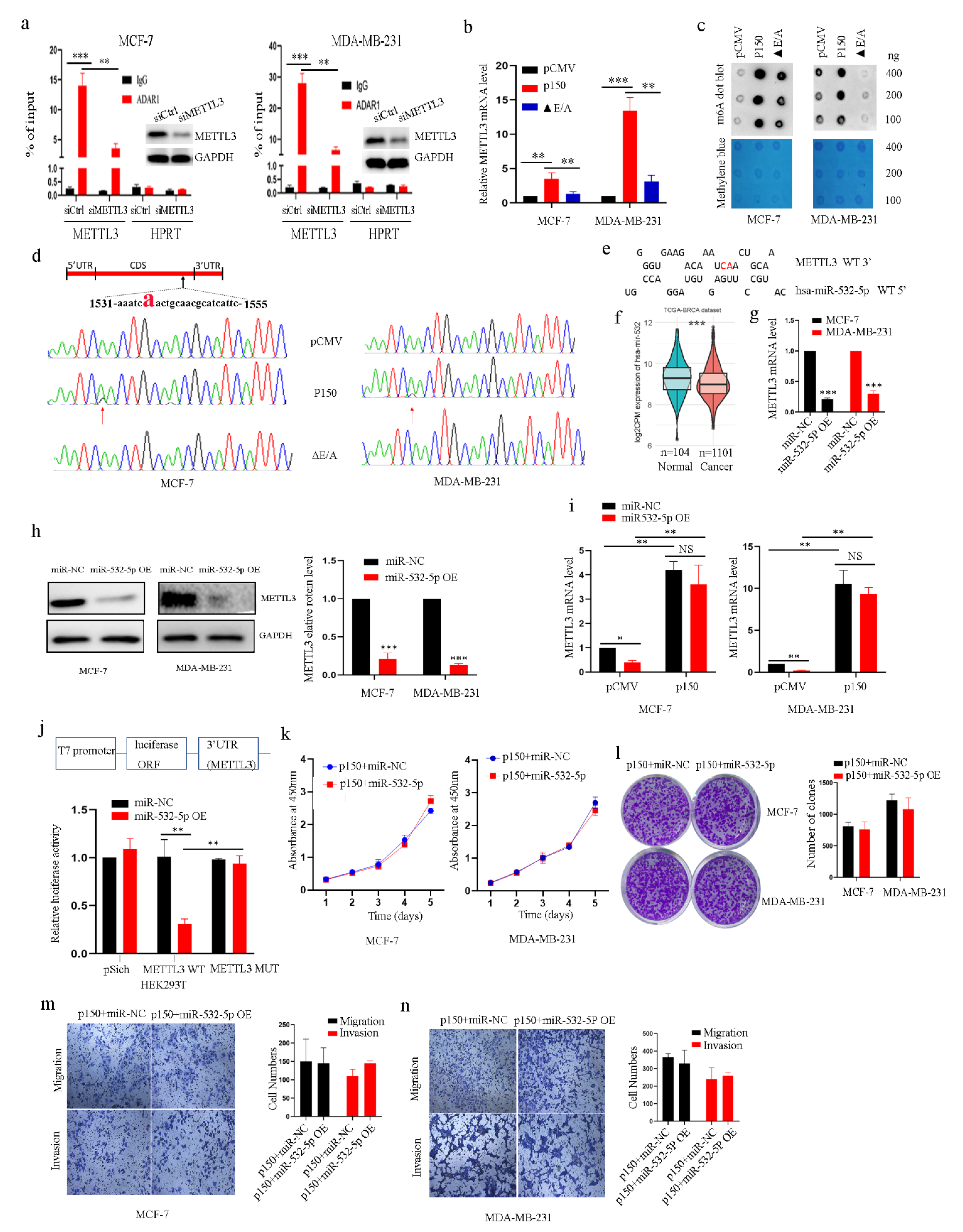
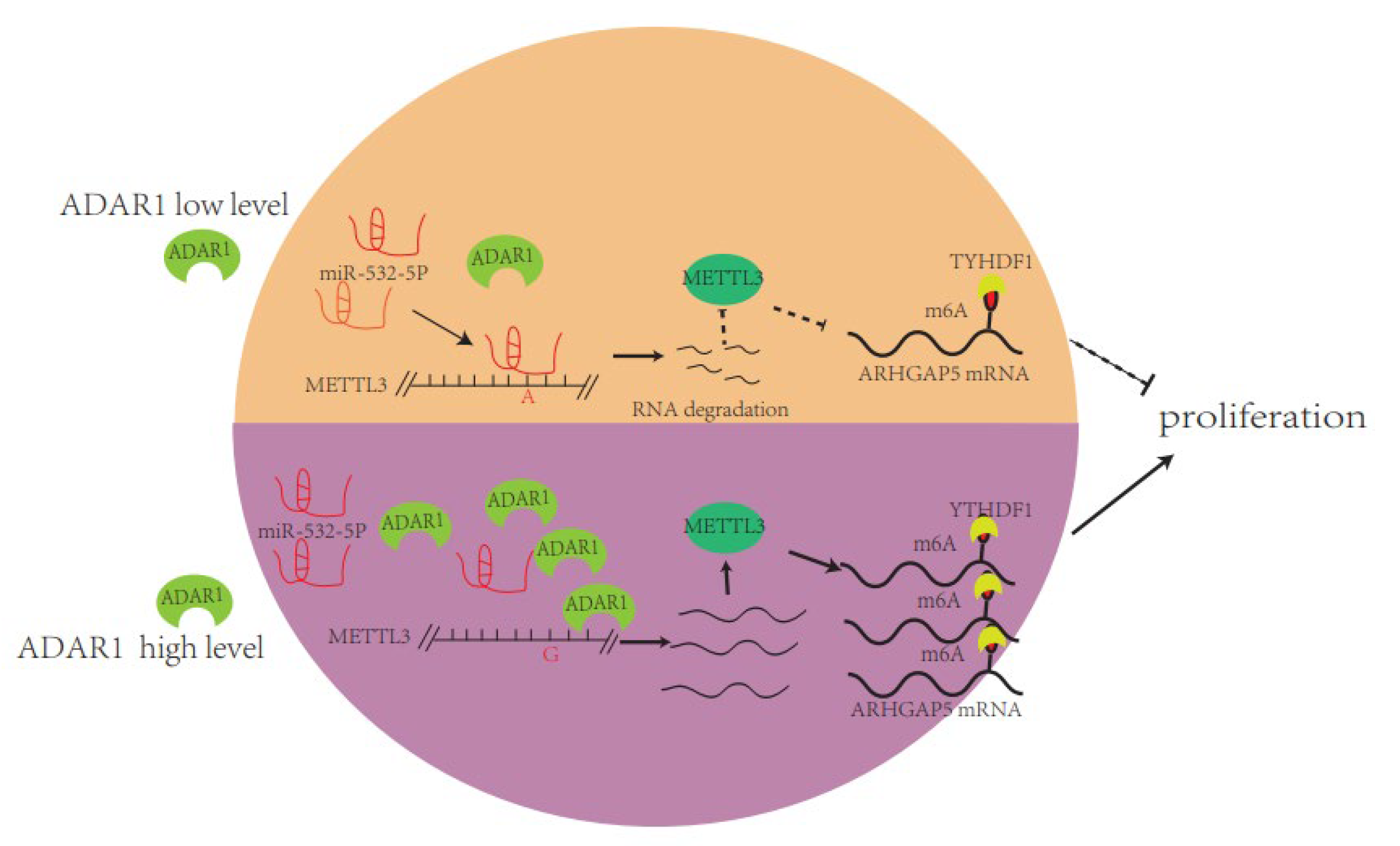
2.4. ADAR1 Intereacts with METTL3 mRNA in an RNA Editing-Dependent Manner and Alters METTL3 Binding Site of miR-532-5p
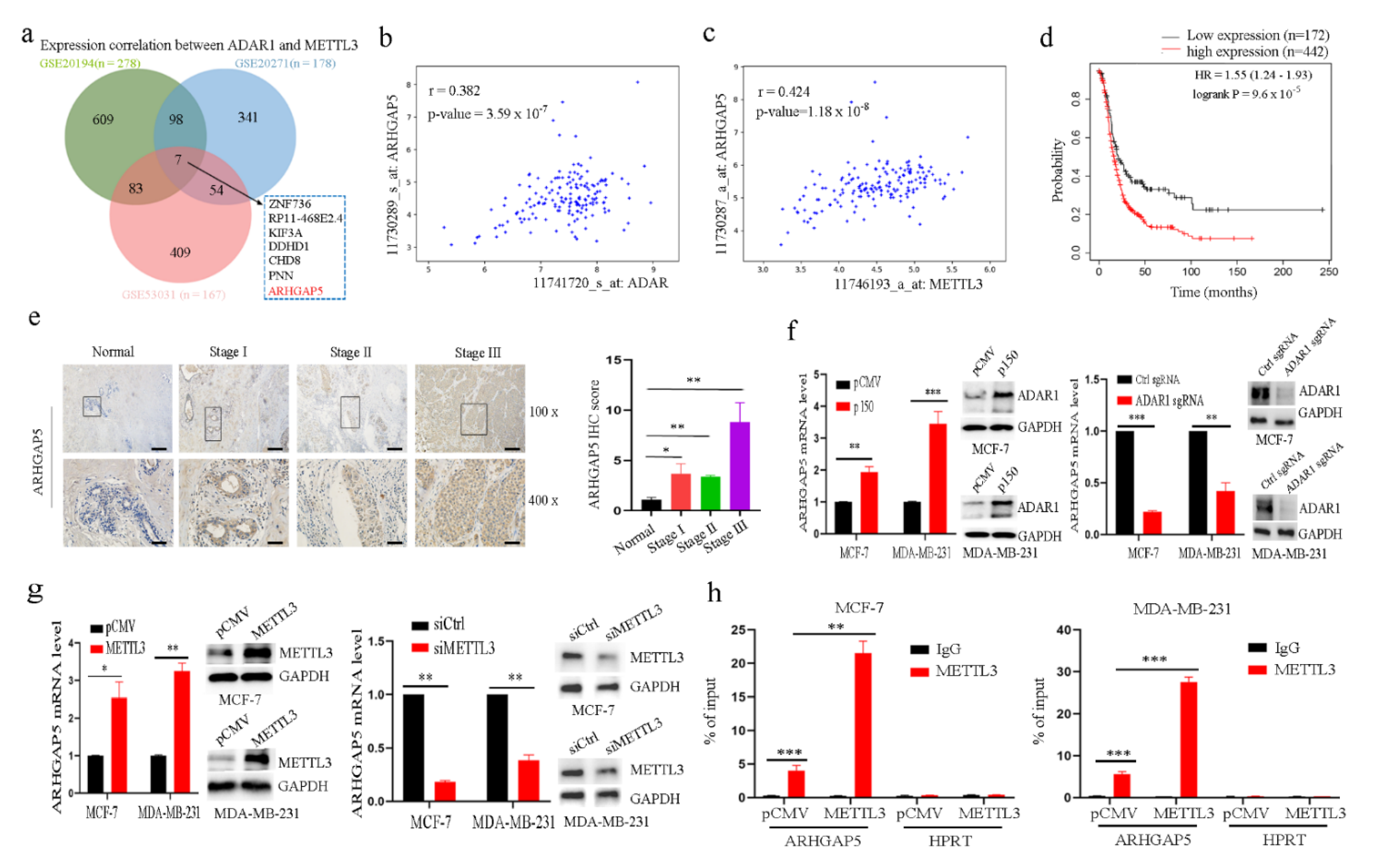
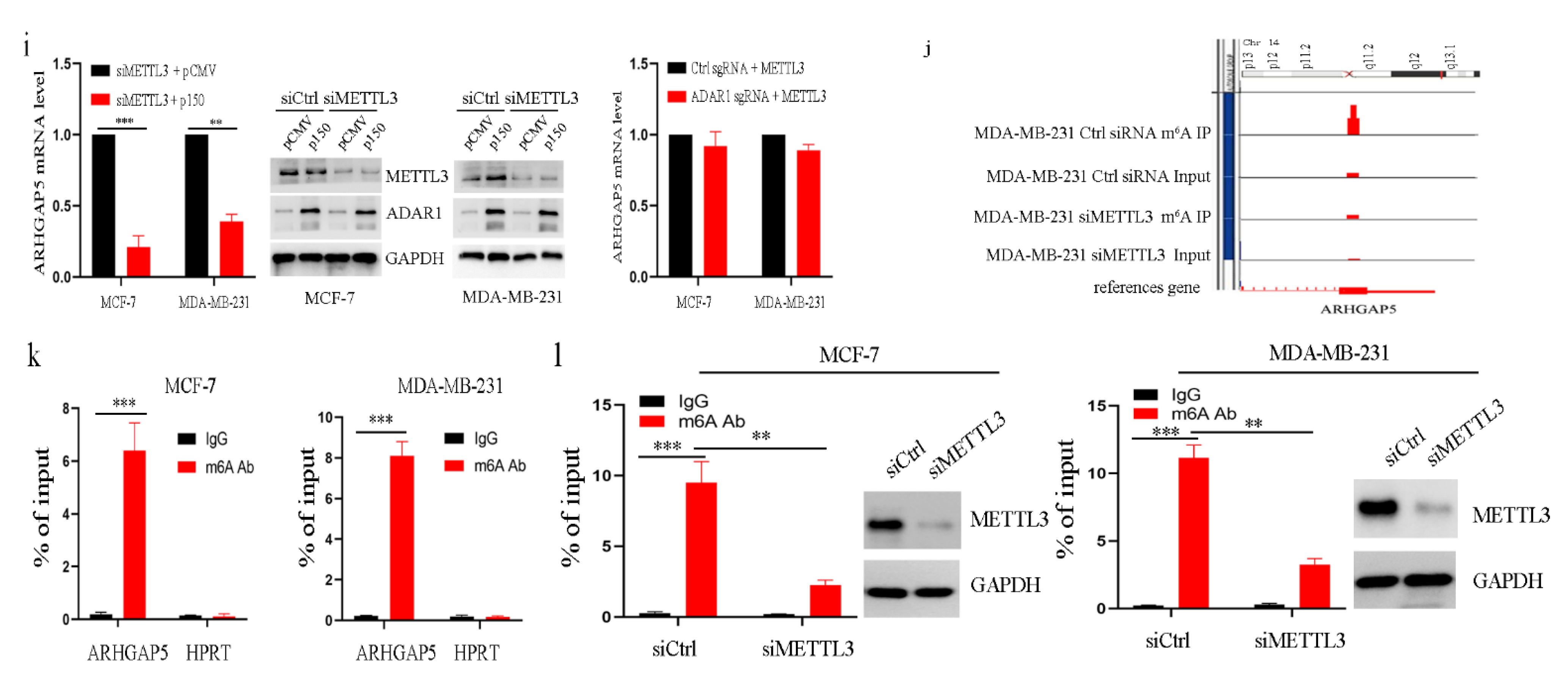
2.5. ADAR1 Upregulates ARHGAP5 Expression through METTL3
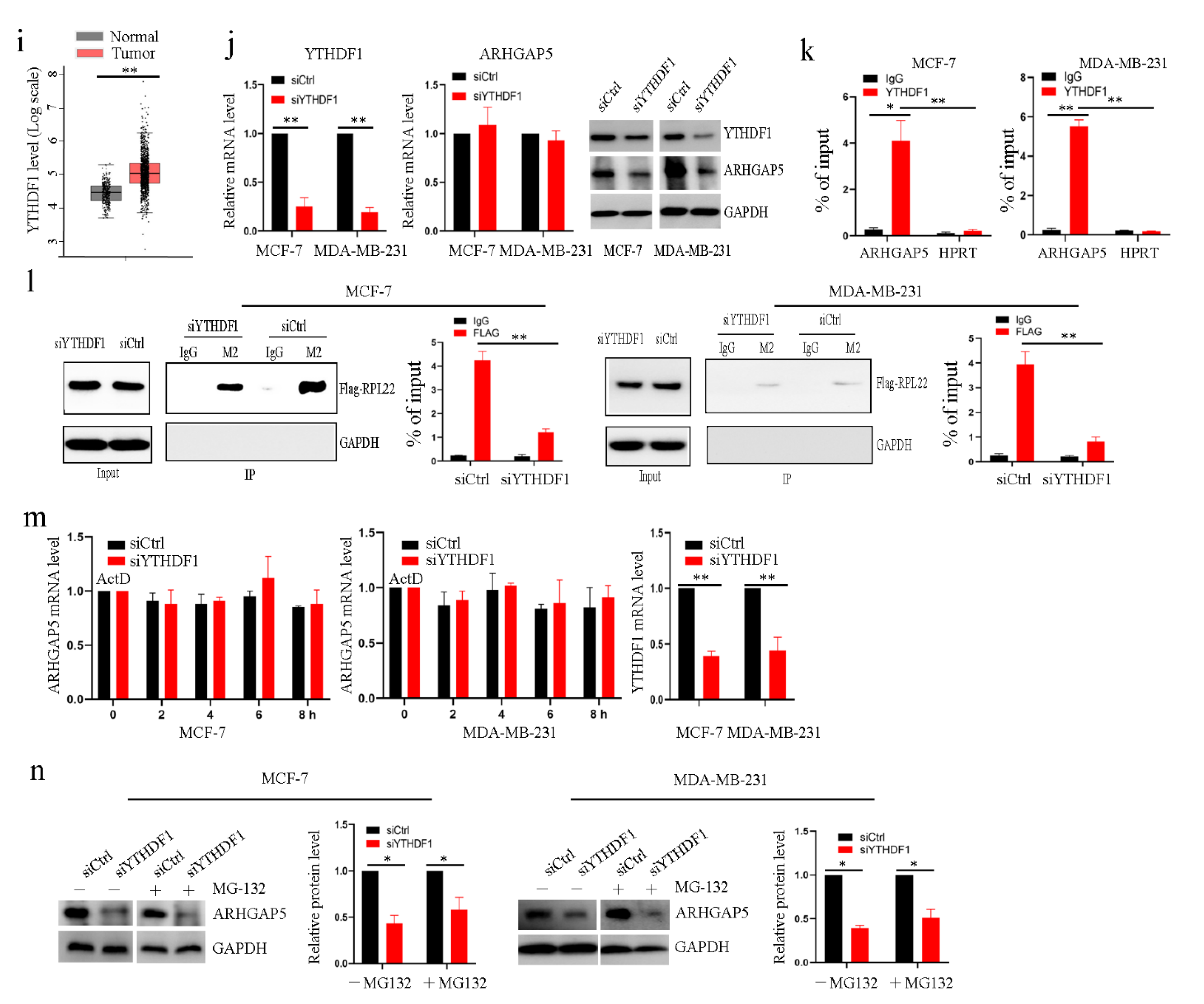
2.6. The Reader Protein YTHDF1 Increases ARHGAP5
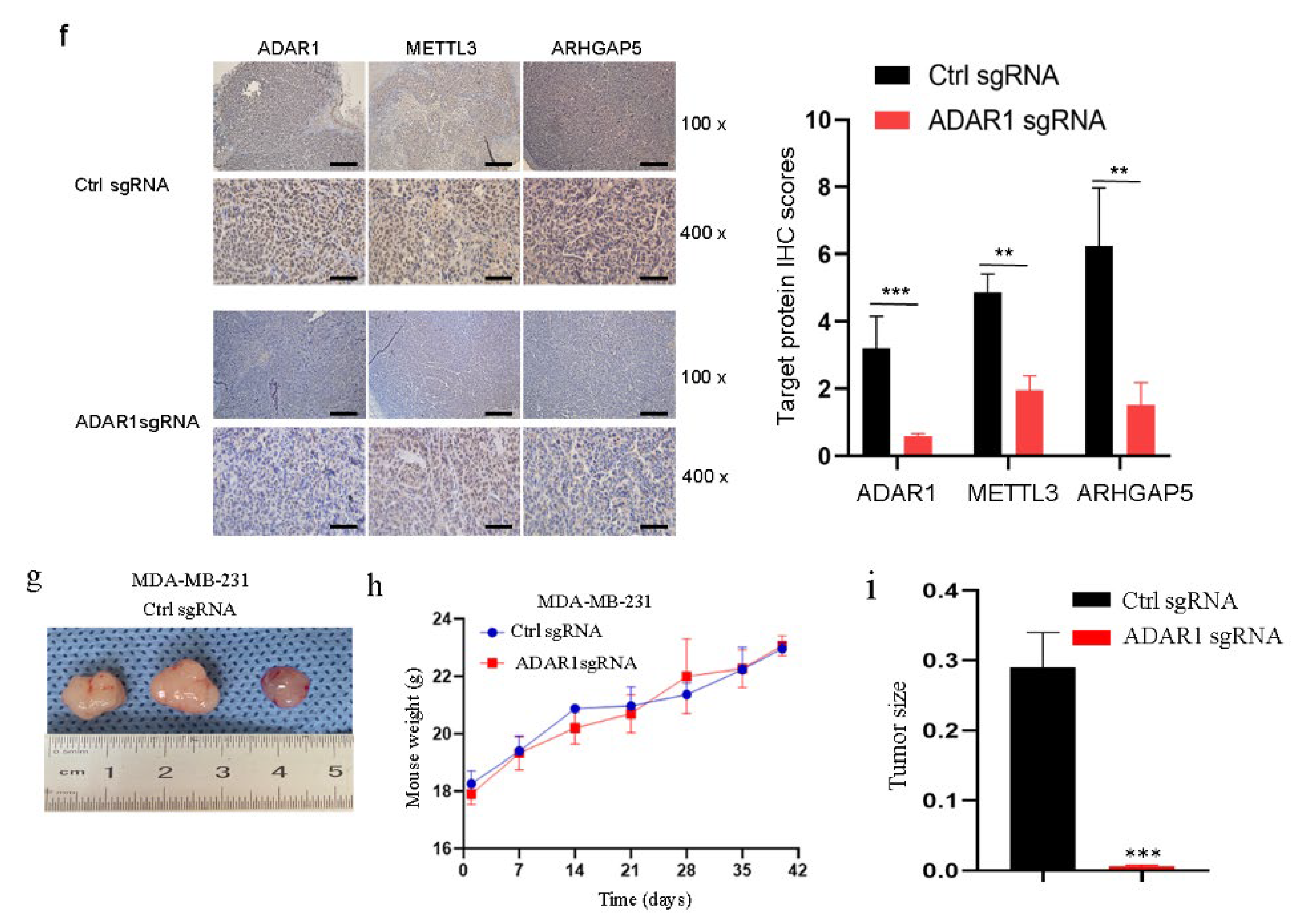
2.7. Loss of ADAR1 Suppresses Breast Cancer Growth and Decreases METTL3 and ARHGAP5 Expression In Vivo
3. Discussion
4. Materials and Methods
4.1. Human Tissues, Cell Lines and Cell Culture
4.2. Plasmids, miRNA Mimics, siRNAs and Transfection
4.3. Cell Proliferation and Colony Formation Assays
4.4. Cell Migration and Invasion Assays
4.5. RNA Extraction, Real-Time PCR
4.6. RNA m6A Dot Blot Assay
4.7. Western Blotting Analysis
4.8. RNA Immunoprecipitation Assay (RIP)
4.9. MeRIP and MeRIP-Seq
4.10. Dual-Luciferase Reporter Assay
4.11. Immunohistochemistry (IHC)
4.12. Actinomycin D Treatment for RNA Stability Assay
4.13. MG132 Treatment
4.14. Transfected Stable Cell Lines
4.15. In Vivo Experiments
4.16. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilkinson, E.; Cui, Y.H.; He, Y.Y. Roles of RNA Modifications in Diverse Cellular Functions. Front. Cell Dev. Biol. 2022, 10, 828683. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.R.; Slack, F.J. ADAR1 and its implications in cancer development and treatment. Trends Genet. TIG 2022, 38, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, Y.; Jin, L.; Yang, F.; Ding, H.; Zhang, L.; Li, L.; Pan, T. The Emerging Role of N6-Methyladenosine RNA Methylation as Regulators in Cancer Therapy and Drug Resistance. Front. Pharmacol. 2022, 13, 873030. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, E.A.; Mahapatra, A.; Mukherjee, P.; Yang, B.; Hundley, H.A. To protect and modify double-stranded RNA—The critical roles of ADARs in development, immunity and oncogenesis. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 54–87. [Google Scholar] [CrossRef]
- Patterson, J.B.; Samuel, C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol. Cell. Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Keegan, L.P.; Lafarga, M.; Berciano, M.T.; O’Connell, M.; Carmo-Fonseca, M. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 2003, 116 Pt 9, 1805–1818. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, D.; Gacquer, D.; Rothé, F.; Lefort, A.; Libert, F.; Brown, D.; Kheddoumi, N.; Shlien, A.; Konopka, T.; Salgado, R.; et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell Rep. 2015, 13, 277–289. [Google Scholar] [CrossRef]
- Han, L.; Diao, L.; Yu, S.; Xu, X.; Li, J.; Zhang, R.; Yang, Y.; Werner, H.M.J.; Eterovic, A.K.; Yuan, Y.; et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28, 515–528. [Google Scholar] [CrossRef]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef]
- Peng, X.; Xu, X.; Wang, Y.; Hawke, D.H.; Yu, S.; Han, L.; Zhou, Z.; Mojumdar, K.; Jeong, K.J.; Labrie, M.; et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33, 817–828.e7. [Google Scholar] [CrossRef] [Green Version]
- Anantharaman, A.; Gholamalamdari, O.; Khan, A.; Yoon, J.H.; Jantsch, M.F.; Hartner, J.C.; Gorospe, M.; Prasanth, S.G.; Prasanth, K.V. RNA-editing enzymes ADAR1 and ADAR2 coordinately regulate the editing and expression of Ctn RNA. FEBS Lett. 2017, 591, 2890–2904. [Google Scholar] [CrossRef] [PubMed]
- Gumireddy, K.; Li, A.; Kossenkov, A.V.; Sakurai, M.; Yan, J.; Li, Y.; Xu, H.; Wang, J.; Zhang, P.J.; Zhang, L.; et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nat. Commun. 2016, 7, 10715. [Google Scholar] [CrossRef]
- Binothman, N.; Hachim, I.Y.; Lebrun, J.J.; Ali, S. CPSF6 is a Clinically Relevant Breast Cancer Vulnerability Target: Role of CPSF6 in Breast Cancer. EBioMedicine 2017, 21, 65–78. [Google Scholar] [CrossRef]
- Dave, B.; Gonzalez, D.D.; Liu, Z.B.; Li, X.; Wong, H.; Granados, S.; Ezzedine, N.E.; Sieglaff, D.H.; Ensor, J.E.; Miller, K.D.; et al. Role of RPL39 in Metaplastic Breast Cancer. J. Natl. Cancer Instig. 2017, 109, djw292. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Fukami, T.; Gotoh, S.; Nakajima, M. A-to-I RNA Editing Up-regulates Human Dihydrofolate Reductase in Breast Cancer. J. Biol. Chem. 2017, 292, 4873–4884. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.Y.; Yang, W.Y.; Zhang, L.H.; Li, L.; Xie, F.; Li, H.Y.; Chen, X.Y.; Tu, Z.; Li, Y.; Chen, Y.; et al. 8-Chloro-Adenosine Inhibits Proliferation of MDA-MB-231 and SK-BR-3 Breast Cancer Cells by Regulating ADAR1/p53 Signaling Pathway. Cell Transplant. 2020, 29, 963689720958656. [Google Scholar] [CrossRef]
- Balacco, D.L.; Soller, M. The m(6)A Writer: Rise of a Machine for Growing Tasks. Biochemistry 2019, 58, 363–378. [Google Scholar] [CrossRef]
- Lence, T.; Paolantoni, C.; Worpenberg, L.; Roignant, J.Y. Mechanistic insights into m(6)A RNA enzymes. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 222–229. [Google Scholar] [CrossRef]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Z.; Yang, F.; Zhou, C.C.; Liu, F.; Yuan, J.H.; Wang, F.; Wang, T.T.; Xu, Q.G.; Zhou, W.P.; Sun, S.H. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6)-methyladenosine-dependent primary MicroRNA processing. Hepatology 2017, 65, 529–543. [Google Scholar] [CrossRef]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.G.; et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef]
- Liu, J.; Ren, D.; Du, Z.; Wang, H.; Zhang, H.; Jin, Y. m(6)A demethylase FTO facilitates tumor progression in lung squamous cell carcinoma by regulating MZF1 expression. Biochem. Biophys. Res. Commun. 2018, 502, 456–464. [Google Scholar] [CrossRef]
- Lin, X.; Chai, G.; Wu, Y.; Li, J.; Chen, F.; Liu, J.; Luo, G.; Tauler, J.; Du, J.; Lin, S.; et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat. Commun. 2019, 10, 2065. [Google Scholar] [CrossRef]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Weng, H.; Chen, J. m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yu, Y.; Yang, M.; Huang, H.; Ma, S.; Hu, J.; Xi, Z.; Guo, H.; Yao, G.; Yang, L.; et al. YTHDF1 promotes breast cancer progression by facilitating FOXM1 translation in an m6A-dependent manner. Cell Biosci. 2022, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Rapino, F.; Delaunay, S.; Rambow, F.; Zhou, Z.; Tharun, L.; De Tullio, P.; Sin, O.; Shostak, K.; Schmitz, S.; Piepers, J.; et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 2018, 558, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, V.; Cesarini, V.; Tomaselli, S.; Ianniello, Z.; Silvestris, D.A.; Ginistrelli, L.C.; Martini, M.; De Angelis, B.; De Luca, G.; Vitiani, L.R.; et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021, 22, 51. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Cottrell, K.A.; Ryu, S.; Bramel, E.R.; Kladney, R.D.; Bao, E.A.; Freeman, E.C.; Sabloak, T.; Maggi, L., Jr.; Weber, J.D. Evaluating the therapeutic potential of ADAR1 inhibition for triple-negative breast cancer. Oncogene 2021, 40, 189–202. [Google Scholar] [CrossRef]
- Terajima, H.; Lu, M.; Zhang, L.; Cui, Q.; Shi, Y.; Li, J.; He, C. N6-methyladenosine promotes induction of ADAR1-mediated A-to-I RNA editing to suppress aberrant antiviral innate immune responses. PLoS Biol. 2021, 19, e3001292. [Google Scholar] [CrossRef]
- Xiang, J.F.; Yang, Q.; Liu, C.X.; Wu, M.; Chen, L.L.; Yang, L. N(6)-Methyladenosines Modulate A-to-I RNA Editing. Mol. Cell 2018, 69, 126–135.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Qi, X.; Liu, L.; Ma, S.; Liu, J.; Wu, J. Epigenetic Regulation of m(6)A Modifications in Human Cancer. Mol. Ther. Nucleic Acids 2020, 19, 405–412. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Wang, Y.P.; Liu, J.; Liu, D.; Wang, X.D.; Bian, A.M.; Fang, D.Z.; Hui, X.B. MiR-532-5p acts as a tumor suppressor and inhibits glioma cell proliferation by targeting CSF1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7206. [Google Scholar] [PubMed]
- Hu, J.; Wang, L.; Guan, C. MiR-532-5p Suppresses Migration and Invasion of Lung Cancer Cells Through Inhibiting CCR4. Cancer Biother. Radiopharm. 2020, 35, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tang, X.; Shi, X.; Su, L. miR-532-5p promotes breast cancer proliferation and migration by targeting RERG. Exp. Ther. Med. 2020, 19, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef]
- Wang, H.; Xu, B.; Shi, J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene 2020, 722, 144076. [Google Scholar] [CrossRef]
- Li, X.; Tang, J.; Huang, W.; Wang, F.; Li, P.; Qin, C.; Qin, Z.; Zou, Q.; Wei, J.; Hua, L.; et al. The M6A methyltransferase METTL3: Acting as a tumor suppressor in renal cell carcinoma. Oncotarget 2017, 8, 96103–96116. [Google Scholar] [CrossRef]
- Deng, R.; Cheng, Y.; Ye, S.; Zhang, J.; Huang, R.; Li, P.; Liu, H.; Deng, Q.; Wu, X.; Lan, P.; et al. m(6)A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. OncoTargets Ther. 2019, 12, 4391–4402. [Google Scholar] [CrossRef]
- Zhu, L.; Zhu, Y.; Han, S.; Chen, M.; Song, P.; Dai, D.; Xu, W.; Jiang, T.; Feng, L.; Shin, V.Y.; et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. 2019, 10, 383. [Google Scholar] [CrossRef]
- Rosman, D.S.; Phukan, S.; Huang, C.C.; Pasche, B. TGFBR1*6A enhances the migration and invasion of MCF-7 breast cancer cells through RhoA activation. Cancer Res. 2008, 68, 1319–1328. [Google Scholar] [CrossRef]
- Yang, S.; Deng, P.; Zhu, Z.; Zhu, J.; Wang, G.; Zhang, L.; Chen, A.F.; Wang, T.; Sarkar, S.N.; Billiar, T.R.; et al. Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J. Immunol. 2014, 193, 3436–3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.Y.; Yang, S.Y.; Ruan, X.J.; Ding, H.Y.; Wang, N.X.; Liu, F.; Li, J.C.; Li, Y. MsrB1 Promotes Proliferation and Invasion of Colorectal Cancer Cells via GSK-3β/β-catenin Signaling Axis. Cell Transplant. 2021, 30, 9636897211053203. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Li, Y.; An, G.S.; Ni, J.H.; Jia, H.T.; Li, S.Y. DNA Damage-Response Pathway Heterogeneity of Human Lung Cancer A549 and H1299 Cells Determines Sensitivity to 8-Chloro-Adenosine. Int. J. Mol. Sci. 2018, 19, 1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wang, N.-X.; Yin, C.; Jiang, S.-S.; Li, J.-C.; Yang, S.-Y. RNA Editing Enzyme ADAR1 Regulates METTL3 in an Editing Dependent Manner to Promote Breast Cancer Progression via METTL3/ARHGAP5/YTHDF1 Axis. Int. J. Mol. Sci. 2022, 23, 9656. https://doi.org/10.3390/ijms23179656
Li Y, Wang N-X, Yin C, Jiang S-S, Li J-C, Yang S-Y. RNA Editing Enzyme ADAR1 Regulates METTL3 in an Editing Dependent Manner to Promote Breast Cancer Progression via METTL3/ARHGAP5/YTHDF1 Axis. International Journal of Molecular Sciences. 2022; 23(17):9656. https://doi.org/10.3390/ijms23179656
Chicago/Turabian StyleLi, Yi, Ning-Xi Wang, Chuan Yin, Shan-Shan Jiang, Jia-Chu Li, and Sheng-Yong Yang. 2022. "RNA Editing Enzyme ADAR1 Regulates METTL3 in an Editing Dependent Manner to Promote Breast Cancer Progression via METTL3/ARHGAP5/YTHDF1 Axis" International Journal of Molecular Sciences 23, no. 17: 9656. https://doi.org/10.3390/ijms23179656