
Combination of Whole Genome Sequencing and Metagenomics for Microbiological Diagnostics


Abstract
:1. Introduction
2. Focus on Individual Microorganisms
3. Focus on Bacterial Communities
4. An Integrated Approach of WGS and Metagenomic Sequencing
5. Conclusions and Future Trends
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharpton, T.J. An Introduction to the Analysis of Shotgun Metagenomic Data. Front. Plant. Sci. 2014, 5, 209. [Google Scholar] [CrossRef]
- Kuper, K.M.; Boles, D.M.; Mohr, J.F.; Wanger, A. Antimicrobial Susceptibility Testing: A Primer for Clinicians. Pharmacotherapy 2009, 29, 1326–1343. [Google Scholar] [CrossRef] [PubMed]
- Van Belkum, A.; Bachmann, T.T.; Lüdke, G.; Lisby, J.G.; Kahlmeter, G.; Mohess, A.; Becker, K.; Hays, J.P.; Woodford, N.; Mitsakakis, K.; et al. Developmental Roadmap for Antimicrobial Susceptibility Testing Systems. Nat. Rev. Microbiol. 2019, 17, 51–62. [Google Scholar] [CrossRef]
- Smith, K.P.; Kirby, J.E. Rapid Susceptibility Testing Methods. Clin. Lab. Med. 2019, 39, 333–344. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The Rebirth of Culture in Microbiology through the Example of Culturomics to Study Human Gut Microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef]
- Corona, F.; Martinez, J.L. Phenotypic Resistance to Antibiotics. Antibiotics 2013, 2, 237–255. [Google Scholar] [CrossRef]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M. Whole-Genome Random Sequencing and Assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef]
- Hasman, H.; Saputra, D.; Sicheritz-Ponten, T.; Lund, O.; Svendsen, C.A.; Frimodt-Møller, N.; Aarestrup, F.M. Rapid Whole-Genome Sequencing for Detection and Characterization of Microorganisms Directly from Clinical Samples. J. Clin. Microbiol. 2014, 52, 139–146. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Miller, S.A. Clinical Metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Eyre, D.W. Infection Prevention and Control Insights from a Decade of Pathogen Whole-Genome Sequencing. J. Hosp. Infect. 2022, 122, 180–186. [Google Scholar] [CrossRef]
- Brown, B.; Allard, M.; Bazaco, M.C.; Blankenship, J.; Minor, T. An Economic Evaluation of the Whole Genome Sequencing Source Tracking Program in the U.S. PLoS ONE 2021, 16, e0258262. [Google Scholar] [CrossRef] [PubMed]
- Dymond, A.; Davies, H.; Mealing, S.; Pollit, V.; Coll, F.; Brown, N.M.; Peacock, S.J. Genomic Surveillance of Methicillin-Resistant Staphylococcus aureus: A Mathematical Early Modeling Study of Cost-Effectiveness. Clin. Infect. Dis. 2020, 70, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sepulveda, B.M.; Heavens, D.; Pulford, C.V.; Predeus, A.V.; Low, R.; Webster, H.; Dykes, G.F.; Schudoma, C.; Rowe, W.; Lipscombe, J.; et al. An Accessible, Efficient and Global Approach for the Large-Scale Sequencing of Bacterial Genomes. Genome Biol. 2021, 22, 349. [Google Scholar] [CrossRef]
- Loman, N.J.; Constantinidou, C.; Chan, J.Z.M.; Halachev, M.; Sergeant, M.; Penn, C.W.; Robinson, E.R.; Pallen, M.J. High-Throughput Bacterial Genome Sequencing: An Embarrassment of Choice, a World of Opportunity. Nat. Rev. Microbiol. 2012, 10, 599–606. [Google Scholar] [CrossRef]
- Smetana, J.; Brož, P. National Genome Initiatives in Europe and the United Kingdom in the Era of Whole-Genome Sequencing: A Comprehensive Review. Genes 2022, 13, 556. [Google Scholar] [CrossRef]
- Van Belkum, A.; Rochas, O. Laboratory-Based and Point-of-Care Testing for MSSA/MRSA Detection in the Age of Whole Genome Sequencing. Front. Microbiol. 2018, 9, 1437. [Google Scholar] [CrossRef]
- Bonsall, D.; Golubchik, T.; de Cesare, M.; Limbada, M.; Kosloff, B.; MacIntyre-Cockett, G.; Hall, M.; Wymant, C.; Ansari, M.A.; Abeler-Dörner, L.; et al. A Comprehensive Genomics Solution for HIV Surveillance and Clinical Monitoring in Low-Income Settings. J. Clin. Microbiol. 2020, 58, e00382-20. [Google Scholar] [CrossRef]
- Aarestrup, F.M.; Bonten, M.; Koopmans, M. Pandemics- One Health Preparedness for the next. Lancet Reg. Health Eur. 2021, 9, 100210. [Google Scholar] [CrossRef]
- Wegner, F.; Roloff, T.; Huber, M.; Cordey, S.; Ramette, A.; Gerth, Y.; Bertelli, C.; Stange, M.; Seth-Smith, H.M.B.; Mari, A.; et al. External Quality Assessment of SARS-CoV-2 Sequencing: An ESGMD-SSM Pilot Trial across 15 European Laboratories. J. Clin. Microbiol. 2022, 60, e0169821. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Xie, H.; Zhong, Q.; Xia, Y. Analysis and Evaluation of Different Sequencing Depths from 5 to 20 Million Reads in Metagenome Shotgun Sequencing, with Optimal Minimum Depth Recommended. Genome 2022. [Google Scholar] [CrossRef]
- Tagini, F.; Greub, G. Bacterial Genome Sequencing in Clinical Microbiology: A Pathogen-Oriented Review. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2007–2020. [Google Scholar] [CrossRef]
- Wu, X.; Tan, G.; Sha, W.; Liu, H.; Yang, J.; Guo, Y.; Shen, X.; Wu, Z.; Shen, H.; Yu, F. Use of Whole-Genome Sequencing to Predict Complex Drug Resistance from Early Positive Liquid Cultures. Microbiol. Spectr. 2022, 10, e0251621. [Google Scholar] [CrossRef] [PubMed]
- Doyle, R.M.; Burgess, C.; Williams, R.; Gorton, R.; Booth, H.; Brown, J.; Bryant, J.M.; Chan, J.; Creer, D.; Holdstock, J.; et al. Direct Whole-Genome Sequencing of Sputum Accurately Identifies Drug-Resistant Mycobacterium tuberculosis Faster than MGIT Culture Sequencing. J. Clin. Microbiol. 2018, 56, e00666-18. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, D.; Lang, D.; Müller, N.F.; Neher, R.A.; Stadler, T.; Egli, A. Evaluation of Two Workflows for Whole Genome Sequencing-Based Typing of Influenza A viruses. J. Virol. Methods 2019, 266, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Stange, M.; Mari, A.; Roloff, T.; Seth-Smith, H.M.; Schweitzer, M.; Brunner, M.; Leuzinger, K.; Søgaard, K.K.; Gensch, A.; Tschudin-Sutter, S.; et al. SARS-CoV-2 Outbreak in a Tri-National Urban Area Is Dominated by a B.1 Lineage Variant Linked to a Mass Gathering Event. PLoS Pathog. 2021, 17, e1009374. [Google Scholar] [CrossRef]
- Seth-Smith, H.M.B.; Harris, S.R.; Skilton, R.J.; Radebe, F.M.; Golparian, D.; Shipitsyna, E.; Duy, P.T.; Scott, P.; Cutcliffe, L.T.; O’Neill, C.; et al. Whole-Genome Sequences of Chlamydia trachomatis Directly from Clinical Samples without Culture. Genome Res. 2013, 23, 855–866. [Google Scholar] [CrossRef]
- Lomsadze, A.; Li, T.; Rajeevan, M.S.; Unger, E.R.; Borodovsky, M. Bioinformatics Pipeline for Human papillomavirus Short Read Genomic Sequences Classification Using Support Vector Machine. Viruses 2020, 12, 710. [Google Scholar] [CrossRef]
- Brown, J.R.; Roy, S.; Ruis, C.; Yara Romero, E.; Shah, D.; Williams, R.; Breuer, J. Norovirus Whole-Genome Sequencing by SureSelect Target Enrichment: A Robust and Sensitive Method. J. Clin. Microbiol. 2016, 54, 2530–2537. [Google Scholar] [CrossRef]
- Mäklin, T.; Kallonen, T.; Alanko, J.; Samuelsen, Ø.; Hegstad, K.; Mäkinen, V.; Corander, J.; Heinz, E.; Honkela, A. Bacterial Genomic Epidemiology with Mixed Samples. Microb. Genom. 2021, 7, 691. [Google Scholar] [CrossRef]
- Gautam, S.S.; Kc, R.; Leong, K.W.; Mac Aogáin, M.; O’Toole, R.F. A Step-by-Step Beginner’s Protocol for Whole Genome Sequencing of Human Bacterial Pathogens. J. Biol. Methods 2019, 6, e110. [Google Scholar] [CrossRef] [Green Version]
- Smits, T.H.M. The Importance of Genome Sequence Quality to Microbial Comparative Genomics. BMC Genom. 2019, 20, 662. [Google Scholar] [CrossRef] [PubMed]
- Rossen, J.W.A.; Friedrich, A.W.; Moran-Gilad, J. ESCMID Study Group for Genomic and Molecular Diagnostics (ESGMD) Practical Issues in Implementing Whole-Genome-Sequencing in Routine Diagnostic Microbiology. Clin. Microbiol. Infect. 2018, 24, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Timme, R.E.; Wolfgang, W.J.; Balkey, M.; Venkata, S.L.G.; Randolph, R.; Allard, M.; Strain, E. Optimizing Open Data to Support One Health: Best Practices to Ensure Interoperability of Genomic Data from Bacterial Pathogens. One Health Outlook 2020, 2, 20. [Google Scholar] [CrossRef]
- Higgs, C.; Sherry, N.L.; Seemann, T.; Horan, K.; Walpola, H.; Kinsella, P.; Bond, K.; Williamson, D.A.; Marshall, C.; Kwong, J.C.; et al. Optimising Genomic Approaches for Identifying Vancomycin-Resistant Enterococcus faecium Transmission in Healthcare Settings. Nat. Commun. 2022, 13, 509. [Google Scholar] [CrossRef]
- Shropshire, W.C.; Dinh, A.Q.; Earley, M.; Komarow, L.; Panesso, D.; Rydell, K.; Gómez-Villegas, S.I.; Miao, H.; Hill, C.; Chen, L.; et al. Accessory Genomes Drive Independent Spread of Carbapenem-Resistant Klebsiella pneumoniae Clonal Groups 258 and 307 in Houston, TX. MBio 2022, 13, e0049722. [Google Scholar] [CrossRef] [PubMed]
- Craddock, H.A.; Motro, Y.; Zilberman, B.; Khalfin, B.; Bardenstein, S.; Moran-Gilad, J. Long-Read Sequencing and Hybrid Assembly for Genomic Analysis of Clinical Isolates. Microorganisms 2022, 10, 619. [Google Scholar] [CrossRef]
- Uelze, L.; Borowiak, M.; Deneke, C.; Szabó, I.; Fischer, J.; Tausch, S.H.; Malorny, B. Performance and Accuracy of Four Open-Source Tools for Serotyping of Spp. Based on Whole-Genome Short-Read Sequencing Data. Appl. Environ. Microbiol. 2020, 86, e02265-19. [Google Scholar] [CrossRef]
- Browne, P.D.; Nielsen, T.K.; Kot, W.; Aggerholm, A.; Gilbert, M.T.P.; Puetz, L.; Rasmussen, M.; Zervas, A.; Hansen, L.H. GC Bias Affects Genomic and Metagenomic Reconstructions, Underrepresenting GC-Poor Organisms. Gigascience 2020, 9, giaa008. [Google Scholar] [CrossRef]
- Seth-Smith, H.M.B.; Bonfiglio, F.; Cuénod, A.; Reist, J.; Egli, A.; Wüthrich, D. Evaluation of Rapid Library Preparation Protocols for Whole Genome Sequencing Based Outbreak Investigation. Front. Public Health 2019, 7, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Wyres, K.L.; Holt, K.E. Recovery of Small Plasmid Sequences via Oxford Nanopore Sequencing. Microb. Genom. 2021, 7, 631. [Google Scholar] [CrossRef]
- Pightling, A.W.; Pettengill, J.B.; Wang, Y.; Rand, H.; Strain, E. Within-Species Contamination of Bacterial Whole-Genome Sequence Data Has a Greater Influence on Clustering Analyses than between-Species Contamination. Genome Biol. 2019, 20, 286. [Google Scholar] [CrossRef] [PubMed]
- Deneke, C.; Brendebach, H.; Uelze, L.; Borowiak, M.; Malorny, B.; Tausch, S.H. Species-Specific Quality Control, Assembly and Contamination Detection in Microbial Isolate Sequences with AQUAMIS. Genes 2021, 12, 644. [Google Scholar] [CrossRef] [PubMed]
- Riojas, M.A.; McGough, K.J.; Rider-Riojas, C.J.; Rastogi, N.; Hazbón, M.H. Phylogenomic Analysis of the Species of the Mycobacterium tuberculosis Complex Demonstrates That Mycobacterium africanum, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium microti and Mycobacterium pinnipedii Are Later Heterotypic Synonyms of Mycobacterium tuberculosis. Int. J. Syst. Evol. Microbiol. 2018, 68, 324–332. [Google Scholar] [PubMed]
- Tortoli, E.; Brown-Elliott, B.A.; Chalmers, J.D.; Cirillo, D.M.; Daley, C.L.; Emler, S.; Floto, R.A.; Garcia, M.J.; Hoefsloot, W.; Koh, W.-J.; et al. Same Meat, Different Gravy: Ignore the New Names of Mycobacteria. Eur. Respir. J. 2019, 54, 1900795. [Google Scholar] [CrossRef]
- International Code of Nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2019, 69, S1–S111. [CrossRef]
- Oren, A.; Arahal, D.R.; Rosselló-Móra, R.; Sutcliffe, I.C.; Moore, E.R.B. Preparing a Revision of the International Code of Nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 4598. [Google Scholar] [CrossRef]
- Oren, A.; Arahal, D.R.; Rosselló-Móra, R.; Sutcliffe, I.C.; Moore, E.R.B. Public Discussion on a Proposed Revision of the International Code of Nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 4918. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef]
- Khieu, V.; Ananta, P.; Kaewprasert, O.; Laohaviroj, M.; Namwat, W.; Faksri, K. Whole-Genome Sequencing Analysis to Identify Infection with Multiple Species of Nontuberculous Mycobacteria. Pathogens 2021, 10, 879. [Google Scholar] [CrossRef]
- Dylus, D.; Pillonel, T.; Opota, O.; Wüthrich, D.; Seth-Smith, H.M.B.; Egli, A.; Leo, S.; Lazarevic, V.; Schrenzel, J.; Laurent, S.; et al. NGS-Based Typing and Outbreak Analysis in Clinical Microbiology Laboratories: Lessons Learned From a Swiss-Wide Proficiency Test. Front. Microbiol. 2020, 11, 591093. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, A.; Andersen, P.S.; Bletz, S.; Friedrich, A.W.; Kohl, T.A.; Lilje, B.; Niemann, S.; Prior, K.; Rossen, J.W.; Harmsen, D. High Interlaboratory Reproducibility and Accuracy of Next-Generation-Sequencing-Based Bacterial Genotyping in a Ring Trial. J. Clin. Microbiol. 2017, 55, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Meinel, D.M.; Kuehl, R.; Zbinden, R.; Boskova, V.; Garzoni, C.; Fadini, D.; Dolina, M.; Blümel, B.; Weibel, T.; Tschudin-Sutter, S.; et al. Outbreak Investigation for Toxigenic Corynebacterium diphtheriae Wound Infections in Refugees from Northeast Africa and Syria in Switzerland and Germany by Whole Genome Sequencing. Clin. Microbiol. Infect. 2016, 22, e1003.e1–e1003.e8. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, D.; Gautsch, S.; Spieler-Denz, R.; Dubuis, O.; Gaia, V.; Moran-Gilad, J.; Hinic, V.; Seth-Smith, H.M.; Nickel, C.H.; Tschudin-Sutter, S.; et al. Air-Conditioner Cooling Towers as Complex Reservoirs and Continuous Source of Infection Evidenced by a Genomic Analysis Study in 2017, Switzerland. Euro Surveill. 2019, 24, 1800192. [Google Scholar] [CrossRef] [PubMed]
- Köser, C.U.; Holden, M.T.G.; Ellington, M.J.; Cartwright, E.J.P.; Brown, N.M.; Ogilvy-Stuart, A.L.; Hsu, L.Y.; Chewapreecha, C.; Croucher, N.J.; Harris, S.R.; et al. Rapid Whole-Genome Sequencing for Investigation of a Neonatal MRSA Outbreak. N. Engl. J. Med. 2012, 366, 2267–2275. [Google Scholar] [CrossRef]
- Wassilew, N.; Seth-Smith, H.M.; Rolli, E.; Fietze, Y.; Casanova, C.; Führer, U.; Egli, A.; Marschall, J.; Buetti, N. Outbreak of Vancomycin-Resistant Clone ST796, Switzerland, December 2017 to April 2018. Euro Surveill. 2018, 23, 1800351. [Google Scholar] [CrossRef]
- Timme, R.E.; Sanchez Leon, M.; Allard, M.W. Utilizing the Public GenomeTrakr Database for Foodborne Pathogen Traceback. Methods Mol. Biol. 2019, 1918, 201–212. [Google Scholar]
- Egli, A.; Blanc, D.S.; Greub, G.; Keller, P.M.; Lazarevic, V.; Lebrand, A.; Leib, S.; Neher, R.A.; Perreten, V.; Ramette, A.; et al. Improving the Quality and Workflow of Bacterial Genome Sequencing and Analysis: Paving the Way for a Switzerland-Wide Molecular Epidemiological Surveillance Platform. Swiss Med. Wkly. 2018, 148, w14693. [Google Scholar] [CrossRef] [PubMed]
- Xanthopoulou, K.; Peter, S.; Tobys, D.; Behnke, M.; Dinkelacker, A.G.; Eisenbeis, S.; Falgenhauer, J.; Falgenhauer, L.; Fritzenwanker, M.; Gölz, H.; et al. Vancomycin-Resistant Enterococcus faecium Colonizing Patients on Hospital Admission in Germany: Prevalence and Molecular Epidemiology. J. Antimicrob. Chemother. 2020, 75, 2743–2751. [Google Scholar] [CrossRef]
- Lee, X.J.; Elliott, T.M.; Harris, P.N.A.; Douglas, J.; Henderson, B.; Watson, C.; Paterson, D.L.; Schofield, D.S.; Graves, N.; Gordon, L.G. Clinical and Economic Outcomes of Genome Sequencing Availability on Containing a Hospital Outbreak of Resistant Escherichia coli in Australia. Value Health 2020, 23, 994–1002. [Google Scholar] [CrossRef]
- Snyder, L.A.; Loman, N.J.; Faraj, L.A.; Levi, K.; Weinstock, G.; Boswell, T.C.; Pallen, M.J.; Ala’Aldeen, D.A. Epidemiological Investigation of Pseudomonas aeruginosa Isolates from a Six-Year-Long Hospital Outbreak Using High-Throughput Whole Genome Sequencing. Euro Surveill. 2013, 18, 20611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, T.; Loman, N.J.; Bingle, L.; Jumaa, P.; Weinstock, G.M.; Mortiboy, D.; Pallen, M.J. High-Throughput Whole-Genome Sequencing to Dissect the Epidemiology of Acinetobacter baumannii Isolates from a Hospital Outbreak. J. Hosp. Infect. 2010, 75, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Song, J.E.; Jeong, H.; Lim, Y.S.; Ha, E.J.; Jung, I.Y.; Jeong, W.; Choi, H.; Jeong, S.J.; Ku, N.S.; Park, E.S.; et al. An Outbreak of KPC-Producing Linked with an Index Case of Community-Acquired KPC-Producing Isolate: Epidemiological Investigation and Whole Genome Sequencing Analysis. Microb. Drug. Resist. 2019, 25, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.; Bosio, M.; Gross, C.; Bezdan, D.; Gutierrez, J.; Oberhettinger, P.; Liese, J.; Vogel, W.; Dörfel, D.; Berger, L.; et al. Tracking of Antibiotic Resistance Transfer and Rapid Plasmid Evolution in a Hospital Setting by Nanopore Sequencing. mSphere 2020, 5, e00525-20. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.M.; Merker, M.; Kohl, T.A.; Crook, D.W.; Niemann, S.; Peto, T.E.A. Whole Genome Sequencing for M/XDR Tuberculosis Surveillance and for Resistance Testing. Clin. Microbiol. Infect. 2017, 23, 161–166. [Google Scholar] [CrossRef]
- Ellington, M.J.; Ekelund, O.; Aarestrup, F.M.; Canton, R.; Doumith, M.; Giske, C.; Grundman, H.; Hasman, H.; Holden, M.T.G.; Hopkins, K.L.; et al. The Role of Whole Genome Sequencing in Antimicrobial Susceptibility Testing of Bacteria: Report from the EUCAST Subcommittee. Clin. Microbiol. Infect. 2017, 23, 2–22. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Mahfouz, N.; Ferreira, I.; Beisken, S.; von Haeseler, A.; Posch, A.E. Large-Scale Assessment of Antimicrobial Resistance Marker Databases for Genetic Phenotype Prediction: A Systematic Review. J. Antimicrob. Chemother. 2020, 75, 3099–3108. [Google Scholar] [CrossRef]
- Papp, M.; Solymosi, N. Review and Comparison of Antimicrobial Resistance Gene Databases. Antibiotics 2022, 11, 339. [Google Scholar] [CrossRef]
- Doyle, R.M.; O’Sullivan, D.M.; Aller, S.D.; Bruchmann, S.; Clark, T.; Coello Pelegrin, A.; Cormican, M.; Diez Benavente, E.; Ellington, M.J.; McGrath, E.; et al. Discordant Bioinformatic Predictions of Antimicrobial Resistance from Whole-Genome Sequencing Data of Bacterial Isolates: An Inter-Laboratory Study. Microb. Genom. 2020, 6, e000335. [Google Scholar] [CrossRef]
- Hunt, M.; Bradley, P.; Lapierre, S.G.; Heys, S.; Thomsit, M.; Hall, M.B.; Malone, K.M.; Wintringer, P.; Walker, T.M.; Cirillo, D.M.; et al. Antibiotic Resistance Prediction for from Genome Sequence Data with Mykrobe. Wellcome Open Res. 2019, 4, 191. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Zhou, J.; Zhang, T.; Li, C.; Chen, J.; Fan, J.; Qu, L.; Su, X. Comparative Genomics of the Sequential Pseudomonas aeruginosa Isolates Obtained from the Continuous Imipenem Stress Evolution. Appl. Microbiol. Biotechnol. 2020, 104, 10655–10667. [Google Scholar] [CrossRef]
- Babouee Flury, B.; Ellington, M.J.; Hopkins, K.L.; Turton, J.F.; Doumith, M.; Loy, R.; Staves, P.; Hinic, V.; Frei, R.; Woodford, N. Association of Novel Nonsynonymous Single Nucleotide Polymorphisms in ampD with Cephalosporin Resistance and Phylogenetic Variations in ampC, ampR, ompF, and ompC in Enterobacter cloacae Isolates That Are Highly Resistant to Carbapenems. Antimicrob. Agents Chemother. 2016, 60, 2383–2390. [Google Scholar] [CrossRef]
- Hu, D.; Fuller, N.R.; Caterson, I.D.; Holmes, A.J.; Reeves, P.R. Single-Gene Long-Read Sequencing Illuminates Escherichia coli Strain Dynamics in the Human Intestinal Microbiome. Cell Rep. 2022, 38, 110239. [Google Scholar] [CrossRef]
- Yang, C.; Yang, R.-F.; Cui, Y. Bacterial Genome-Wide Association Study: Methodologies and Applications. Yi Chuan 2018, 40, 57–65. [Google Scholar]
- Allen, J.P.; Snitkin, E.; Pincus, N.B.; Hauser, A.R. Forest and Trees: Exploring Bacterial Virulence with Genome-Wide Association Studies and Machine Learning. Trends Microbiol. 2021, 29, 621–633. [Google Scholar] [CrossRef]
- Weber, R.E.; Fuchs, S.; Layer, F.; Sommer, A.; Bender, J.K.; Thürmer, A.; Werner, G.; Strommenger, B. Corrigendum: Genome-Wide Association Studies for the Detection of Genetic Variants Associated With Daptomycin and Ceftaroline Resistance in Staphylococcus aureus. Front. Microbiol. 2021, 12, 686197. [Google Scholar] [CrossRef]
- Biggel, M.; Xavier, B.B.; Johnson, J.R.; Nielsen, K.L.; Frimodt-Møller, N.; Matheeussen, V.; Goossens, H.; Moons, P.; Van Puyvelde, S. Horizontally Acquired papGII-Containing Pathogenicity Islands Underlie the Emergence of Invasive Uropathogenic Escherichia coli Lineages. Nat. Commun. 2020, 11, 5968. [Google Scholar] [CrossRef]
- Denamur, E.; Condamine, B.; Esposito-Farèse, M.; Royer, G.; Clermont, O.; Laouenan, C.; Lefort, A.; de Lastours, V.; Galardini, M.; The Colibafi; et al. Genome Wide Association Study of Escherichia coli Bloodstream Infection Isolates Identifies Genetic Determinants for the Portal of Entry but Not Fatal Outcome. PLoS Genet. 2022, 18, e1010112. [Google Scholar] [CrossRef]
- Coll, F.; Gouliouris, T.; Bruchmann, S.; Phelan, J.; Raven, K.E.; Clark, T.G.; Parkhill, J.; Peacock, S.J. PowerBacGWAS: A Computational Pipeline to Perform Power Calculations for Bacterial Genome-Wide Association Studies. Commun. Biol. 2022, 5, 266. [Google Scholar] [CrossRef]
- Gygli, S.M.; Keller, P.M.; Ballif, M.; Blöchliger, N.; Hömke, R.; Reinhard, M.; Loiseau, C.; Ritter, C.; Sander, P.; Borrell, S.; et al. Whole-Genome Sequencing for Drug Resistance Profile Prediction in. Antimicrob. Agents Chemother. 2019, 63, e02175-18. [Google Scholar] [CrossRef]
- Juraschek, K.; Borowiak, M.; Tausch, S.H.; Malorny, B.; Käsbohrer, A.; Otani, S.; Schwarz, S.; Meemken, D.; Deneke, C.; Hammerl, J.A. Outcome of Different Sequencing and Assembly Approaches on the Detection of Plasmids and Localization of Antimicrobial Resistance Genes in Commensal. Microorganisms 2021, 9, 598. [Google Scholar] [CrossRef]
- Wareth, G.; Linde, J.; Hammer, P.; Nguyen, N.H.; Nguyen, T.N.M.; Splettstoesser, W.D.; Makarewicz, O.; Neubauer, H.; Sprague, L.D.; Pletz, M.W. Phenotypic and WGS-Derived Antimicrobial Resistance Profiles of Clinical and Non-Clinical Acinetobacter baumannii Isolates from Germany and Vietnam. Int. J. Antimicrob. Agents 2020, 56, 106127. [Google Scholar] [CrossRef]
- Case, R.J.; Boucher, Y.; Dahllöf, I.; Holmström, C.; Doolittle, W.F.; Kjelleberg, S. Use of 16S rRNA and rpoB Genes as Molecular Markers for Microbial Ecology Studies. Appl. Environ. Microbiol. 2007, 73, 278–288. [Google Scholar] [CrossRef]
- Stebner, A.; Ensser, A.; Geißdörfer, W.; Bozhkov, Y.; Lang, R. Molecular Diagnosis of Polymicrobial Brain Abscesses with 16S-rDNA-Based next-Generation Sequencing. Clin. Microbiol. Infect. 2021, 27, 76–82. [Google Scholar] [CrossRef]
- Ishihara, T.; Watanabe, N.; Inoue, S.; Aoki, H.; Tsuji, T.; Yamamoto, B.; Yanagi, H.; Oki, M.; Kryukov, K.; Nakagawa, S.; et al. Usefulness of next-Generation DNA Sequencing for the Diagnosis of Urinary Tract Infection. Drug Discov. Ther. 2020, 14, 42–49. [Google Scholar] [CrossRef]
- Fida, M.; Wolf, M.J.; Hamdi, A.; Vijayvargiya, P.; Esquer Garrigos, Z.; Khalil, S.; Greenwood-Quaintance, K.E.; Thoendel, M.J.; Patel, R. Detection of Pathogenic Bacteria From Septic Patients Using 16S Ribosomal RNA Gene-Targeted Metagenomic Sequencing. Clin. Infect. Dis. 2021, 73, 1165–1172. [Google Scholar] [CrossRef]
- Oberauner, L.; Zachow, C.; Lackner, S.; Högenauer, C.; Smolle, K.-H.; Berg, G. The Ignored Diversity: Complex Bacterial Communities in Intensive Care Units Revealed by 16S Pyrosequencing. Sci. Rep. 2013, 3, 1413. [Google Scholar] [CrossRef]
- Edgar, R.C. Updating the 97% Identity Threshold for 16S Ribosomal RNA OTUs. Bioinformatics 2018, 34, 2371–2375. [Google Scholar] [CrossRef]
- Mysara, M.; Leys, N.; Raes, J.; Monsieurs, P. IPED: A Highly Efficient Denoising Tool for Illumina MiSeq Paired-End 16S rRNA Gene Amplicon Sequencing Data. BMC Bioinform. 2016, 17, 192. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Navas-Molina, J.A.; Mercier, C.; Xu, Z.Z.; Mahé, F.; He, Y.; Zhou, H.-W.; Rognes, T.; Caporaso, J.G.; Knight, R. Open-Source Sequence Clustering Methods Improve the State Of the Art. mSystems 2016, 1, e00003-15. [Google Scholar] [CrossRef]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A Perspective on 16S rRNA Operational Taxonomic Unit Clustering Using Sequence Similarity. NPJ Biofilms Microbiomes 2016, 2, 16004. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA Gene Sequencing for Species and Strain-Level Microbiome Analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- Cai, H.; Archambault, M.; Prescott, J.F. 16S Ribosomal RNA Sequence-Based Identification of Veterinary Clinical Bacteria. J. Vet. Diagn. Investig. 2003, 15, 465–469. [Google Scholar] [CrossRef]
- Hoffman, C.; Siddiqui, N.Y.; Fields, I.; Gregory, W.T.; Simon, H.M.; Mooney, M.A.; Wolfe, A.J.; Karstens, L. Species-Level Resolution of Female Bladder Microbiota from 16S rRNA Amplicon Sequencing. mSystems 2021, 6, e0051821. [Google Scholar] [CrossRef]
- Meola, M.; Rifa, E.; Shani, N.; Delbès, C.; Berthoud, H.; Chassard, C. DAIRYdb: A Manually Curated Reference Database for Improved Taxonomy Annotation of 16S rRNA Gene Sequences from Dairy Products. BMC Genomics 2019, 20, 560. [Google Scholar] [CrossRef]
- Peker, N.; Garcia-Croes, S.; Dijkhuizen, B.; Wiersma, H.H.; van Zanten, E.; Wisselink, G.; Friedrich, A.W.; Kooistra-Smid, M.; Sinha, B.; Rossen, J.W.A.; et al. A Comparison of Three Different Bioinformatics Analyses of the 16S-23S rRNA Encoding Region for Bacterial Identification. Front. Microbiol. 2019, 10, 620. [Google Scholar] [CrossRef]
- Seol, D.; Lim, J.S.; Sung, S.; Lee, Y.H.; Jeong, M.; Cho, S.; Kwak, W.; Kim, H. Microbial Identification Using rRNA Operon Region: Database and Tool for Metataxonomics with Long-Read Sequence. Microbiol. Spectr. 2022, 10, e0201721. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Bowman, J.S.; Ducklow, H.W. Microbial Communities Can Be Described by Metabolic Structure: A General Framework and Application to a Seasonally Variable, Depth-Stratified Microbial Community from the Coastal West Antarctic Peninsula. PLoS ONE 2015, 10, e0135868. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting Functional Profiles from Metagenomic 16S rRNA Data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Laroche, O.; Pochon, X.; Wood, S.A.; Keeley, N. Beyond Taxonomy: Validating Functional Inference Approaches in the Context of Fish-Farm Impact Assessments. Mol. Ecol. Resour. 2021, 21, 2264–2277. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun Metagenomics, from Sampling to Analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Gomez-Valero, L.; Buchrieser, C. Metagenomic Approaches in Microbial Ecology: An Update on Whole-Genome and Marker Gene Sequencing Analyses. Microb. Genom. 2020, 6, mgen000409. [Google Scholar] [CrossRef]
- Junier, T.; Huber, M.; Schmutz, S.; Kufner, V.; Zagordi, O.; Neuenschwander, S.; Ramette, A.; Kubacki, J.; Bachofen, C.; Qi, W.; et al. Viral Metagenomics in the Clinical Realm: Lessons Learned from a Swiss-Wide Ring Trial. Genes 2019, 10, 655. [Google Scholar] [CrossRef]
- De Vries, J.J.C.; Brown, J.R.; Fischer, N.; Sidorov, I.A.; Morfopoulou, S.; Huang, J.; Munnink, B.B.O.; Sayiner, A.; Bulgurcu, A.; Rodriguez, C.; et al. Benchmark of Thirteen Bioinformatic Pipelines for Metagenomic Virus Diagnostics Using Datasets from Clinical Samples. J. Clin. Virol. 2021, 141, 104908. [Google Scholar] [CrossRef]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C.; et al. Clinical Metagenomic Sequencing for Diagnosis of Meningitis and Encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Shishido, A.A.; Noe, M.; Saharia, K.; Luethy, P. Clinical Impact of a Metagenomic Microbial Plasma Cell-Free DNA next-Generation Sequencing Assay on Treatment Decisions: A Single-Center Retrospective Study. BMC Infect. Dis. 2022, 22, 372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhuang, Y.; Xiao, Z.-H.; Li, C.-Y.; Zhang, F.; Huang, W.-Q.; Zhang, M.; Peng, X.-M.; Liu, C. Diagnosis and Surveillance of Neonatal Infections by Metagenomic Next-Generation Sequencing. Front. Microbiol. 2022, 13, 855988. [Google Scholar] [CrossRef]
- Li, D.; Gai, W.; Zhang, J.; Cheng, W.; Cui, N.; Wang, H. Metagenomic Next-Generation Sequencing for the Microbiological Diagnosis of Abdominal Sepsis Patients. Front. Microbiol. 2022, 13, 816631. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Fang, K.; Shi, X.; Yang, D.; Zhao, L.; Yu, W.; Zheng, Y.; Xu, Y.; Ma, X.; Chen, L.; et al. Enhanced DNA and RNA Pathogen Detection via Metagenomic Sequencing in Patients with Pneumonia. J. Transl. Med. 2022, 20, 195. [Google Scholar] [CrossRef] [PubMed]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore Metagenomics Enables Rapid Clinical Diagnosis of Bacterial Lower Respiratory Infection. Nat. Biotechnol. 2019, 37, 783–792. [Google Scholar] [CrossRef]
- Sanabria, A.; Hjerde, E.; Johannessen, M.; Sollid, J.E.; Simonsen, G.S.; Hanssen, A.-M. Shotgun-Metagenomics on Positive Blood Culture Bottles Inoculated With Prosthetic Joint Tissue: A Proof of Concept Study. Front. Microbiol. 2020, 11, 1687. [Google Scholar] [CrossRef]
- Noone, J.C.; Helmersen, K.; Leegaard, T.M.; Skråmm, I.; Aamot, H.V. Rapid Diagnostics of Orthopaedic-Implant-Associated Infections Using Nanopore Shotgun Metagenomic Sequencing on Tissue Biopsies. Microorganisms 2021, 9, 97. [Google Scholar] [CrossRef]
- Lamoureux, C.; Surgers, L.; Fihman, V.; Gricourt, G.; Demontant, V.; Trawinski, E.; N’Debi, M.; Gomart, C.; Royer, G.; Launay, N.; et al. Prospective Comparison Between Shotgun Metagenomics and Sanger Sequencing of the 16S rRNA Gene for the Etiological Diagnosis of Infections. Front. Microbiol. 2022, 13, 761873. [Google Scholar] [CrossRef]
- Gu, W.; Deng, X.; Lee, M.; Sucu, Y.D.; Arevalo, S.; Stryke, D.; Federman, S.; Gopez, A.; Reyes, K.; Zorn, K.; et al. Rapid Pathogen Detection by Metagenomic next-Generation Sequencing of Infected Body Fluids. Nat. Med. 2021, 27, 115–124. [Google Scholar] [CrossRef]
- Martin, S.; Heavens, D.; Lan, Y.; Horsfield, S.; Clark, M.D.; Leggett, R.M. Nanopore Adaptive Sampling: A Tool for Enrichment of Low Abundance Species in Metagenomic Samples. Genome Biol. 2022, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Gan, M.; Wu, B.; Yan, G.; Li, G.; Sun, L.; Lu, G.; Zhou, W. Combined Nanopore Adaptive Sequencing and Enzyme-Based Host Depletion Efficiently Enriched Microbial Sequences and Identified Missing Respiratory Pathogens. BMC Genom. 2021, 22, 732. [Google Scholar] [CrossRef] [PubMed]
- Marquet, M.; Zöllkau, J.; Pastuschek, J.; Viehweger, A.; Schleußner, E.; Makarewicz, O.; Pletz, M.W.; Ehricht, R.; Brandt, C. Evaluation of Microbiome Enrichment and Host DNA Depletion in Human Vaginal Samples Using Oxford Nanopore’s Adaptive Sequencing. Sci. Rep. 2022, 12, 4000. [Google Scholar] [CrossRef]
- Arikawa, K.; Ide, K.; Kogawa, M.; Saeki, T.; Yoda, T.; Endoh, T.; Matsuhashi, A.; Takeyama, H.; Hosokawa, M. Recovery of Strain-Resolved Genomes from Human Microbiome through an Integration Framework of Single-Cell Genomics and Metagenomics. Microbiome 2021, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Vicedomini, R.; Quince, C.; Darling, A.E.; Chikhi, R. Strainberry: Automated Strain Separation in Low-Complexity Metagenomes Using Long Reads. Nat. Commun. 2021, 12, 4485. [Google Scholar] [CrossRef]
- Quince, C.; Nurk, S.; Raguideau, S.; James, R.; Soyer, O.S.; Summers, J.K.; Limasset, A.; Eren, A.M.; Chikhi, R.; Darling, A.E. STRONG: Metagenomics Strain Resolution on Assembly Graphs. Genome Biol. 2021, 22, 214. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-Scale Sequence Alignment Catalyses Viral Discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef]
- Zayed, A.A.; Wainaina, J.M.; Dominguez-Huerta, G.; Pelletier, E.; Guo, J.; Mohssen, M.; Tian, F.; Pratama, A.A.; Bolduc, B.; Zablocki, O.; et al. Cryptic and Abundant Marine Viruses at the Evolutionary Origins of Earth’s RNA Virome. Science 2022, 376, 156–162. [Google Scholar] [CrossRef]
- Djemiel, C.; Maron, P.-A.; Terrat, S.; Dequiedt, S.; Cottin, A.; Ranjard, L. Inferring Microbiota Functions from Taxonomic Genes: A Review. Gigascience 2022, 11, giab090. [Google Scholar] [CrossRef]
- Belcour, A.; Frioux, C.; Aite, M.; Bretaudeau, A.; Hildebrand, F.; Siegel, A. Metage2Metabo, Microbiota-Scale Metabolic Complementarity for the Identification of Key Species. Elife 2020, 9, e61968. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Chng, K.R.; Li, C.; Bertrand, D.; Ng, A.H.Q.; Kwah, J.S.; Low, H.M.; Tong, C.; Natrajan, M.; Zhang, M.H.; Xu, L.; et al. Cartography of Opportunistic Pathogens and Antibiotic Resistance Genes in a Tertiary Hospital Environment. Nat. Med. 2020, 26, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Urban, L.; Holzer, A.; Baronas, J.J.; Hall, M.B.; Braeuninger-Weimer, P.; Scherm, M.J.; Kunz, D.J.; Perera, S.N.; Martin-Herranz, D.E.; Tipper, E.T.; et al. Freshwater Monitoring by Nanopore Sequencing. Elife 2021, 10, e61504. [Google Scholar] [CrossRef]
- Mallapaty, S. How Sewage Could Reveal True Scale of Coronavirus Outbreak. Nature 2020, 580, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, R.S.; Munk, P.; Njage, P.; van Bunnik, B.; McNally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J.; et al. Global Monitoring of Antimicrobial Resistance Based on Metagenomics Analyses of Urban Sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Perry, M.R.; Lepper, H.C.; McNally, L.; Wee, B.A.; Munk, P.; Warr, A.; Moore, B.; Kalima, P.; Philip, C.; de Roda Husman, A.M.; et al. Secrets of the Hospital Underbelly: Patterns of Abundance of Antimicrobial Resistance Genes in Hospital Wastewater Vary by Specific Antimicrobial and Bacterial Family. Front. Microbiol. 2021, 12, 703560. [Google Scholar] [CrossRef]
- Röttjers, L.; Faust, K. Fast and Flexible Analysis of Linked Microbiome Data with Mako. Nat. Methods 2022, 19, 51–54. [Google Scholar] [CrossRef]
- Kishikawa, T.; Maeda, Y.; Nii, T.; Motooka, D.; Matsumoto, Y.; Matsushita, M.; Matsuoka, H.; Yoshimura, M.; Kawada, S.; Teshigawara, S.; et al. Metagenome-Wide Association Study of Gut Microbiome Revealed Novel Aetiology of Rheumatoid Arthritis in the Japanese Population. Ann. Rheum. Dis. 2020, 79, 103–111. [Google Scholar] [CrossRef]
- Wang, D.; Doestzada, M.; Chen, L.; Andreu-Sánchez, S.; van den Munckhof, I.C.L.; Augustijn, H.E.; Koehorst, M.; Ruiz-Moreno, A.J.; Bloks, V.W.; Riksen, N.P.; et al. Characterization of Gut Microbial Structural Variations as Determinants of Human Bile Acid Metabolism. Cell Host Microbe 2021, 29, 1802–1814.e5. [Google Scholar] [CrossRef]
- Tomofuji, Y.; Maeda, Y.; Oguro-Igashira, E.; Kishikawa, T.; Yamamoto, K.; Sonehara, K.; Motooka, D.; Matsumoto, Y.; Matsuoka, H.; Yoshimura, M.; et al. Metagenome-Wide Association Study Revealed Disease-Specific Landscape of the Gut Microbiome of Systemic Lupus Erythematosus in Japanese. Ann. Rheum. Dis. 2021, 80, 1575–1583. [Google Scholar] [CrossRef]
- Kishikawa, T.; Tomofuji, Y.; Inohara, H.; Okada, Y. OMARU: A Robust and Multifaceted Pipeline for Metagenome-Wide Association Study. NAR Genom Bioinform 2022, 4, lqac019. [Google Scholar] [CrossRef] [PubMed]
- Vega, L.; Herrera, G.; Muñoz, M.; Patarroyo, M.A.; Ramírez, J.D. Occurrence of in Patients with Infection. Pathogens 2020, 9, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casimiro-Soriguer, C.S.; Loucera, C.; Peña-Chilet, M.; Dopazo, J. Towards a Metagenomics Machine Learning Interpretable Model for Understanding the Transition from Adenoma to Colorectal Cancer. Sci. Rep. 2022, 12, 450. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.-N.; Jiao, N.; Tan, J.-C.; Wang, Z.; Wu, D.; Wang, A.-J.; Chen, J.; Tao, L.; Zhou, C.; Fang, W.; et al. Multi-Kingdom Microbiota Analyses Identify Bacterial-Fungal Interactions and Biomarkers of Colorectal Cancer across Cohorts. Nat. Microbiol. 2022, 7, 238–250. [Google Scholar] [CrossRef]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic Analysis of Colorectal Cancer Datasets Identifies Cross-Cohort Microbial Diagnostic Signatures and a Link with Choline Degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef]
- Hayase, E.; Jenq, R.R. Role of the Intestinal Microbiome and Microbial-Derived Metabolites in Immune Checkpoint Blockade Immunotherapy of Cancer. Genome Med. 2021, 13, 107. [Google Scholar] [CrossRef]
- Dekker, J.P. Metagenomics for Clinical Infectious Disease Diagnostics Steps Closer to Reality. J. Clin. Microbiol. 2018, 56, e00850-18. [Google Scholar] [CrossRef]
- Wang, W.-L.; Xu, S.-Y.; Ren, Z.-G.; Tao, L.; Jiang, J.-W.; Zheng, S.-S. Application of Metagenomics in the Human Gut Microbiome. World J. Gastroenterol. 2015, 21, 803–814. [Google Scholar] [CrossRef]
- Ahn, T.-H.; Chai, J.; Pan, C. Sigma: Strain-Level Inference of Genomes from Metagenomic Analysis for Biosurveillance. Bioinformatics 2015, 31, 170–177. [Google Scholar] [CrossRef]
- Ko, K.K.K.; Chng, K.R.; Nagarajan, N. Metagenomics-Enabled Microbial Surveillance. Nat. Microbiol 2022, 7, 486–496. [Google Scholar] [CrossRef]
- Sukhum, K.V.; Diorio-Toth, L.; Dantas, G. Genomic and Metagenomic Approaches for Predictive Surveillance of Emerging Pathogens and Antibiotic Resistance. Clin. Pharmacol. Ther. 2019, 106, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Pulido-Tamayo, S.; Sánchez-Rodríguez, A.; Swings, T.; Van den Bergh, B.; Dubey, A.; Steenackers, H.; Michiels, J.; Fostier, J.; Marchal, K. Frequency-Based Haplotype Reconstruction from Deep Sequencing Data of Bacterial Populations. Nucleic Acids Res. 2015, 43, e105. [Google Scholar] [CrossRef] [PubMed]
- Tringe, S.G.; Rubin, E.M. Metagenomics: DNA Sequencing of Environmental Samples. Nat. Rev. Genet. 2005, 6, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Tringe, S.G.; von Mering, C.; Kobayashi, A.; Salamov, A.A.; Chen, K.; Chang, H.W.; Podar, M.; Short, J.M.; Mathur, E.J.; Detter, J.C.; et al. Comparative Metagenomics of Microbial Communities. Science 2005, 308, 554–557. [Google Scholar] [CrossRef]
- Roberts, L.W.; Harris, P.N.A.; Forde, B.M.; Ben Zakour, N.L.; Catchpoole, E.; Stanton-Cook, M.; Phan, M.-D.; Sidjabat, H.E.; Bergh, H.; Heney, C.; et al. Integrating Multiple Genomic Technologies to Investigate an Outbreak of Carbapenemase-Producing Enterobacter hormaechei. Nat. Commun. 2020, 11, 466. [Google Scholar] [CrossRef]
- Roberts, L.W.; Forde, B.M.; Hurst, T.; Ling, W.; Nimmo, G.R.; Bergh, H.; George, N.; Hajkowicz, K.; McNamara, J.F.; Lipman, J.; et al. Genomic Surveillance, Characterization and Intervention of a Polymicrobial Multidrug-Resistant Outbreak in Critical Care. Microb. Genom. 2021, 7, mgen000530. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, S.; Yin, Y.; Zhang, H.; Needham, D.M.; Evans, E.D.; Dai, C.L.; Lu, P.J.; Alm, E.J.; Weitz, D.A. High-Throughput, Single-Microbe Genomics with Strain Resolution, Applied to a Human Gut Microbiome. Science 2022, 376, eabm1483. [Google Scholar] [CrossRef]
- Anyansi, C.; Straub, T.J.; Manson, A.L.; Earl, A.M.; Abeel, T. Computational Methods for Strain-Level Microbial Detection in Colony and Metagenome Sequencing Data. Front. Microbiol. 2020, 11, 1925. [Google Scholar] [CrossRef]
- Votintseva, A.A.; Bradley, P.; Pankhurst, L.; Del Ojo Elias, C.; Loose, M.; Nilgiriwala, K.; Chatterjee, A.; Smith, E.G.; Sanderson, N.; Walker, T.M.; et al. Same-Day Diagnostic and Surveillance Data for Tuberculosis via Whole-Genome Sequencing of Direct Respiratory Samples. J. Clin. Microbiol. 2017, 55, 1285–1298. [Google Scholar] [CrossRef]
- Marcelino, V.R.; Holmes, E.C.; Sorrell, T.C. The Use of Taxon-Specific Reference Databases Compromises Metagenomic Classification. BMC Genom. 2020, 21, 184. [Google Scholar] [CrossRef] [PubMed]
- Zlitni, S.; Bishara, A.; Moss, E.L.; Tkachenko, E.; Kang, J.B.; Culver, R.N.; Andermann, T.M.; Weng, Z.; Wood, C.; Handy, C.; et al. Strain-Resolved Microbiome Sequencing Reveals Mobile Elements That Drive Bacterial Competition on a Clinical Timescale. Genome Med. 2020, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, V.; Chernevskaya, E.; Vasiluev, P.; Ivanov, A.; Tolstoganov, I.; Shafranskaya, D.; Ulyantsev, V.; Korobeynikov, A.; Razin, S.V.; Beloborodova, N.; et al. Hi-C Metagenomics in the ICU: Exploring Clinically Relevant Features of Gut Microbiome in Chronically Critically Ill Patients. Front. Microbiol. 2021, 12, 770323. [Google Scholar] [CrossRef]
- Li, H.; Homer, N. A Survey of Sequence Alignment Algorithms for next-Generation Sequencing. Brief. Bioinform. 2010, 11, 473–483. [Google Scholar] [CrossRef]
- Hong, C.; Manimaran, S.; Shen, Y.; Perez-Rogers, J.F.; Byrd, A.L.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. PathoScope 2.0: A Complete Computational Framework for Strain Identification in Environmental or Clinical Sequencing Samples. Microbiome 2014, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Clement, N.L.; Snell, Q.; Clement, M.J.; Hollenhorst, P.C.; Purwar, J.; Graves, B.J.; Cairns, B.R.; Johnson, W.E. The GNUMAP Algorithm: Unbiased Probabilistic Mapping of Oligonucleotides from next-Generation Sequencing. Bioinformatics 2010, 26, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for Enhanced Metagenomic Taxonomic Profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Nayfach, S.; Rodriguez-Mueller, B.; Garud, N.; Pollard, K.S. An Integrated Metagenomics Pipeline for Strain Profiling Reveals Novel Patterns of Bacterial Transmission and Biogeography. Genome Res. 2016, 26, 1612–1625. [Google Scholar] [CrossRef]
- Tu, Q.; He, Z.; Zhou, J. Strain/species Identification in Metagenomes Using Genome-Specific Markers. Nucleic Acids Res. 2014, 42, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudadio, I.; Fulci, V.; Palone, F.; Stronati, L.; Cucchiara, S.; Carissimi, C. Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. OMICS 2018, 22, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.A.; Hasan, N.A.; Klaassen, M.; Karathia, H.; Colwell, R.R.; Shrout, J.D. Prosthetic Joint Infections Present Diverse and Unique Microbial Communities Using Combined Whole-Genome Shotgun Sequencing and Culturing Methods. J. Med. Microbiol. 2019, 68, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Brumfield, K.D.; Huq, A.; Colwell, R.R.; Olds, J.L.; Leddy, M.B. Microbial Resolution of Whole Genome Shotgun and 16S Amplicon Metagenomic Sequencing Using Publicly Available NEON Data. PLoS ONE 2020, 15, e0228899. [Google Scholar] [CrossRef] [PubMed]
- Araos, R.; Battaglia, T.; Ugalde, J.A.; Rojas-Herrera, M.; Blaser, M.J.; D’Agata, E.M.C. Fecal Microbiome Characteristics and the Resistome Associated With Acquisition of Multidrug-Resistant Organisms Among Elderly Subjects. Front. Microbiol. 2019, 10, 2260. [Google Scholar] [CrossRef]


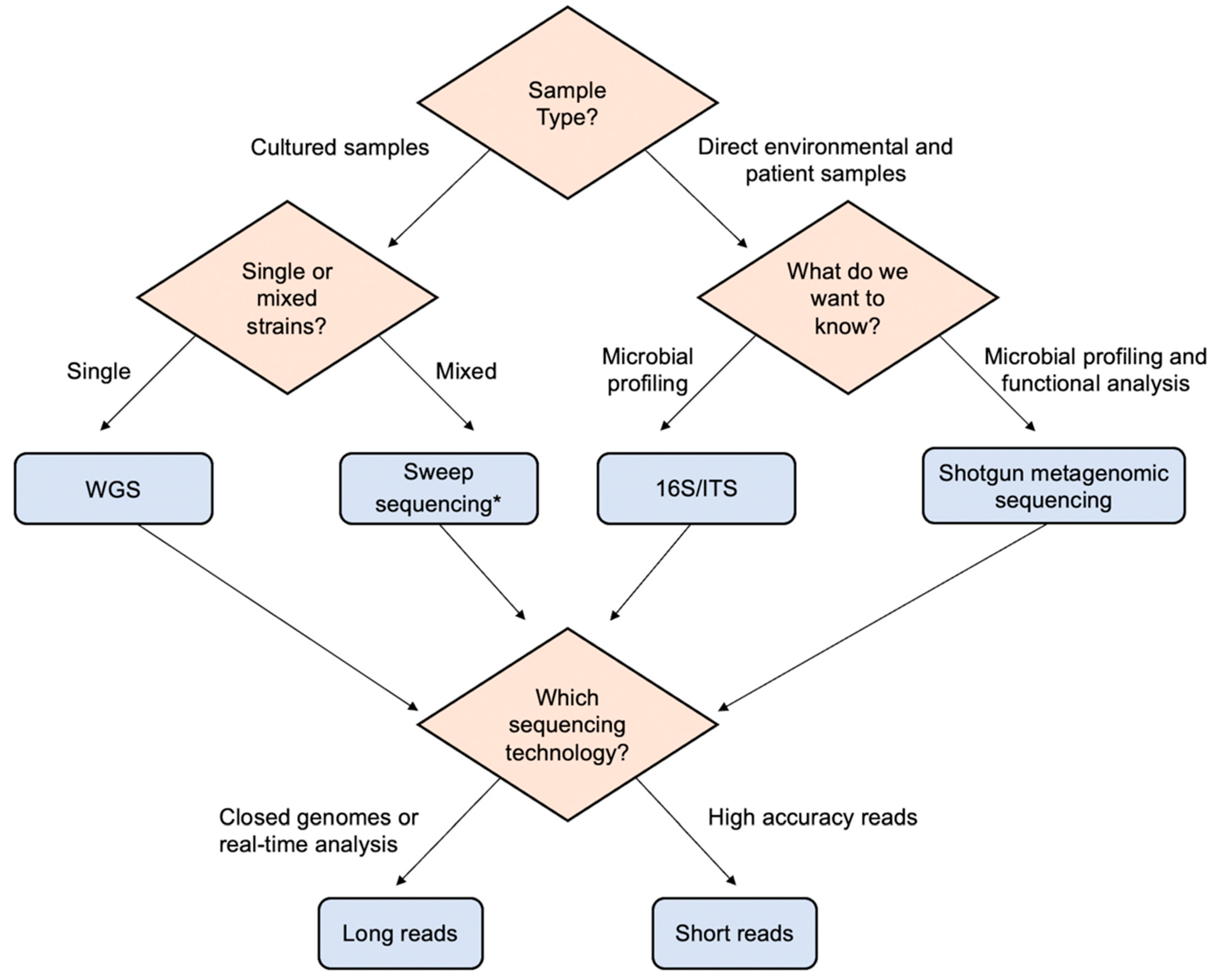{kind=link}
| Parameters | WGS | 16S/ITS | Shotgun Metagenomic Sequencing |
|---|---|---|---|
| Sample | Cultured or enriched microorganisms | Swabs from body sites, stool samples, body fluids or tissue samples, and sewage | Swabs from body sites, stool samples, body fluids or tissue samples fecal matter, and sewage |
| Species identification | Yes | Yes | Yes |
| Degree of resolution | Species-Strain level | Genus-Species level | Species-Strain level |
| Complete genome | Complete genome possible depending on sequencing platforms | No | Near complete to gapped genomes. |
| SNP analysis | Yes | No | Yes |
| GWAS | Yes | No | Yes |
| Identification of virulence factors and resistance genes | Yes | No | Yes |
| Microbial community profiling | No | Yes | Yes |
| Cost | $$ | $ | $$$ |
| Turnaround Time (TAT) | + | ++ | +++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purushothaman, S.; Meola, M.; Egli, A. Combination of Whole Genome Sequencing and Metagenomics for Microbiological Diagnostics. Int. J. Mol. Sci. 2022, 23, 9834. https://doi.org/10.3390/ijms23179834
Purushothaman S, Meola M, Egli A. Combination of Whole Genome Sequencing and Metagenomics for Microbiological Diagnostics. International Journal of Molecular Sciences. 2022; 23(17):9834. https://doi.org/10.3390/ijms23179834
Chicago/Turabian StylePurushothaman, Srinithi, Marco Meola, and Adrian Egli. 2022. "Combination of Whole Genome Sequencing and Metagenomics for Microbiological Diagnostics" International Journal of Molecular Sciences 23, no. 17: 9834. https://doi.org/10.3390/ijms23179834
APA StylePurushothaman, S., Meola, M., & Egli, A. (2022). Combination of Whole Genome Sequencing and Metagenomics for Microbiological Diagnostics. International Journal of Molecular Sciences, 23(17), 9834. https://doi.org/10.3390/ijms23179834