
The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma
 ,
,  ,
,  ,
,  ,
, 
 ,
, 
Abstract
:
1. Introduction
2. Results
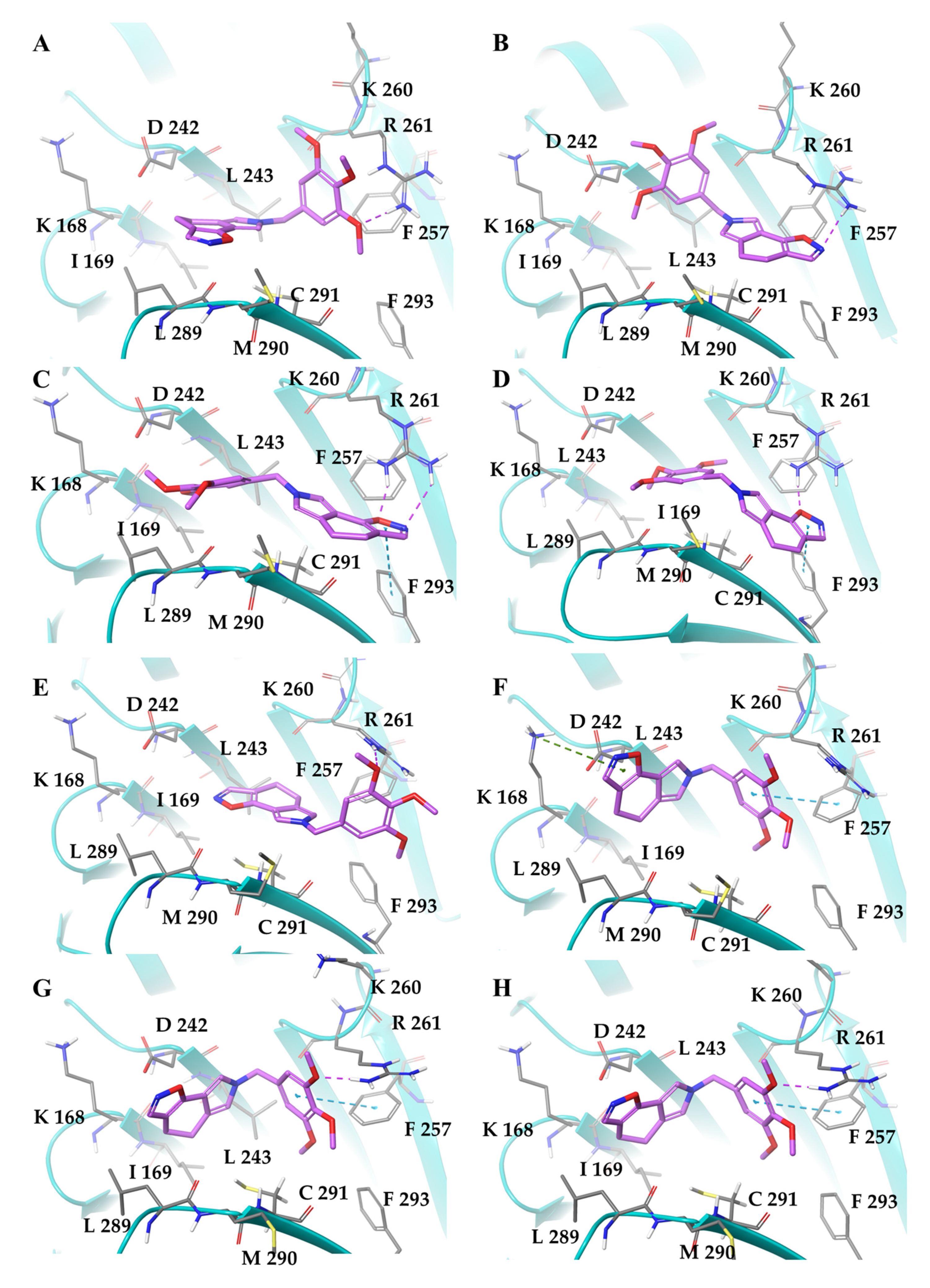
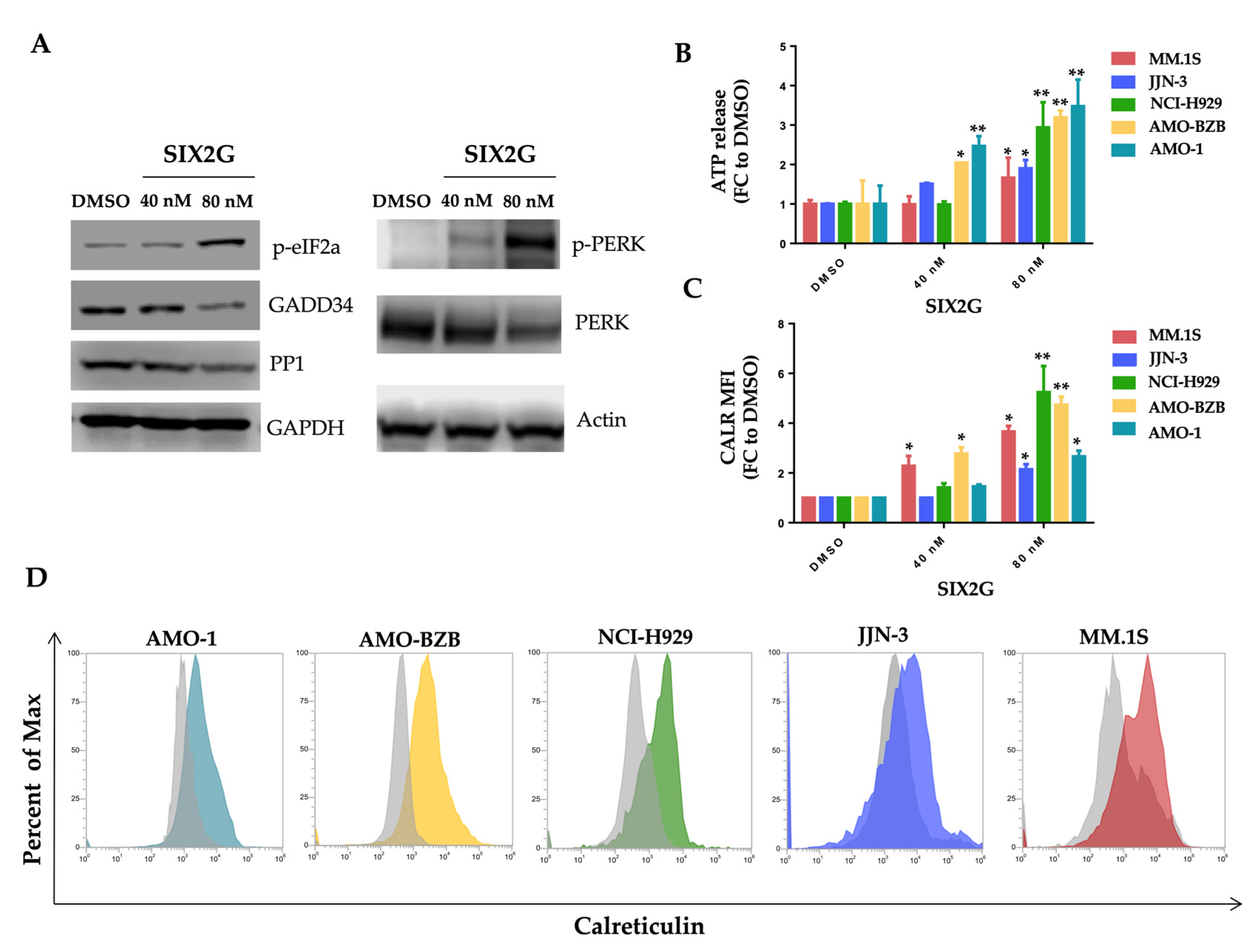
2.1. SIX2G Could Sterically Prevent PP1/GADD34 Association by Inducing Immunogenic Cell Death
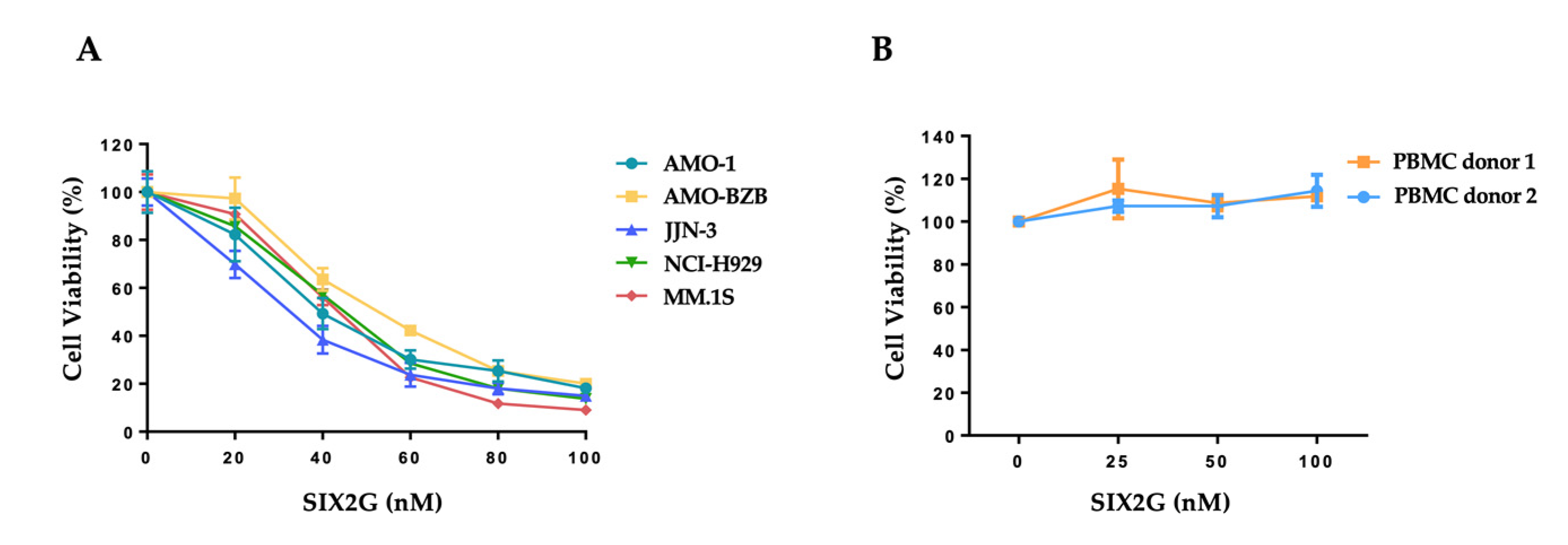
2.2. SIX2G Inhibits Cell Viability and Induces Apoptosis of Multiple Myeloma Cells
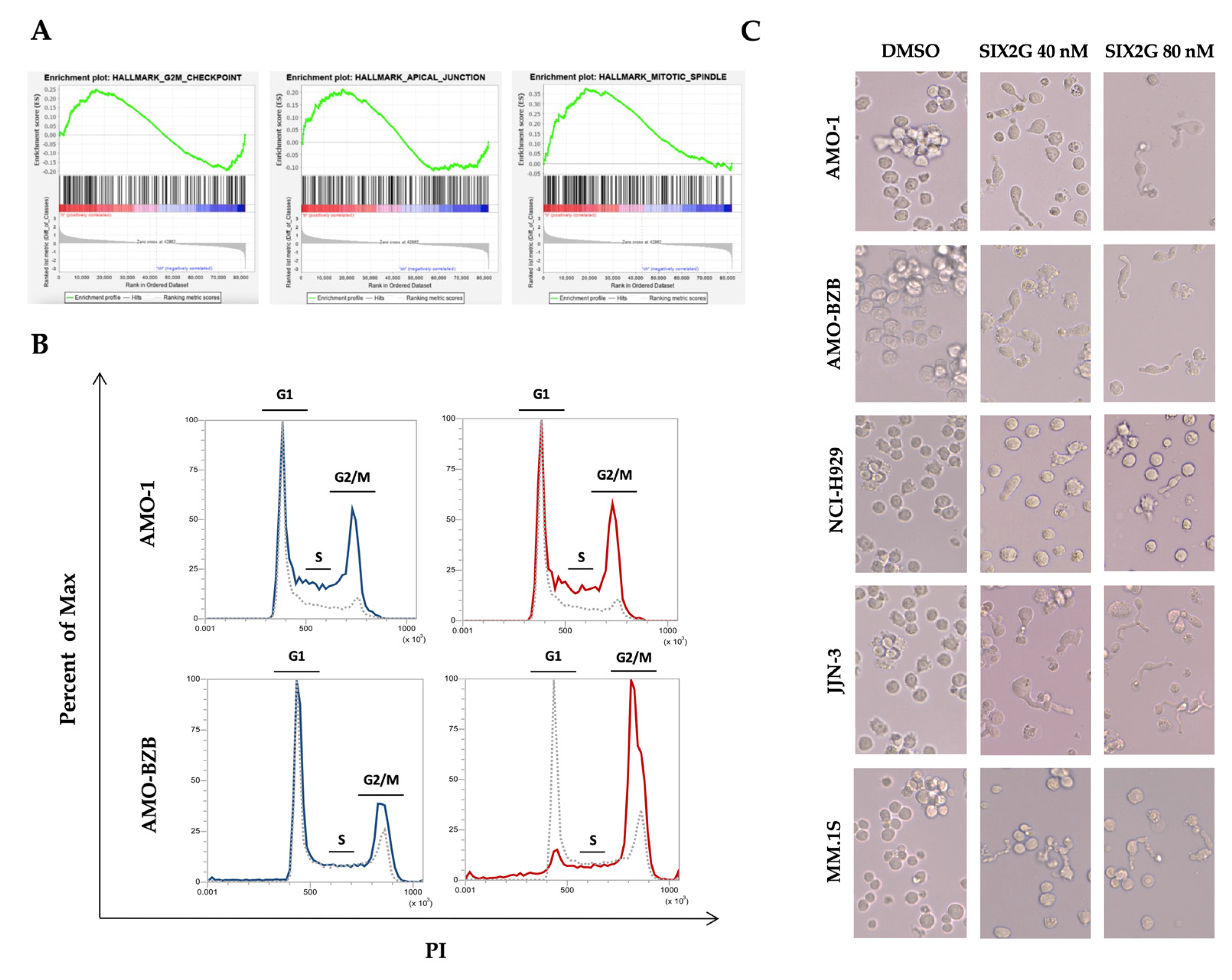
2.3. SIX2G Induces Alteration in Mitotic-Spindle and Apical Junction Pathways and Triggers Cell Cycle Arrest in G2/M Phases
2.4. SIX2G Induces Calreticulin Exposure and ATP Release by Activating an Immunogenic Cascade
2.5. SIX2G Treatment on Tumor Cells Promotes DC Maturation, Activation and Cross-Priming
3. Discussion
4. Materials and Methods
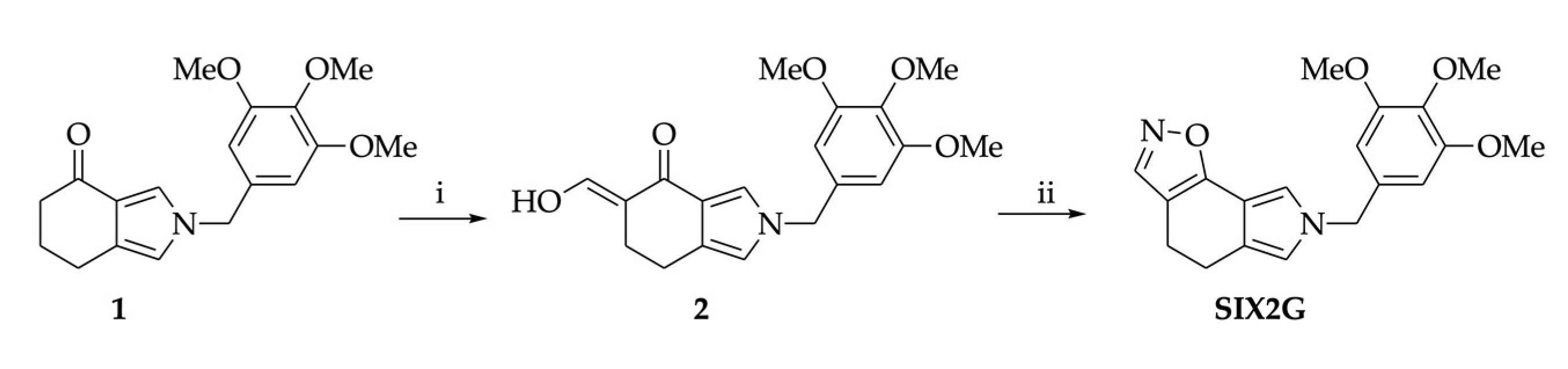
4.1. Chemical Synthesis of SIX2G
4.2. Docking Simulations
4.3. Cell Cultures
4.4. Dentric Cells Generation
4.5. Cell Viability, Apoptosis and Cell Cycle Analysis
4.6. Gene Expression Profiling
4.7. Western Blot Analysis
4.8. Calreticulin Exposure and ATP Release Analyses
4.9. DC Maturation and Phagocytosis Assay
4.10. DC Cross-Priming
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Wordeman, L.; Vicente, J.J. Microtubule Targeting Agents in Disease: Classic Drugs, Novel Roles. Cancers 2021, 13, 5650. [Google Scholar] [CrossRef]
- Barreca, M.; Stathis, A.; Barraja, P.; Bertoni, F. An overview on anti-tubulin agents for the treatment of lymphoma patients. Pharmacol. Ther. 2020, 211, 107552. [Google Scholar] [CrossRef]
- Cermak, V.; Dostal, V.; Jelinek, M.; Libusova, L.; Kovar, J.; Rosel, D.; Brabek, J. Microtubule-targeting agents and their impact on cancer treatment. Eur. J. Cell Biol. 2020, 99, 151075. [Google Scholar] [CrossRef]
- Bocci, G.; Di Paolo, A.; Danesi, R. The pharmacological bases of the antiangiogenic activity of paclitaxel. Angiogenesis 2013, 16, 481–492. [Google Scholar] [CrossRef]
- Bijman, M.N.; van Nieuw Amerongen, G.P.; Laurens, N.; van Hinsbergh, V.W.; Boven, E. Microtubule-targeting agents inhibit angiogenesis at subtoxic concentrations, a process associated with inhibition of Rac1 and Cdc42 activity and changes in the endothelial cytoskeleton. Mol. Cancer Ther. 2006, 5, 2348–2357. [Google Scholar] [CrossRef]
- Lu, H.; Murtagh, J.; Schwartz, E.L. The microtubule binding drug laulimalide inhibits vascular endothelial growth factor-induced human endothelial cell migration and is synergistic when combined with docetaxel (taxotere). Mol. Pharmacol. 2006, 69, 1207–1215. [Google Scholar] [CrossRef]
- Kamath, K.; Smiyun, G.; Wilson, L.; Jordan, M.A. Mechanisms of inhibition of endothelial cell migration by taxanes. Cytoskeleton 2014, 71, 46–60. [Google Scholar] [CrossRef]
- Fong, A.; Durkin, A.; Lee, H. The Potential of Combining Tubulin-Targeting Anticancer Therapeutics and Immune Therapy. Int. J. Mol. Sci. 2019, 20, 586. [Google Scholar] [CrossRef]
- Serpico, A.F.; Visconti, R.; Grieco, D. Exploiting immune-dependent effects of microtubule-targeting agents to improve efficacy and tolerability of cancer treatment. Cell Death Dis. 2020, 11, 361. [Google Scholar] [CrossRef]
- Sato-Kaneko, F.; Wang, X.; Yao, S.; Hosoya, T.; Lao, F.S.; Messer, K.; Pu, M.; Shukla, N.M.; Cottam, H.B.; Chan, M.; et al. Discovery of a Novel Microtubule Targeting Agent as an Adjuvant for Cancer Immunotherapy. BioMed Res. Int. 2018, 2018, 8091283. [Google Scholar] [CrossRef]
- Novais, P.; Silva, P.M.A.; Amorim, I.; Bousbaa, H. Second-Generation Antimitotics in Cancer Clinical Trials. Pharmaceutics 2021, 13, 1011. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal chemistry of combretastatin A4: Present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Schmidt, J.M.; Hogan, F. Antineoplastic agents. 291. Isolation and synthesis of combretastatins A-4, A-5, and A-6(1a). J. Med. Chem. 1995, 38, 1666–1672. [Google Scholar] [CrossRef]
- Perez-Perez, M.J.; Priego, E.M.; Bueno, O.; Martins, M.S.; Canela, M.D.; Liekens, S. Blocking Blood Flow to Solid Tumors by Destabilizing Tubulin: An Approach to Targeting Tumor Growth. J. Med. Chem. 2016, 59, 8685–8711. [Google Scholar] [CrossRef]
- Sun, C.M.; Lin, L.G.; Yu, H.J.; Cheng, C.Y.; Tsai, Y.C.; Chu, C.W.; Din, Y.H.; Chau, Y.P.; Don, M.J. Synthesis and cytotoxic activities of 4,5-diarylisoxazoles. Bioorg. Med. Chem. Lett. 2007, 17, 1078–1081. [Google Scholar] [CrossRef]
- Kaffy, J.; Pontikis, R.; Carrez, D.; Croisy, A.; Monneret, C.; Florent, J.C. Isoxazole-type derivatives related to combretastatin A-4, synthesis and biological evaluation. Bioorg. Med. Chem. 2006, 14, 4067–4077. [Google Scholar] [CrossRef]
- Barraja, P.; Caracausi, L.; Diana, P.; Spano, V.; Montalbano, A.; Carbone, A.; Parrino, B.; Cirrincione, G. Synthesis and antiproliferative activity of the ring system [1,2]oxazolo [4,5-g]indole. ChemMedChem 2012, 7, 1901–1904. [Google Scholar] [CrossRef]
- Spano, V.; Pennati, M.; Parrino, B.; Carbone, A.; Montalbano, A.; Cilibrasi, V.; Zuco, V.; Lopergolo, A.; Cominetti, D.; Diana, P.; et al. Preclinical Activity of New [1,2]Oxazolo [5,4-e]isoindole Derivatives in Diffuse Malignant Peritoneal Mesothelioma. J. Med. Chem. 2016, 59, 7223–7238. [Google Scholar] [CrossRef]
- Spano, V.; Pennati, M.; Parrino, B.; Carbone, A.; Montalbano, A.; Lopergolo, A.; Zuco, V.; Cominetti, D.; Diana, P.; Cirrincione, G.; et al. [1,2]Oxazolo [5,4-e]isoindoles as promising tubulin polymerization inhibitors. Eur. J. Med. Chem. 2016, 124, 840–851. [Google Scholar] [CrossRef]
- Spano, V.; Rocca, R.; Barreca, M.; Giallombardo, D.; Montalbano, A.; Carbone, A.; Raimondi, M.V.; Gaudio, E.; Bortolozzi, R.; Bai, R.; et al. Pyrrolo [2′,3′:3,4]cyclohepta [1,2-d][1,2]oxazoles, a New Class of Antimitotic Agents Active against Multiple Malignant Cell Types. J. Med. Chem. 2020, 63, 12023–12042. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Raimondi, M.V.; Montalbano, A.; Bai, R.; Gaudio, E.; Alcaro, S.; Hamel, E.; Bertoni, F.; Barraja, P. Evaluation of [1,2]oxazolo [5,4-e]isoindoles in lymphoma cells. Eur. J. Cancer 2020, 138 (Suppl. S2), S35–S36. [Google Scholar] [CrossRef]
- Van de Donk, N.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef]
- Bianchi, G.; Richardson, P.G.; Anderson, K.C. Promising therapies in multiple myeloma. Blood 2015, 126, 300–310. [Google Scholar] [CrossRef]
- Caracciolo, D.; Scionti, F.; Juli, G.; Altomare, E.; Golino, G.; Todoerti, K.; Grillone, K.; Riillo, C.; Arbitrio, M.; Iannone, M.; et al. Exploiting MYC-induced PARPness to target genomic instability in multiple myeloma. Haematologica 2021, 106, 185–195. [Google Scholar] [CrossRef]
- Burger, R.; Le Gouill, S.; Tai, Y.T.; Shringarpure, R.; Tassone, P.; Neri, P.; Podar, K.; Catley, L.; Hideshima, T.; Chauhan, D.; et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol. Cancer Ther. 2009, 8, 26–35. [Google Scholar] [CrossRef]
- Caracciolo, D.; Riillo, C.; Juli, G.; Scionti, F.; Todoerti, K.; Polera, N.; Grillone, K.; Fiorillo, L.; Arbitrio, M.; Di Martino, M.T.; et al. miR-22 Modulates Lenalidomide Activity by Counteracting MYC Addiction in Multiple Myeloma. Cancers 2021, 13, 4365. [Google Scholar] [CrossRef]
- Rossi, M.; Di Martino, M.T.; Morelli, E.; Leotta, M.; Rizzo, A.; Grimaldi, A.; Misso, G.; Tassone, P.; Caraglia, M. Molecular targets for the treatment of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 757–767. [Google Scholar] [CrossRef]
- Offidani, M.; Corvatta, L.; More, S.; Olivieri, A. Novel Experimental Drugs for Treatment of Multiple Myeloma. J. Exp. Pharmacol. 2021, 13, 245–264. [Google Scholar] [CrossRef]
- Fabian, K.P.; Wolfson, B.; Hodge, J.W. From Immunogenic Cell Death to Immunogenic Modulation: Select Chemotherapy Regimens Induce a Spectrum of Immune-Enhancing Activities in the Tumor Microenvironment. Front. Oncol. 2021, 11, 728018. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Senovilla, L.; Menger, L.; Chaput, N.; Kroemer, G. Immunogenic tumor cell death for optimal anticancer therapy: The calreticulin exposure pathway. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 3100–3104. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Kepp, O.; Galluzzi, L.; Giordanetto, F.; Tesniere, A.; Vitale, I.; Martins, I.; Schlemmer, F.; Adjemian, S.; Zitvogel, L.; Kroemer, G. Disruption of the PP1/GADD34 complex induces calreticulin exposure. Cell Cycle 2009, 8, 3971–3977. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.; Wu, H.X.; Xu, R.H. Advancing to the era of cancer immunotherapy. Cancer Commun. 2021, 41, 803–829. [Google Scholar] [CrossRef]
- Caracciolo, D.; Riillo, C.; Arbitrio, M.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Error-prone DNA repair pathways as determinants of immunotherapy activity: An emerging scenario for cancer treatment. Int. J. Cancer 2020, 147, 2658–2668. [Google Scholar] [CrossRef]
- Di Martino, M.T.; Riillo, C.; Scionti, F.; Grillone, K.; Polera, N.; Caracciolo, D.; Arbitrio, M.; Tagliaferri, P.; Tassone, P. miRNAs and lncRNAs as Novel Therapeutic Targets to Improve Cancer Immunotherapy. Cancers 2021, 13, 1587. [Google Scholar] [CrossRef]
- Lin, X.; Ammosova, T.; Choy, M.S.; Pietzsch, C.A.; Ivanov, A.; Ahmad, A.; Saygideger, Y.; Kumari, N.; Kovalskyy, D.; Uren, A.; et al. Targeting the Non-catalytic RVxF Site of Protein Phosphatase-1 With Small Molecules for Ebola Virus Inhibition. Front. Microbiol. 2019, 10, 2145. [Google Scholar] [CrossRef] [PubMed]
- Ammosova, T.; Platonov, M.; Yedavalli, V.R.; Obukhov, Y.; Gordeuk, V.R.; Jeang, K.T.; Kovalskyy, D.; Nekhai, S. Small molecules targeted to a non-catalytic “RVxF” binding site of protein phosphatase-1 inhibit HIV-1. PLoS ONE 2012, 7, e39481. [Google Scholar] [CrossRef] [PubMed]
- Ammosova, T.; Pietzsch, C.A.; Saygideger, Y.; Ilatovsky, A.; Lin, X.; Ivanov, A.; Kumari, N.; Jerebtsova, M.; Kulkarni, A.; Petukhov, M.; et al. Protein Phosphatase 1-Targeting Small-Molecule C31 Inhibits Ebola Virus Replication. J. Infect. Dis. 2018, 218 (Suppl. S5), S627–S635. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Sauvat, A.; Humeau, J.; Gomes-da-Silva, L.C.; Iribarren, K.; Forveille, S.; Garcia, P.; Zhao, L.; Liu, P.; Zitvogel, L.; et al. eIF2alpha phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ. 2018, 25, 1375–1393. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Nakamura, K.; Smyth, M.J.; Martinet, L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood 2020, 136, 2731–2740. [Google Scholar] [CrossRef]
- Krejcik, J.; Barnkob, M.B.; Nyvold, C.G.; Larsen, T.S.; Barington, T.; Abildgaard, N. Harnessing the Immune System to Fight Multiple Myeloma. Cancers 2021, 13, 4546. [Google Scholar] [CrossRef]
- Uckun, F.M. Overcoming the Immunosuppressive Tumor Microenvironment in Multiple Myeloma. Cancers 2021, 13, 2018. [Google Scholar] [CrossRef]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Malvestiti, S.; Prabhala, R.H.; Talluri, S.; et al. Bortezomib induces anti-multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov. 2021, 2, 468–483. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kroemer, G. Bortezomib Induces Immunogenic Cell Death in Multiple Myeloma. Blood Cancer Discov. 2021, 2, 405–407. [Google Scholar] [CrossRef]
- Velankar, S.; Burley, S.K.; Kurisu, G.; Hoch, J.C.; Markley, J.L. The Protein Data Bank Archive. Methods Mol. Biol. 2021, 2305, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Kelker, M.S.; Page, R.; Peti, W. Crystal structures of protein phosphatase-1 bound to nodularin-R and tautomycin: A novel scaffold for structure-based drug design of serine/threonine phosphatase inhibitors. J. Mol. Biol. 2009, 385, 11–21. [Google Scholar] [CrossRef]
- Ragusa, M.J.; Dancheck, B.; Critton, D.A.; Nairn, A.C.; Page, R.; Peti, W. Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat. Struct. Mol. Biol. 2010, 17, 459–464. [Google Scholar] [CrossRef]
- Choy, M.S.; Yusoff, P.; Lee, I.C.; Newton, J.C.; Goh, C.W.; Page, R.; Shenolikar, S.; Peti, W. Structural and Functional Analysis of the GADD34:PP1 eIF2alpha Phosphatase. Cell Rep. 2015, 11, 1885–1891. [Google Scholar] [CrossRef]
- Schrödinger. Schrödinger Suites; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger. Protein Preparation Wizard; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Shivakumar, D.; Harder, E.; Damm, W.; Friesner, R.A.; Sherman, W. Improving the Prediction of Absolute Solvation Free Energies Using the Next Generation OPLS Force Field. J. Chem. Theory Comput. 2012, 8, 2553–2558. [Google Scholar] [CrossRef]
- Schrödinger. Maestro; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger. LigPrep; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger. Prime; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Maruca, A.; Ambrosio, A.F.; Lupia, A.; Romeo, I.; Rocca, R.; Moraca, F.; Talarico, C.; Bagetta, D.; Catalano, R.; Costa, G.; et al. Computer-based techniques for lead identification and optimization I: Basics. Phys. Sci. Rev. 2019, 4, 113. [Google Scholar] [CrossRef]
- Caracciolo, D.; Riillo, C.; Ballerini, A.; Gaipa, G.; Lhermitte, L.; Rossi, M.; Botta, C.; Duroyon, E.; Grillone, K.; Gallo Cantafio, M.E.; et al. Therapeutic afucosylated monoclonal antibody and bispecific T-cell engagers for T-cell acute lymphoblastic leukemia. J. Immunother. Cancer 2021, 9, e002026. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Model | ΔGbind | ΔGCoul | ΔGLipo | ΔGSolv | ΔGvdW |
|---|---|---|---|---|---|
| 3E7A chain A | −51.72 | −17.78 | −29.04 | 27.20 | −29.61 |
| 3E7A chain B | −46.90 | −13.12 | −25.99 | 23.24 | −30.87 |
| 3E7B chain A | −42.50 | −7.76 | −28.00 | 22.98 | −30.99 |
| 3E7B chain B | −39.70 | −1.79 | −28.62 | 18.86 | −31.61 |
| 4XPN chain A | −41.56 | −6.96 | −22.45 | 14.25 | −22.46 |
| 4XPN chain B | −53.17 | −13.14 | −31.37 | 23.57 | −30.97 |
| 3EGG chain A | −57.66 | −11.89 | −34.15 | 20.25 | −32.64 |
| 3EGG chain B | −57.33 | −12.70 | −33.59 | 21.36 | −30.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grillone, K.; Riillo, C.; Rocca, R.; Ascrizzi, S.; Spanò, V.; Scionti, F.; Polerà, N.; Maruca, A.; Barreca, M.; Juli, G.; et al. The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 10222. https://doi.org/10.3390/ijms231810222
Grillone K, Riillo C, Rocca R, Ascrizzi S, Spanò V, Scionti F, Polerà N, Maruca A, Barreca M, Juli G, et al. The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma. International Journal of Molecular Sciences. 2022; 23(18):10222. https://doi.org/10.3390/ijms231810222
Chicago/Turabian StyleGrillone, Katia, Caterina Riillo, Roberta Rocca, Serena Ascrizzi, Virginia Spanò, Francesca Scionti, Nicoletta Polerà, Annalisa Maruca, Marilia Barreca, Giada Juli, and et al. 2022. "The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma" International Journal of Molecular Sciences 23, no. 18: 10222. https://doi.org/10.3390/ijms231810222
APA StyleGrillone, K., Riillo, C., Rocca, R., Ascrizzi, S., Spanò, V., Scionti, F., Polerà, N., Maruca, A., Barreca, M., Juli, G., Arbitrio, M., Di Martino, M. T., Caracciolo, D., Tagliaferri, P., Alcaro, S., Montalbano, A., Barraja, P., & Tassone, P. (2022). The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma. International Journal of Molecular Sciences, 23(18), 10222. https://doi.org/10.3390/ijms231810222