

Mucins and CFTR: Their Close Relationship


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Affected Regions in CF Lung Disease
2.1. Role of CFTR in the Lungs
2.2. Structural and Regional Specificity of the Lungs
2.3. Clinical Observations Related to Small Airway Diseases in CF
2.4. Airflow, Shear Forces, and Mucus Clearance in the Small Airways
3. Regional Distribution of Secretory Mucins in the Conducting Airways
3.1. Mucin Concentrations in Health and Disease
3.2. Airway Mucins and Their Functions
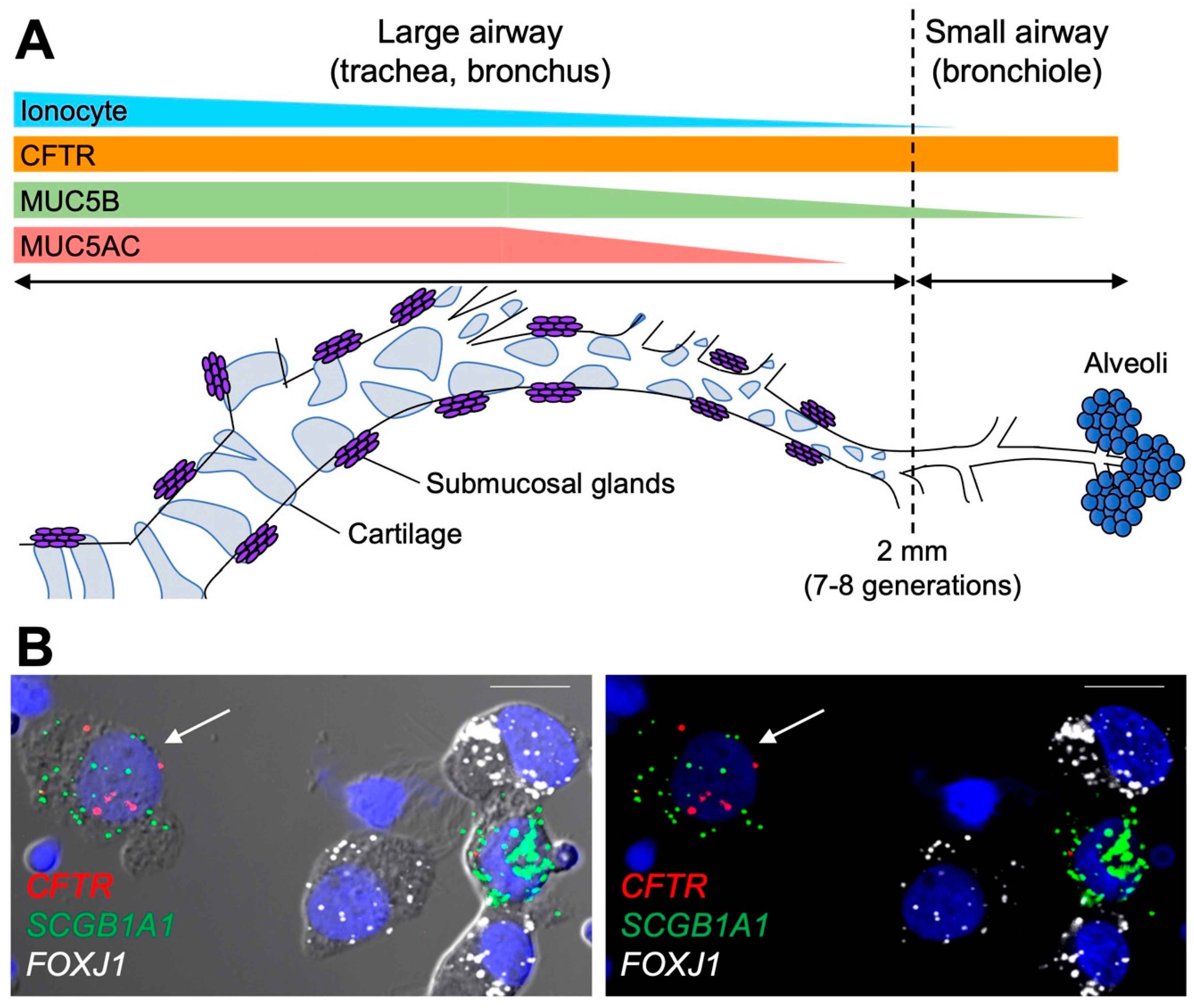
3.3. MUC5AC and MUC5B Distribution in the Respiratory Tree
4. Cell Types Expressing CFTR in Human Conducting Airways
4.1. Early Findings on Cell Types Expressing CFTR
4.2. New Insights with Single-Cell Transcriptional Profiling
4.3. The Role of Secretory Cells in the Small Airways
5. CFTR Malfunction Affects Mucus Properties
5.1. Effects of Inflammation on Mucus
5.2. Low HCO3− and pH Influence Mucin Network Organization
5.3. Mucin Hyperconcentration Alters Mucus Viscous and Elastic Behavior
6. Biochemical Alterations Impair Mucus Transport
6.1. In Vitro CF Models Replicate Impaired Mucociliary Transport
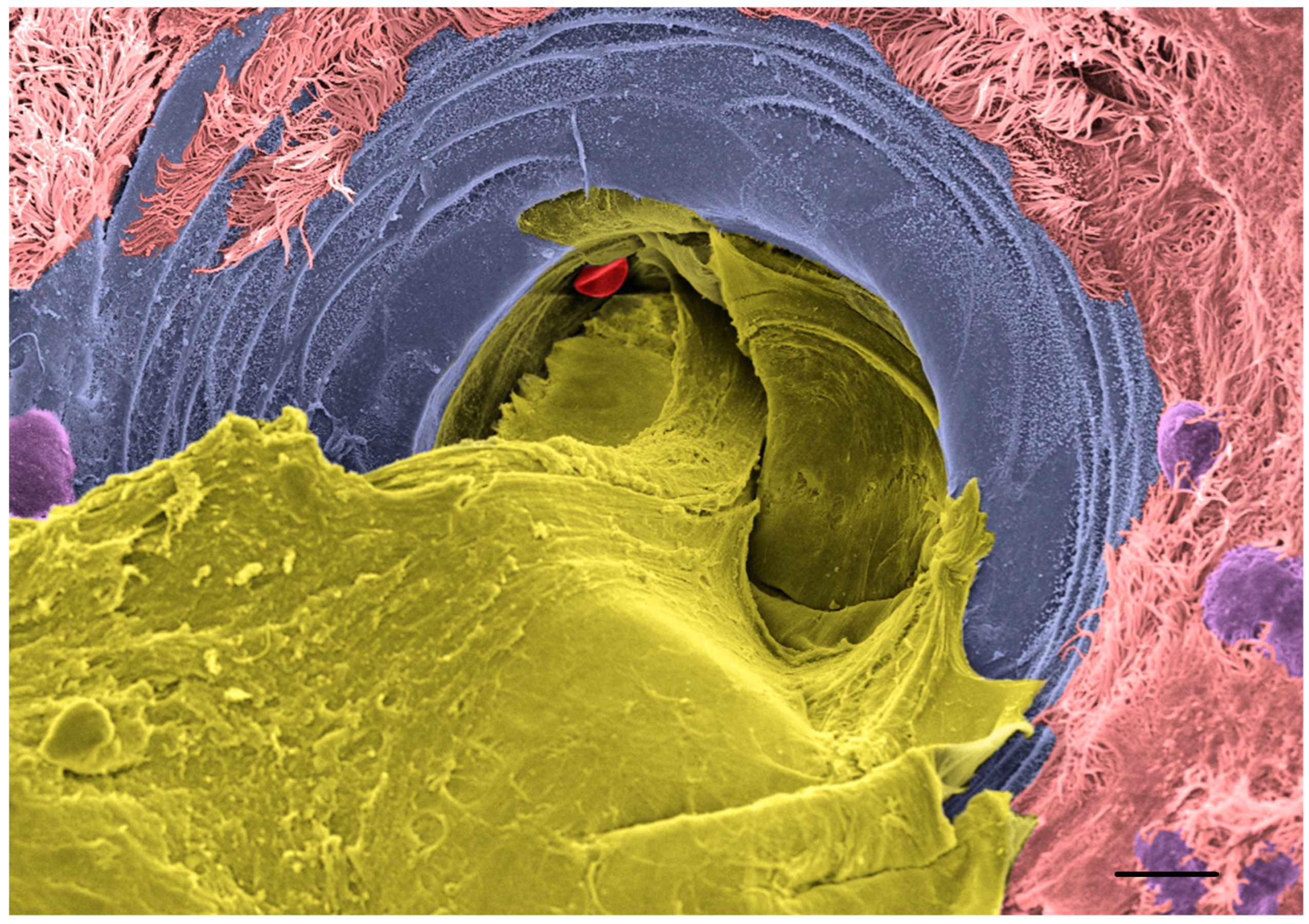
6.2. Defective Functioning of the CF Submucosal Glands
6.3. CFTR Rescue Reverses Mucus Abnormality In Vitro
7. Discussion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ehre, C.; Ridley, C.; Thornton, D.J. Cystic fibrosis: An inherited disease affecting mucin-producing organs. Int. J. Biochem. Cell Biol. 2014, 52, 136–145. [Google Scholar] [CrossRef]
- Morrison, C.B.; Markovetz, M.R.; Ehre, C. Mucus, mucins, and cystic fibrosis. Pediatric Pulmonol. 2019, 54 (Suppl. 3), S84–S96. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C. Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 2007, 261, 5–16. [Google Scholar] [CrossRef]
- Quinton, P.M. Cystic fibrosis: Impaired bicarbonate secretion and mucoviscidosis. Lancet 2008, 372, 415–417. [Google Scholar] [CrossRef]
- Andersen, D.H. Cystic Fibrosis of the Pancreas and Its Relation to Celiac Disease: A Clinical and Pathologic Study. Am. J. Dis. Child. 1938, 56, 344–399. [Google Scholar] [CrossRef]
- Navarro, S. Historical compilation of cystic fibrosis. Gastroenterol. Hepatol. 2016, 39, 36–42. [Google Scholar] [CrossRef]
- Pinto, M.C.; Silva, I.A.L.; Figueira, M.F.; Amaral, M.D.; Lopes-Pacheco, M. Pharmacological Modulation of Ion Channels for the Treatment of Cystic Fibrosis. J. Exp. Pharmacol. 2021, 13, 693–723. [Google Scholar] [CrossRef]
- Davis, P.B. Cystic fibrosis since 1938. Am. J. Respir. Crit. Care Med. 2006, 173, 475–482. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Skilton, M.; Krishan, A.; Patel, S.; Sinha, I.P.; Southern, K.W.; Cochrane Cystic Fibrosis and Genetic Disorders Group. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst. Rev. 2019, 2019, CD009841. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Rowe, S.M.; Accurso, F.J.; Aitken, M.L.; Amin, R.S.; Ashlock, M.A.; Ballmann, M.; Boyle, M.P.; Bronsveld, I.; Campbell, P.W.; et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012, 67, 12–18. [Google Scholar] [CrossRef]
- Gramegna, A.; Contarini, M.; Aliberti, S.; Casciaro, R.; Blasi, F.; Castellani, C. From Ivacaftor to Triple Combination: A Systematic Review of Efficacy and Safety of CFTR Modulators in People with Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 5882. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T.; VX11-661-101 Study Group. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.D.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Esther, C.R., Jr.; Muhlebach, M.S.; Ehre, C.; Hill, D.B.; Wolfgang, M.C.; Kesimer, M.; Ramsey, K.A.; Markovetz, M.R.; Garbarine, I.C.; Forest, M.G.; et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med. 2019, 11, eaav3488. [Google Scholar] [CrossRef]
- Henderson, A.G.; Ehre, C.; Button, B.; Abdullah, L.H.; Cai, L.-H.; Leigh, M.W.; DeMaria, G.C.; Matsui, H.; Donaldson, S.H.; Davis, C.W.; et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Investig. 2014, 124, 3047–3060. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.X.; Ostedgaard, L.S.; Hoegger, M.J.; Moninger, T.O.; Karp, P.H.; McMenimen, J.D.; Choudhury, B.; Varki, A.; Stoltz, D.A.; Welsh, M.J. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J. Clin. Investig. 2016, 126, 879–891. [Google Scholar] [PubMed]
- Shah, V.S.; Meyerholz, D.K.; Tang, X.X.; Reznikov, L.; Abou Alaiwa, M.; Ernst, S.E.; Karp, P.H.; Wohlford-Lenane, C.L.; Heilmann, K.P.; Leidinger, M.R.; et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science 2016, 351, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Puvvadi, R.; Borisov, S.M.; Shaw, N.C.; Klimant, I.; Berry, L.J.; Montgomery, S.T.; Nguyen, T.; Kreda, S.M.; Kicic, A.; et al. Airway surface liquid pH is not acidic in children with cystic fibrosis. Nat. Commun. 2017, 8, 1409. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R. Chapter II–Organization of the Human Lung. In Morphometry of the Human Lung; Academic Press: New York, NY, USA, 1963; pp. 4–9. [Google Scholar]
- Weibel, E.R. Morphometry of the human lung: The state of the art after two decades. Bull. Eur. Physiopathol. Respir. 1979, 15, 999–1013. [Google Scholar]
- Spencer, H. Morphometry of the Human Lung. J. Anat. 1964, 98, 457. [Google Scholar]
- Widdicombe, J.H.; Wine, J.J. Airway Gland Structure and Function. Physiol. Rev. 2015, 95, 1241–1319. [Google Scholar]
- Wickstrom, C.; Davies, J.; Eriksen, G.V.; Veerman, E.C.I.; Carlstedt, I. MUC5B is a major gel-forming, oligomeric mucin from human salivary gland, respiratory tract and endocervix: Identification of glycoforms and C-terminal cleavage. Biochem. J. 1998, 334, 685–693. [Google Scholar] [CrossRef]
- Jeong, J.H.; Joo, N.S.; Hwang, P.H.; Wine, J.J. Mucociliary clearance and submucosal gland secretion in the ex vivo ferret trachea. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L83–L93. [Google Scholar] [CrossRef] [PubMed]
- Ermund, A.; Meiss, L.N.; Rodriguez-Pineiro, A.M.; Bähr, A.; Nilsson, H.E.; Trillo-Muyo, S.; Ridley, C.; Thornton, D.J.; Wine, J.J.; Hebert, H.; et al. The normal trachea is cleaned by MUC5B mucin bundles from the submucosal glands coated with the MUC5AC mucin. Biochem. Biophys. Res. Commun. 2017, 492, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Radicioni, G.; Papanikolas, M.J.; Stoychev, G.V.; Markovetz, M.R.; Aoki, K.; Porterfield, M.; Okuda, K.; Cardenas, S.M.B.; Gilmore, R.C.; et al. Mucus concentration–Dependent biophysical abnormalities unify submucosal gland and superficial airway dysfunction in cystic fibrosis. Sci. Adv. 2022, 8, eabm9718. [Google Scholar] [CrossRef]
- Button, B.; Cai, L.-H.; Ehre, C.; Kesimer, M.; Hill, D.B.; Sheehan, J.K.; Boucher, R.C.; Rubinstein, M. A Periciliary Brush Promotes the Lung Health by Separating the Mucus Layer from Airway Epithelia. Science 2012, 337, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.-R.; Montani, D.; Danel, C.; Dusser, D.J.; Nadel, J.A. A morphometric study of mucins and small airway plugging in cystic fibrosis. Thorax 2007, 62, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Zuelzer, W.W.; Newton, W.A., Jr. The pathogenesis of fibrocystic disease of the pancreas; A study of 36 cases with special reference to the pulmonary lesions. Pediatrics 1949, 4, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, M.N. The pathology of cystic fibrosis. In Cystic Fibrosis; Hodson, M.E., Geddes, D.M., Eds.; Chapman and Hall: London, UK, 1995; pp. 131–149. [Google Scholar]
- Khan, T.Z.; Wagener, J.S.; Bost, T.; Martinez, J.; Accurso, F.J.; Riches, D.W. Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1075–1082. [Google Scholar]
- Noah, T.L.; Murphy, P.C.; Alink, J.J.; Leigh, M.W.; Hull, W.M.; Stahlman, M.T.; Whitsett, J.A. Bronchoalveolar Lavage Fluid Surfactant Protein-A and Surfactant Protein-D Are Inversely Related to Inflammation in Early Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 685–691. [Google Scholar] [CrossRef]
- Mellins, R.B. The Site of Airway Obstruction in Cystic Fibrosis. Pediatrics 1969, 44, 315–318. [Google Scholar] [CrossRef]
- Ranganathan, S.C.; Stocks, J.; Dezateux, C.; Bush, A.; Wade, A.; Carr, S.; Castle, R.; Dinwiddie, R.; Hoo, A.-F.; Lum, S.; et al. The Evolution of Airway Function in Early Childhood Following Clinical Diagnosis of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2004, 169, 928–933. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Bennett, W.D.; Zeman, K.L.; Knowles, M.R.; Tarran, R.; Boucher, R.C. Mucus Clearance and Lung Function in Cystic Fibrosis with Hypertonic Saline. N. Engl. J. Med. 2006, 354, 241–250. [Google Scholar] [CrossRef]
- Tiddens, H.A.; Donaldson, S.H.; Rosenfeld, M.; Paré, P.D. Cystic fibrosis lung disease starts in the small airways: Can we treat it more effectively? Pediatric Pulmonol. 2010, 45, 107–117. [Google Scholar] [CrossRef]
- Bennett, W.D.; Olivier, K.N.; Zeman, K.L.; Hohneker, K.W.; Boucher, R.C.; Knowles, M.R. Effect of uridine 5′-triphosphate plus amiloride on mucociliary clearance in adult cystic fibrosis. Am. J. Respir. Crit. Care Med. 1996, 153, 1796–1801. [Google Scholar] [CrossRef]
- Schwiebert, E.M.; Benos, D.J.; Egan, M.E.; Stutts, M.J.; Guggino, W.B. CFTR Is a Conductance Regulator as well as a Chloride Channel. Physiol. Rev. 1999, 79, S145–S166. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K. The cystic fibrosis transmembrane conductance regulator and its function in epithelial transport. Rev. Physiol. Biochem. Pharmacol. 1999, 137, 1–70. [Google Scholar]
- Boon, M.; Verleden, S.E.; Bosch, B.; Lammertyn, E.J.; McDonough, J.E.; Mai, C.; Verschakelen, J.; Kemner-van de Corput, M.; Tiddens, H.A.; Proesmans, M.; et al. Morphometric Analysis of Explant Lungs in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Regnis, J.A.; Zeman, K.L.; Noone, P.G.; Knowles, M.R.; Bennett, W.D. Prolonged airway retention of insoluble particles in cystic fibrosis versus primary ciliary dyskinesia. Exp. Lung Res. 2000, 26, 149–162. [Google Scholar] [PubMed]
- Button, B.; Goodell, H.P.; Atieh, E.; Chen, Y.-C.; Williams, R.; Shenoy, S.; Lackey, E.; Shenkute, N.T.; Cai, L.-H.; Dennis, R.G.; et al. Roles of mucus adhesion and cohesion in cough clearance. Proc. Natl. Acad. Sci. USA 2018, 115, 12501–12506. [Google Scholar] [PubMed] [Green Version]
- Livraghi-Butrico, A.; Grubb, B.R.; Wilkinson, K.J.; Volmer, A.S.; Burns, K.A.; Evans, C.M.; O’Neal, W.K.; Boucher, R.C. Contribution of mucus concentration and secreted mucins Muc5ac and Muc5b to the pathogenesis of muco-obstructive lung disease. Mucosal Immunol. 2017, 10, 395–407. [Google Scholar] [CrossRef]
- Serafini, S.M.; Michaelson, E.D. Length and distribution of cilia in human and canine airways. Bull. Eur. Physiopathol. Respir. 1977, 13, 551–559. [Google Scholar]
- Murthy, P.K.L.; Sontake, V.; Tata, A.; Kobayashi, Y.; Macadlo, L.; Okuda, K.; Conchola, A.S.; Nakano, S.; Gregory, S.; Miller, L.A.; et al. Human distal lung maps and lineage hierarchies reveal a bipotent progenitor. Nature 2022, 604, 111–119. [Google Scholar]
- Smith, D.J.; Gaffney, E.A.; Blake, J.R. Modelling mucociliary clearance. Respir. Physiol. Neurobiol. 2008, 163, 178–188. [Google Scholar] [CrossRef]
- Kesimer, M.; Kirkham, S.; Pickles, R.J.; Henderson, A.G.; Alexis, N.E.; Demaria, G.; Knight, D.; Thornton, D.J.; Sheehan, J.K. Tracheobronchial air-liquid interface cell culture: A model for innate mucosal defense of the upper airways? Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L92–L100. [Google Scholar] [CrossRef]
- Kesimer, M.; Ehre, C.; Burns, K.A.; Davis, C.W.; Sheehan, J.K.; Pickles, R.J. Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal Immunol. 2013, 6, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Buisine, M.-P.; Devisme, L.; Copin, M.-C.; Durand-Réville, M.; Gosselin, B.; Aubert, J.-P.; Porchet, N. Developmental Mucin Gene Expression in the Human Respiratory Tract. Am. J. Respir. Cell Mol. Biol. 1999, 20, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, S.; Sheehan, J.K.; Knight, D.; Richardson, P.S.; Thornton, D.J. Heterogeneity of airways mucus: Variations in the amounts and glycoforms of the major oligomeric mucins MUC5AC and MUC5B. Biochem. J. 2002, 361, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Radicioni, G.; Ceppe, A.; Ford, A.A.; Alexis, N.E.; Barr, R.G.; Bleecker, E.R.; Christenson, S.A.; Cooper, C.B.; Han, M.K.; Hansel, N.N.; et al. Airway mucin MUC5AC and MUC5B concentrations and the initiation and progression of chronic obstructive pulmonary disease: An analysis of the SPIROMICS cohort. Lancet. Respir. Med. 2021, 9, 1241–1254. [Google Scholar] [CrossRef]
- Kesimer, M.; Ford, A.A.; Ceppe, A.; Radicioni, G.; Cao, R.; Davis, C.W.; Doerschuk, C.M.; Alexis, N.E.; Anderson, W.H.; Henderson, A.G.; et al. Airway Mucin Concentration as a Marker of Chronic Bronchitis. N. Engl. J. Med. 2017, 377, 911–922. [Google Scholar] [CrossRef]
- Henke, M.O.; John, G.; Germann, M.; Lindemann, H.; Rubin, B.K. MUC5AC and MUC5B mucins increase in cystic fibrosis airway secretions during pulmonary exacerbation. Am. J. Respir. Crit. Care Med. 2007, 175, 816–821. [Google Scholar]
- Ramsey, K.A.; Chen, A.C.H.; Radicioni, G.; Lourie, R.; Martin, M.; Broomfield, A.; Sheng, Y.H.; Hasnain, S.Z.; Radford-Smith, G.; Simms, L.A.; et al. Airway mucus hyperconcentration in non–cystic fibrosis bronchiectasis. Am. J. Respir. Crit. Care Med. 2020, 201, 661–670. [Google Scholar] [CrossRef]
- Anderson, W.H.; Coakley, R.D.; Button, B.; Henderson, A.G.; Zeman, K.L.; Alexis, N.E.; Peden, D.B.; Lazarowski, E.R.; Davis, C.W.; Bailey, S.; et al. The Relationship of Mucus Concentration (Hydration) to Mucus Osmotic Pressure and Transport in Chronic Bronchitis. Am. J. Respir. Crit. Care Med. 2015, 192, 182–190. [Google Scholar]
- Welsh, K.G.; Rousseau, K.; Fisher, G.; Bonser, L.R.; Bradding, P.; Brightling, C.E.; Thornton, D.J.; Gaillard, E.A. MUC5AC and a Glycosylated Variant of MUC5B Alter Mucin Composition in Children with Acute Asthma. Chest 2017, 152, 771–779. [Google Scholar] [CrossRef] [Green Version]
- Bonser, L.R.; Zlock, L.; Finkbeiner, W.; Erle, D.J. Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma. J. Clin. Investig. 2016, 126, 2367–2371. [Google Scholar] [CrossRef]
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nature 2014, 505, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, D.W.; West, J.D.; Keighren, M.A.; Flockhart, J.H.; Innes, B.A.; Dorin, J.R. Murine Submucosal Glands Are Clonally Derived and Show a Cystic Fibrosis Gene–Dependent Distribution Pattern. Am. J. Respir. Cell Mol. Biol. 1999, 20, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.B.; Edwards, C.E.; Shaffer, K.M.; Araba, K.C.; Wykoff, J.A.; Williams, D.R.; Asakura, T.; Dang, H.; Morton, L.C.; Gilmore, R.C.; et al. SARS-CoV-2 infection of airway cells causes intense viral and cell shedding, two spreading mechanisms affected by IL-13. Proc. Natl. Acad. Sci. USA 2022, 119, e2119680119. [Google Scholar] [CrossRef]
- Carpenter, J.; Wang, Y.; Gupta, R.; Li, Y.; Haridass, P.; Subramani, D.B.; Reidel, B.; Morton, L.; Ridley, C.; O’Neal, W.K.; et al. Assembly and organization of the N-terminal region of mucin MUC5AC: Indications for structural and functional distinction from MUC5B. Proc. Natl. Acad. Sci. USA 2021, 118, e2104490118. [Google Scholar] [CrossRef] [PubMed]
- Ehre, C.; Worthington, E.N.; Liesman, R.M.; Grubb, B.R.; Barbier, D.; O’Neal, W.K.; Sallenave, J.-M.; Pickles, R.J.; Boucher, R.C. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc. Natl. Acad. Sci. USA 2012, 109, 16528–16533. [Google Scholar] [CrossRef]
- Hasnain, S.; Wang, H.; Ghia, J.-E.; Haq, N.; Deng, Y.; Velcich, A.; Grencis, R.K.; Thornton, D.J.; Khan, W.I. Mucin Gene Deficiency in Mice Impairs Host Resistance to an Enteric Parasitic Infection. Gastroenterology 2010, 138, 1763–1771.e5. [Google Scholar] [CrossRef]
- Kreda, S.M.; Davis, C.W.; Rose, M.C. CFTR, Mucins, and Mucus Obstruction in Cystic Fibrosis. Cold Spring Harb. Perspect. Med. 2012, 2, a009589. [Google Scholar] [CrossRef]
- Zhu, Y.; Ehre, C.; Abdullah, L.H.; Sheehan, J.K.; Roy, M.; Evans, C.M.; Dickey, B.F.; Davis, C.W. Munc13-2−/− baseline secretion defect reveals source of oligomeric mucins in mouse airways. J. Physiol. 2008, 586, 1977–1992. [Google Scholar] [CrossRef]
- Evans, C.M.; Williams, O.W.; Tuvim, M.J.; Nigam, R.; Mixides, G.P.; Blackburn, M.R.; DeMayo, F.J.; Burns, A.R.; Smith, C.; Reynolds, S.D.; et al. Mucin Is Produced by Clara Cells in the Proximal Airways of Antigen-Challenged Mice. Am. J. Respir. Cell Mol. Biol. 2004, 31, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Goldfarbmuren, K.C.; Jackson, N.D.; Sajuthi, S.P.; Dyjack, N.; Li, K.S.; Rios, C.L.; Plender, E.G.; Montgomery, M.T.; Everman, J.L.; Bratcher, P.E.; et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun. 2020, 11, 2485. [Google Scholar] [CrossRef]
- Okuda, K.; Chen, G.; Subramani, D.B.; Wolf, M.; Gilmore, R.C.; Kato, T.; Radicioni, G.; Kesimer, M.; Chua, M.; Dang, H.; et al. Localization of Secretory Mucins MUC5AC and MUC5B in Normal/Healthy Human Airways. Am. J. Respir. Crit. Care Med. 2019, 199, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Sun, L.; Kato, T.; Okuda, K.; Martino, M.B.; Abzhanova, A.; Lin, J.M.; Gilmore, R.C.; Batson, B.D.; O’Neal, Y.K.; et al. IL-1β dominates the promucin secretory cytokine profile in cystic fibrosis. J. Clin. Investig. 2019, 129, 4433–4450. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.F.; Yankaskas, J.R.; Ernst, S.A.; Yang, Y.; Marino, C.R.; Boucher, R.C.; Cohn, J.A.; Wilson, J.M. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat. Genet. 1992, 2, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.; Zepeda, M.; Cohn, J.A.; Yankaskas, J.R.; Wilson, J. Expression of the cystic fibrosis gene in adult human lung. J. Clin. Investig. 1994, 93, 737–749. [Google Scholar] [CrossRef]
- Kreda, S.M.; Mall, M.; Mengos, A.; Rochelle, L.; Yankaskas, J.; Riordan, J.R.; Boucher, R.C. Characterization of wild-type and ∆F508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell 2005, 16, 2154–2167. [Google Scholar] [CrossRef]
- Sato, Y.; Mustafina, K.R.; Luo, Y.; Martini, C.; Thomas, D.Y.; Wiseman, P.W.; Hanrahan, J.W. Nonspecific binding of common anti-CFTR antibodies in ciliated cells of human airway epithelium. Sci. Rep. 2021, 11, 23256. [Google Scholar] [CrossRef]
- Kälin, N.; Claaß, A.; Sommer, M.; Puchelle, E.; Tümmler, B. ∆F508 CFTR protein expression in tissues from patients with cystic fibrosis. J. Clin. Investig. 1999, 103, 1379–1389. [Google Scholar] [CrossRef]
- Treutlein, B.; Brownfield, D.; Wu, A.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef]
- Wagner, D.E.; Weinreb, C.; Collins, Z.M.; Briggs, J.A.; Megason, S.G.; Klein, A.M. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 2018, 360, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Nagendran, M.; Riordan, D.P.; Harbury, P.B.; Desai, T.J. Automated cell-type classification in intact tissues by single-cell molecular profiling. eLife 2018, 7, e30510. [Google Scholar] [CrossRef]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am. J. Respir. Crit. Care Med. 2021, 203, 1275–1289. [Google Scholar] [CrossRef]
- Carraro, G.; Langerman, J.; Sabri, S.; Lorenzana, Z.; Purkayastha, A.; Zhang, G.; Konda, B.; Aros, C.J.; Calvert, B.A.; Szymaniak, A.; et al. Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Nat. Med. 2021, 27, 806–814. [Google Scholar] [CrossRef]
- Tokita, E.; Tanabe, T.; Asano, K.; Suzaki, H.; Rubin, B.K. Club cell 10-kDa protein attenuates airway mucus hypersecretion and inflammation. Eur. Respir. J. 2014, 44, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Gamez, A.S.; Gras, D.; Petit, A.; Knabe, L.; Molinari, N.; Vachier, I.; Chanez, P.; Bourdin, A. Supplementing defect in club cell secretory protein attenuates airway inflammation in COPD. Chest 2015, 147, 1467–1476. [Google Scholar] [CrossRef]
- Plopper, C.G.; Cranz, D.L.; Kemp, L.; Serabjit-Singh, C.J.; Philpot, R.M. Immunohistochemical Demonstration of Cytochrome P-450 Monooxygenase in Clara Cells throughout the Tracheobronchial Airways of the Rabbit. Exp. Lung Res. 1987, 13, 59–68. [Google Scholar] [CrossRef]
- Mango, G.W.; Johnston, C.J.; Reynolds, S.D.; Finkelstein, J.N.; Plopper, C.G.; Stripp, B.R. Clara cell secretory protein deficiency increases oxidant stress response in conducting airways. Am. J. Physiol. Cell. Mol. Physiol. 1998, 275, L348–L356. [Google Scholar] [CrossRef]
- Shamsuddin, A.K.M.; Quinton, P.M. Native Small Airways Secrete Bicarbonate. Am. J. Respir. Cell Mol. Biol. 2014, 50, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Shamsuddin, A.K.M.; Quinton, P.M. Concurrent absorption and secretion of airway surface liquids and bicarbonate secretion in human bronchioles. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L953–L960. [Google Scholar] [CrossRef]
- Blouquit, S.; Regnier, A.; Dannhoffer, L.; Fermanian, C.; Naline, E.; Boucher, R.; Chinet, T. Ion and Fluid Transport Properties of Small Airways in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tang, X.X.; Buonfiglio, L.G.V.; Comellas, A.P.; Thornell, I.M.; Ramachandran, S.; Karp, P.H.; Taft, P.J.; Sheets, K.; Abou Alaiwa, M.H.; et al. Electrolyte transport properties in distal small airways from cystic fibrosis pigs with implications for host defense. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L670–L679. [Google Scholar] [CrossRef] [PubMed]
- Van Scott, M.R.; Hester, S.; Boucher, R.C. Ion transport by rabbit nonciliated bronchiolar epithelial cells (Clara cells) in culture. Proc. Natl. Acad. Sci. USA 1987, 84, 5496–5500. [Google Scholar] [CrossRef]
- Kulaksiz, H.; Schmid, A.; Hönscheid, M.; Ramaswamy, A.; Cetin, Y. Clara cell impact in air-side activation of CFTR in small pulmonary airways. Proc. Natl. Acad. Sci. USA 2002, 99, 6796–6801. [Google Scholar] [CrossRef] [PubMed]
- Kogan, I.; Ramjeesingh, M.; Li, C.; Kidd, J.F.; Wang, Y.; Leslie, E.; Cole, S.; Bear, C.E. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J. 2003, 22, 1981–1989. [Google Scholar] [CrossRef]
- Gray, R.D.; Mccullagh, B.N.; McCray, J.P.B. NETs and CF Lung Disease: Current Status and Future Prospects. Antibiotics 2015, 4, 62–75. [Google Scholar] [CrossRef]
- Khan, M.A.; Ali, Z.S.; Sweezey, N.; Grasemann, H.; Palaniyar, N. Progression of Cystic Fibrosis Lung Disease from Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation. Genes 2019, 10, 183. [Google Scholar] [CrossRef]
- Shak, S.; Capon, D.J.; Hellmiss, R.; Marsters, S.A.; Baker, C.L. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 1990, 87, 9188–9192. [Google Scholar] [CrossRef]
- Fuchs, H.J.; Borowitz, D.S.; Christiansen, D.H.; Morris, E.M.; Nash, M.L.; Ramsey, B.W.; Rosenstein, B.J.; Smith, A.L.; Wohl, M.E. Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. N. Engl. J. Med. 1994, 331, 637–642. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Garcia-Caballero, A.; Rasmussen, J.E.; Gaillard, E.; Watson, M.J.; Olsen, J.C.; Donaldson, S.H.; Stutts, M.J.; Tarran, R. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc. Natl. Acad. Sci. USA 2009, 106, 11412–11417. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Hollinger, M.; Lachowicz-Scroggins, M.E.; Kerr, S.C.; Dunican, E.M.; Daniel, B.M.; Ghosh, S.; Erzurum, S.C.; Willard, B.; Hazen, S.L.; et al. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci. Transl. Med. 2015, 7, 276ra27. [Google Scholar] [CrossRef] [PubMed]
- Kesimer, M.; Makhov, A.M.; Griffith, J.D.; Verdugo, P.; Sheehan, J.K. Unpacking a gel-forming mucin: A view of MUC5B organization after granular release. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L15–L22. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, P. Supramolecular dynamics of mucus. Cold Spring Harb. Perspect. Med. 2012, 2, a009597. [Google Scholar] [CrossRef] [PubMed]
- Quinton, P.M. Role of epithelial HCO3− transport in mucin secretion: Lessons from cystic fibrosis. Am. J. Physiol. Cell Physiol. 2010, 299, C1222–C1233. [Google Scholar] [CrossRef]
- Georgiades, P.; Pudney, P.D.A.; Thornton, D.J.; Waigh, T.A. Particle tracking microrheology of purified gastrointestinal mucins. Biopolymers 2014, 101, 366–377. [Google Scholar] [CrossRef]
- Wagner, C.E.; Turner, B.S.; Rubinstein, M.; McKinley, G.H.; Ribbeck, K. A Rheological Study of the Association and Dynamics of MUC5AC Gels. Biomacromolecules 2017, 18, 3654–3664. [Google Scholar] [CrossRef]
- Garland, A.L.; Walton, W.G.; Coakley, R.D.; Tan, C.D.; Gilmore, R.C.; Hobbs, C.A.; Tripathy, A.; Clunes, L.A.; Bencharit, S.; Stutts, M.J.; et al. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2013, 110, 15973–15978. [Google Scholar] [CrossRef]
- Coakley, R.D.; Grubb, B.R.; Paradiso, A.M.; Gatzy, J.T.; Johnson, L.G.; Kreda, S.M.; O’Neal, W.K.; Boucher, R.C. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc. Natl. Acad. Sci. USA 2003, 100, 16083–16088. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.B.; Long, R.F.; Kissner, W.J.; Atieh, E.; Garbarine, I.C.; Markovetz, M.R.; Fontana, N.C.; Christy, M.; Habibpour, M.; Tarran, R.; et al. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur. Respir. J. 2018, 52, 1801297. [Google Scholar] [CrossRef]
- Matsui, H.; Grubb, B.R.; Tarran, R.; Randell, S.H.; Gatzy, J.T.; Davis, C.W.; Boucher, R.C. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998, 95, 1005–1015. [Google Scholar] [CrossRef]
- Ma, J.T.; Tang, C.; Kang, L.; Voynow, J.A.; Rubin, B.K. Cystic Fibrosis Sputum Rheology Correlates with Both Acute and Longitudinal Changes in Lung Function. Chest 2018, 154, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.B.; Button, B.; Rubinstein, M.; Boucher, R.C. Physiology and Pathophysiology of Human Airway Mucus. Physiol. Rev. 2022, 102, 1757–1836. [Google Scholar] [CrossRef]
- Fulcher, M.L.; Randell, S.H. Human Nasal and Tracheo–Bronchial Respiratory Epithelial Cell Culture. Methods Mol. Biol. 2013, 945, 109–121. [Google Scholar] [PubMed]
- Matsui, H.; Randell, S.H.; Peretti, S.W.; Davis, C.W.; Boucher, R.C. Coordinated clearance of periciliary liquid and mucus from airway surfaces. J. Clin. Investig. 1998, 102, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Morrison, C.B.; Shaffer, K.M.; Araba, K.C.; Markovetz, M.R.; Wykoff, J.A.; Quinney, N.L.; Hao, S.; Delion, M.F.; Flen, A.L.; Morton, L.C.; et al. Treatment of cystic fibrosis airway cells with CFTR modulators reverses aberrant mucus properties via hydration. Eur. Respir. J. 2022, 59, 2100185. [Google Scholar] [CrossRef] [PubMed]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef]
- Fischer, A.J.; Pino-Argumedo, M.I.; Hilkin, B.M.; Shanrock, C.R.; Gansemer, N.D.; Chaly, A.L.; Zarei, K.; Allen, P.D.; Ostedgaard, L.S.; Hoffman, E.; et al. Mucus strands from submucosal glands initiate mucociliary transport of large particles. JCI Insight 2019, 4, e124863. [Google Scholar] [CrossRef] [Green Version]
- Ermund, A.; Meiss, L.N.; Dolan, B.; Bähr, A.; Klymiuk, N.; Hansson, G.C. The mucus bundles responsible for airway cleaning are retained in cystic fibrosis and by cholinergic stimulation. Eur. Respir. J. 2018, 52, 1800457. [Google Scholar] [CrossRef]
- Fan, Z.; Perisse, I.V.; Cotton, C.U.; Regouski, M.; Meng, Q.; Domb, C.; Van Wettere, A.J.; Wang, Z.; Harris, A.; White, K.L.; et al. A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight 2018, 3, e123529. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sui, H.; Fisher, J.T.; Yan, Z.; Liu, X.; Cho, H.-J.; Joo, N.S.; Zhang, Y.; Zhou, W.; Yi, Y.; et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J. Clin. Investig. 2010, 120, 3149–3160. [Google Scholar] [CrossRef] [PubMed]
- Birket, S.E.; Davis, J.M.; Fernandez, C.M.; Tuggle, K.L.; Oden, A.M.; Chu, K.K.; Tearney, G.J.; Fanucchi, M.V.; Sorscher, E.J.; Rowe, S.M. Development of an airway mucus defect in the cystic fibrosis rat. JCI Insight 2018, 3, e97199. [Google Scholar] [CrossRef] [PubMed]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; Van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Miller, A.C.; Harris, L.M.; Cavanaugh, J.E.; Alaiwa, M.A.; Stoltz, D.A.; Hornick, D.B.; Polgreen, P.M. The Rapid Reduction of Infection-Related Visits and Antibiotic Use among People with Cystic Fibrosis after Starting Elexacaftor-Tezacaftor-Ivacaftor. Clin. Infect. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okuda, K.; Shaffer, K.M.; Ehre, C. Mucins and CFTR: Their Close Relationship. Int. J. Mol. Sci. 2022, 23, 10232. https://doi.org/10.3390/ijms231810232
Okuda K, Shaffer KM, Ehre C. Mucins and CFTR: Their Close Relationship. International Journal of Molecular Sciences. 2022; 23(18):10232. https://doi.org/10.3390/ijms231810232
Chicago/Turabian StyleOkuda, Kenichi, Kendall M. Shaffer, and Camille Ehre. 2022. "Mucins and CFTR: Their Close Relationship" International Journal of Molecular Sciences 23, no. 18: 10232. https://doi.org/10.3390/ijms231810232
APA StyleOkuda, K., Shaffer, K. M., & Ehre, C. (2022). Mucins and CFTR: Their Close Relationship. International Journal of Molecular Sciences, 23(18), 10232. https://doi.org/10.3390/ijms231810232