Physiological and Molecular Characteristics of Southern Leaf Blight Resistance in Sweet Corn Inbred Lines


Abstract
:1. Introduction
2. Results
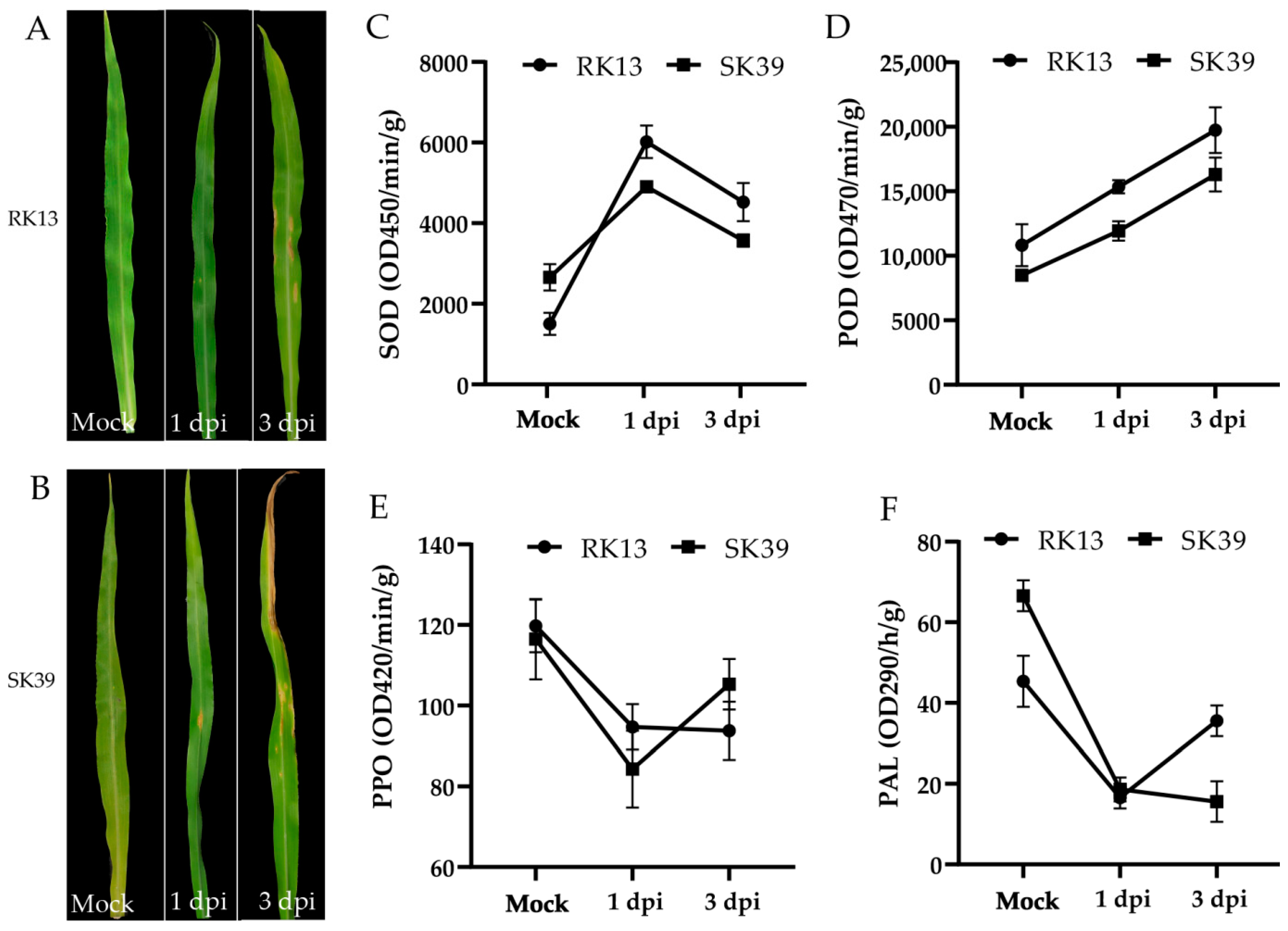
2.1. Morphological Characteristics and Changes in Protective Enzymatic Activity of Sweet Corn Leaves after Inoculation of C. heterostrophus
2.2. Overview of mRNA Sequencing Data
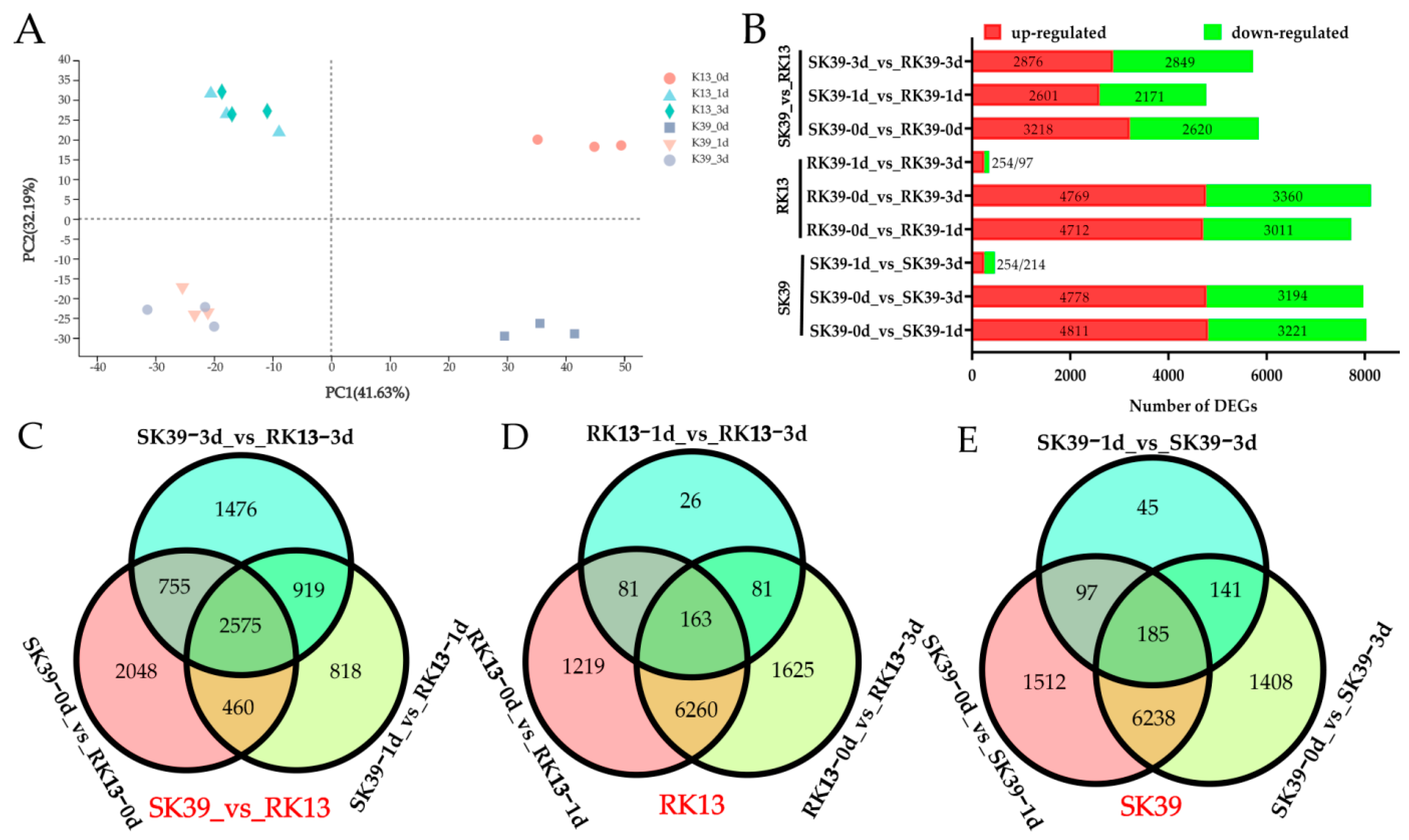
2.3. Identification of Differentially Expressed Genes (DEGs)
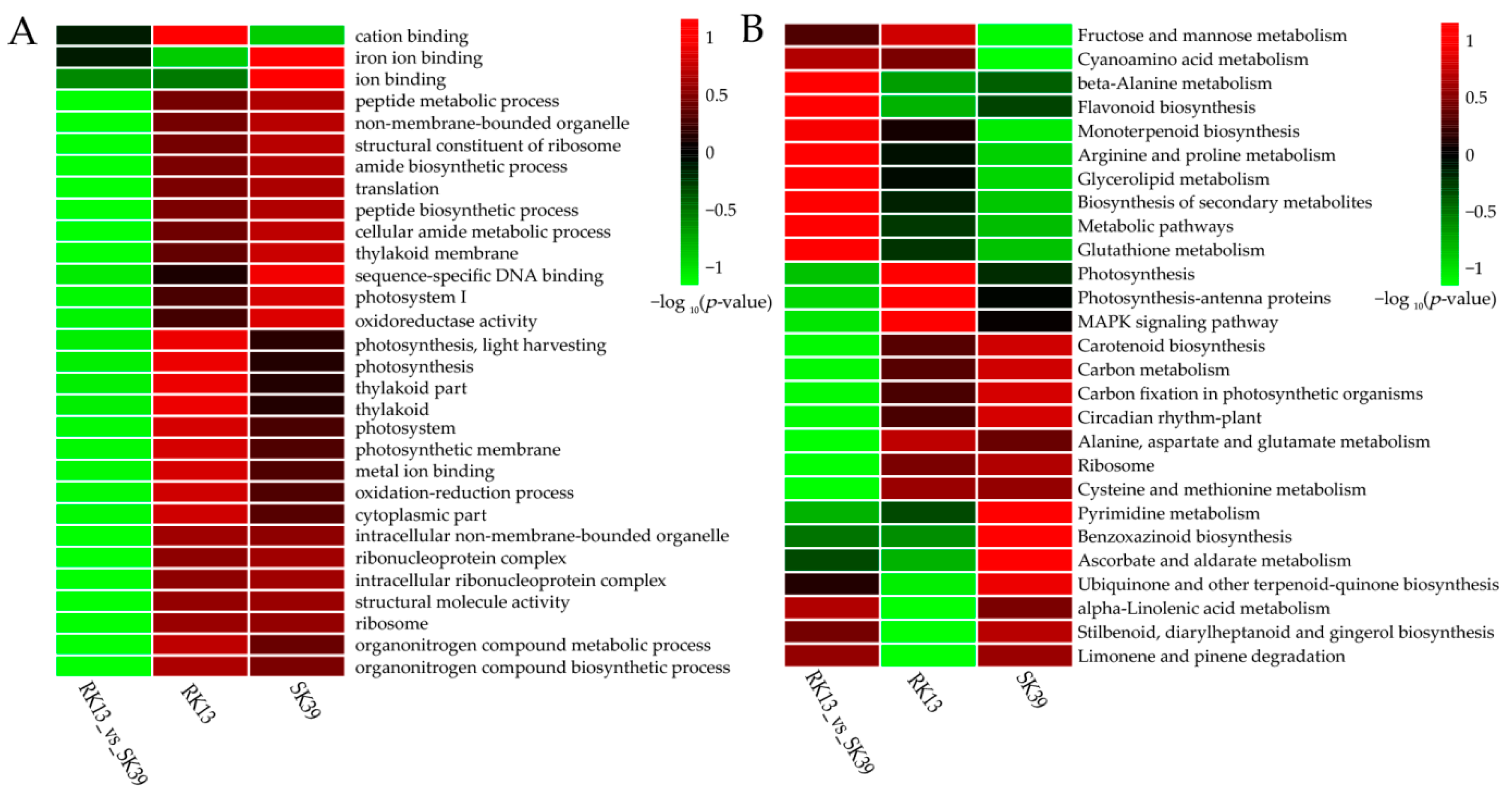
2.4. Functional Classification of Differentially Expressed Genes (DEGs)
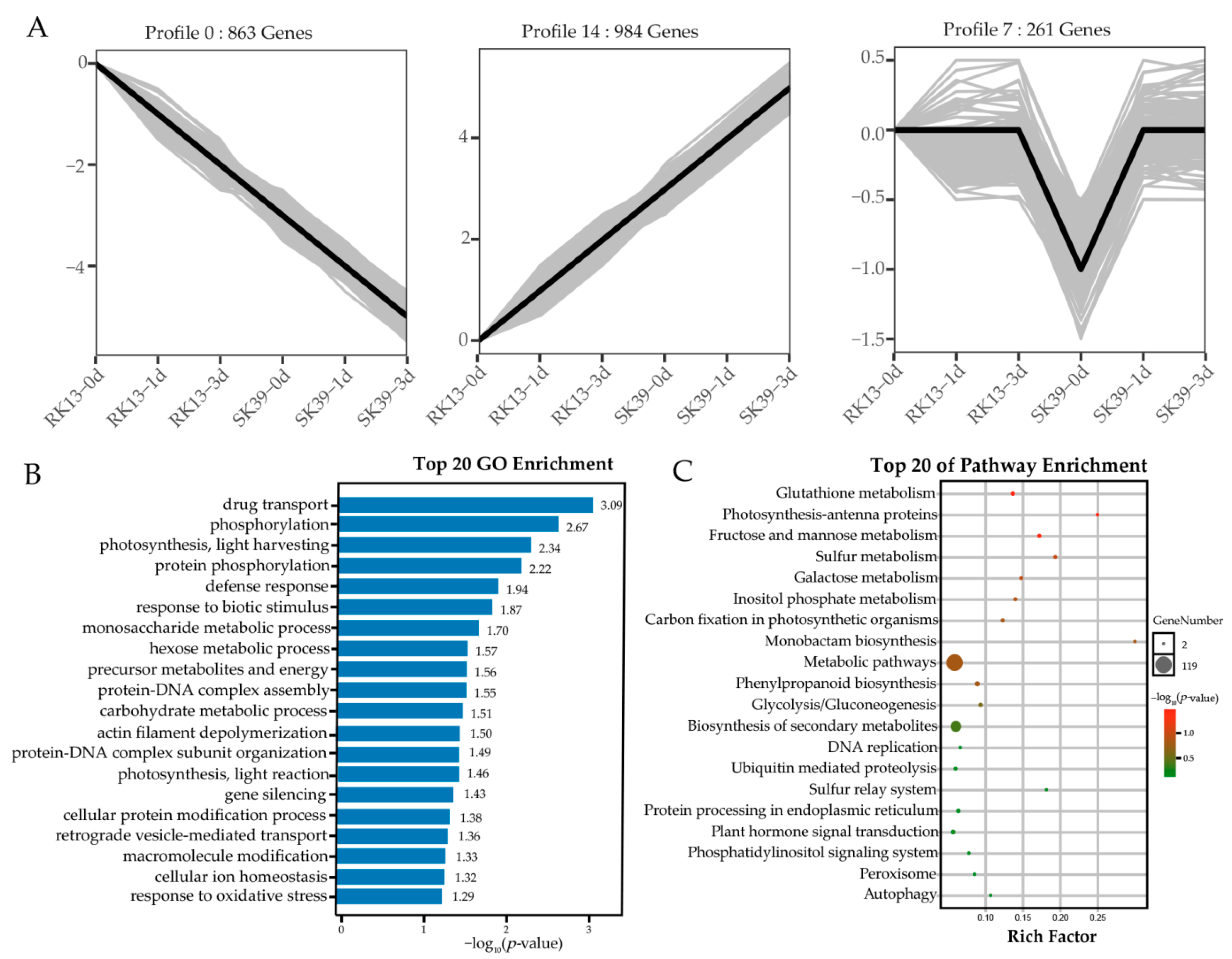
2.5. Clustering Analysis of the Co-Expressed DEGs
2.6. Analysis of Important Pathways in Response to SCLB in Sweet Corn
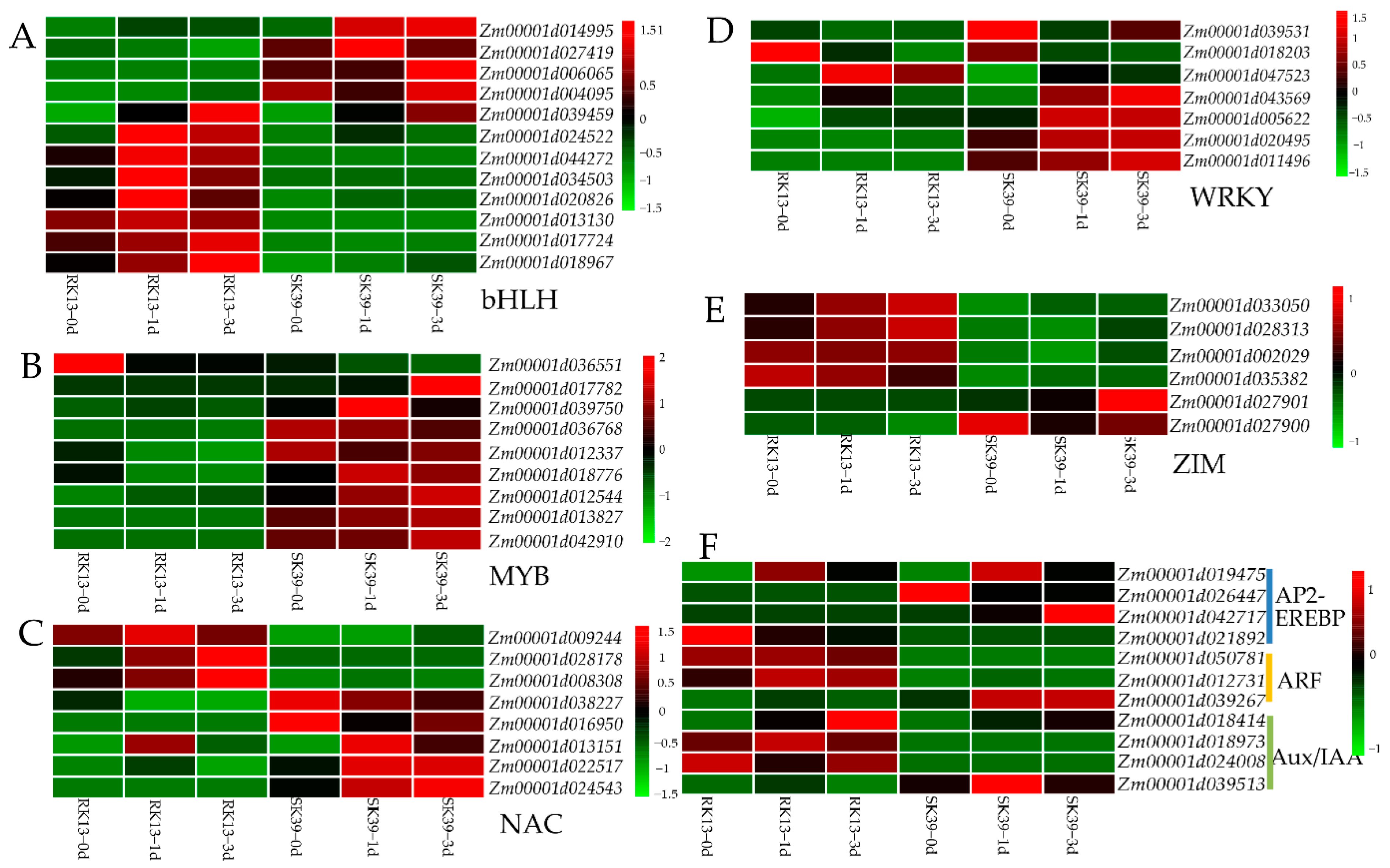
2.7. Expression of Transcription Factors (TFs) in Response to SCLB
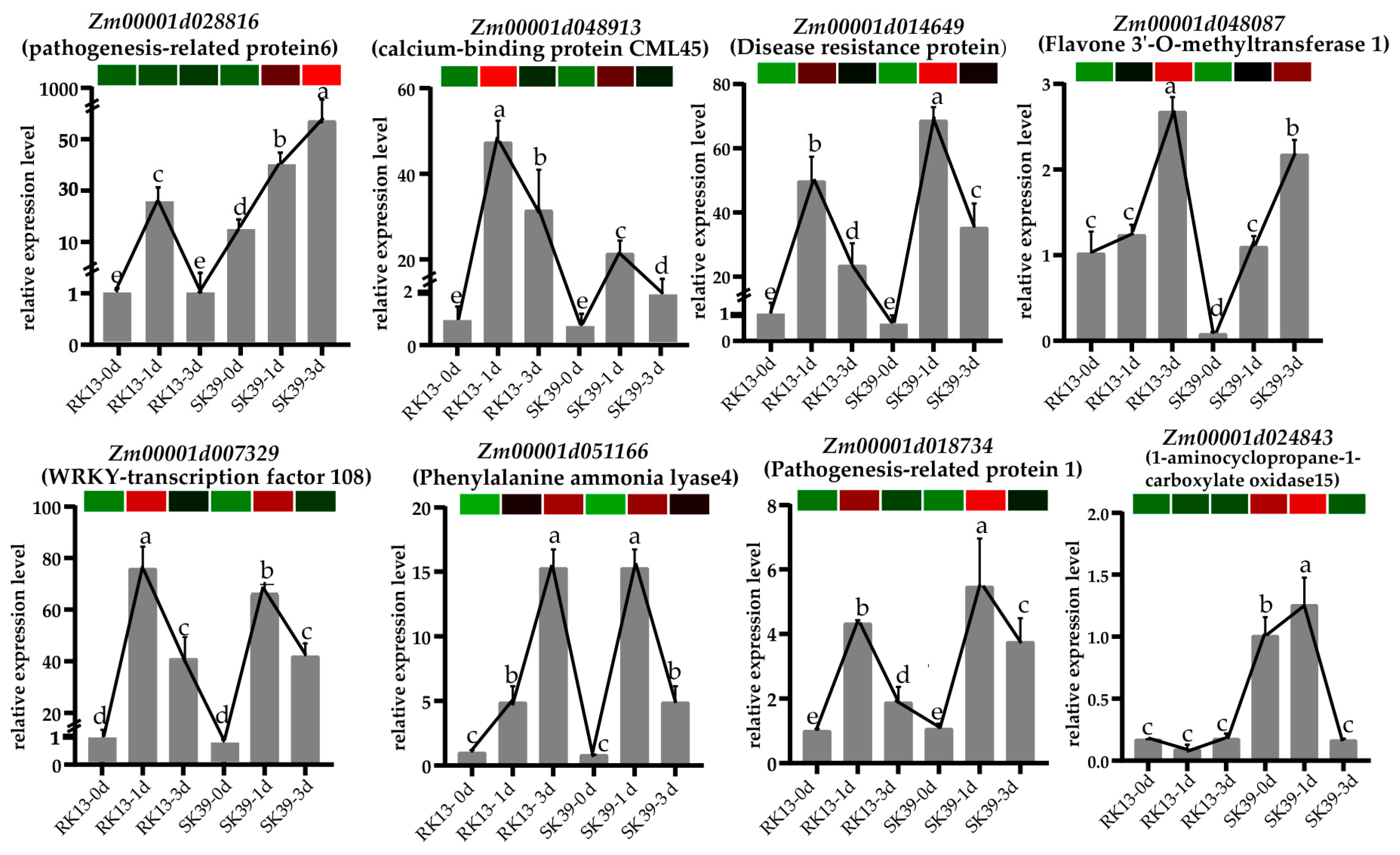
2.8. Verification of DEGs by Quantitative RT-PCR (qRT-PCR)
3. Discussion
4. Materials and Method
4.1. Plant Materials
4.2. Determination of Plant Protective Enzyme Activity
4.3. RNA Extraction and Library Preparation
4.4. Preprocessing of RNA-Seq Data
4.5. Identification of DEGs and Functional Classification
4.6. Real-Time PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABA | abscisic acid |
| DEGs | differentially expressed genes |
| dpi | days post inoculation |
| GO | Gene Ontology |
| JA | jasmonic acid |
| JAZ | JASMONATE ZIM-domain |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MAPK | mitogen-activated protein kinase |
| PAL | phenylalanine ammonia lyase |
| POD | peroxidase |
| PPO | polyphenol oxidase |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| QTL | quantitative trait locus |
| ROS | reactive oxygen species |
| SA | salicylic acid |
| SCLB | southern corn leaf blight |
| SOD | superoxide dismutase |
| TFs | transcription factors |
References
- McDonald, B.A.; McDermott, J.M.; Goodwin, S.B.; Allard, R.W. The population biology of host-pathogen interactions. Annu. Rev. Phytopathol. 1989, 27, 77–94. [Google Scholar] [CrossRef]
- Wang, M.; Ma, J.; Fan, L.L.; Fu, K.H.; Yu, C.J.; Gao, J.X.; Li, Y.Q.; Chen, J. Biological control of southern corn leaf blight by Trichoderma atroviride SG3403. Biocontrol Sci. Technol. 2015, 25, 1133–1146. [Google Scholar] [CrossRef]
- Wang, M.; Wang, S.; Ma, J.; Yu, C.; Gao, J.; Chen, J. Detection of Cochliobolus heterostrophus races in south China. J. Phytopathol. 2017, 165, 681–691. [Google Scholar] [CrossRef]
- Chen, J. Damage symptoms and countermeasures of main diseases and pests of corn. J. Agric. Catastrophol. 2014, 4, 15–20. [Google Scholar] [CrossRef]
- Tatum, L.A. The southern corn leaf blight epidemic. Science 1971, 171, 1113–1118. [Google Scholar] [CrossRef]
- Li, K.; Ma, L.; Du, P.F.; Wang, Q. Gene expression of maize terpenoid phytoalexin metabolism in response to southern leaf blight. Acta Bot. Boreali-Occident. Sin. 2015, 35, 1776–1780. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, S.; Hu, B.J.; Zhang, Y.; Teng, B. Investigation on inhibitory activity of fermentation products of Endophytic fungi from allelopathic rice and their effects on activities of protective enzymes in rice. J. Nuclear Agric. Sci. 2019, 33, 2347–2353. [Google Scholar] [CrossRef]
- Feng, K.; Zheng, Q.S.; Yu, J.H.; Cheng, Y.; Ye, Q.J.; Ruan, M.Y.; Wang, R.Q.; Li, Z.M.; Yao, Z.P.; Yang, Y.J.; et al. The characteristics of superoxide dismutase (SOD) in evolutions and its research in plant resistance. Mol. Plant Breed. 2017, 15, 4498–4505. [Google Scholar] [CrossRef]
- Ding, X.Y.; Cao, J.K. Characteristics of peroxidase from fruit and vegetable research progress. Food Sci. Technol. 2012, 37, 62–66. [Google Scholar] [CrossRef]
- Peng, S.Q. Advance in research of polyphenol oxidase in plants. Chin. J. Trop. Agric. 2000, 3, 61–66. [Google Scholar] [CrossRef]
- Saberi-Riseh, R.; Dashti, H.; Gholizadeh-Vazvani, M.; Dini, A. Changes in the activity of enzymes phenylalanine ammonia-lyase, polyphenol oxidase, and peroxidase in some wheat genotypes against take-all disease. J. Agric. Sci. Technol. 2021, 23, 929–942. [Google Scholar]
- Vanitha, S.C.; Niranjana, S.R.; Umesha, S. Role of phenylalanine ammonia lyase and polyphenol oxidase in host resistance to bacterial wilt of tomato. J. Phytopathol. 2009, 157, 552–557. [Google Scholar] [CrossRef]
- Puttarach, J.; Puddhanon, P.; Siripin, S.; Sangtong, V.; Songchantuek, S. Marker assisted selection for resistance to northern corn leaf blight in sweet corn. SABRAO J. Breed. Genet. 2016, 48, 72–79. [Google Scholar]
- Mushtaq, M.; Bhat, M.A.; Bhat, J.A.; Mushtaq, S. Comparative analysis of genetic diversity of maize inbred lines from Kashmir Valley using agro-morphological and SSR markers. SABRAO J. Breed. Genet. 2016, 48, 518–527. [Google Scholar]
- Zwonitzer, J.C.; Coles, N.D.; Krakowsky, M.D.; Arellano, C.; Holland, J.B.; McMullen, M.D.; Pratt, R.C.; Balint-Kurti, P.J. Mapping resistance quantitative trait loci for three foliar diseases in a maize recombinant inbred line population-evidence for multiple disease resistance? Phytopathology 2010, 100, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.L.; Stuber, C.W.; Senior, M.L. Identification and mapping of quantitative trait loci conditioning resistance to southern leaf blight of maize caused by Cochliobolus heterostrophus race O. Phytopathology 2004, 94, 863–867. [Google Scholar] [CrossRef]
- Balint-Kurti, P.J.; Carson, M.L. Analysis of quantitative trait loci for resistance to southern leaf blight in juvenile maize. Phytopathology 2006, 96, 221–225. [Google Scholar] [CrossRef]
- Balint-Kurti, P.J.; Zwonitzer, J.C.; Wisser, R.J.; Carson, M.L.; Oropeza-Rosas, M.A.; Holland, J.B.; Szalma, S.J. Precise mapping of quantitative trait loci for resistance to southern leaf blight, caused by Cochliobolus heterostrophus race O, and flowering time using advanced intercross maize lines. Genetics 2007, 176, 645–657. [Google Scholar] [CrossRef]
- Belcher, A.R.; Zwonitzer, J.C.; Cruz, J.S.; Krakowsky, M.D.; Chung, C.L.; Nelson, R.; Arellano, C.; Balint-Kurti, P.J. Analysis of quantitative disease resistance to southern leaf blight and of multiple disease resistance in maize, using near-isogenic lines. Theor. Appl. Genet. 2012, 124, 433–445. [Google Scholar] [CrossRef]
- Zhao, Y.Z.; Lu, X.M.; Liu, C.X.; Guan, H.Y.; Zhang, M.; Li, Z.F.; Cai, H.W.; Lai, J.S. Identification and fine mapping of rhm1 locus for resistance to southern corn leaf blight in maize. J. Integr. Plant Biol. 2012, 54, 321–329. [Google Scholar] [CrossRef]
- Cai, H.W.; Gao, Z.S.; Yuyama, N.; Ogawa, N. Identification of AFLP markers closely linked to the rhm gene for resistance to southern corn leaf blight in maize by using bulked segregant analysis. Mol. Genet. Genom. 2003, 269, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Zaitlin, D.; DeMars, S.; Ma, Y. Linkage of rhm, a recessive gene for resistance to southern corn leaf blight, to RFLP marker loci in maize (Zea mays) seedlings. Genome 1993, 36, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.Y.; Chen, J.B.; Yang, A.G.; Yuan, Q.H.; Zhao, W.C.; Xu, T.Y.; Chen, B.W.; Ren, M.; Geng, R.M.; Zong, Z.H.; et al. Comparative transcriptome profiling reveals defense-related genes against Ralstonia solanacearum infection in tobacco. Front. Plant Sci. 2021, 12, 767882. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Hu, X.X.; Chen, J.R.; Wang, W.X.; Xiong, X.Y.; He, C.Z. Integrated mRNA and microRNA transcriptome analysis reveals miRNA regulation in response to PVA in potato. Sci. Rep. 2017, 7, 16925. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.W.; Li, S.P.; Xiao, Y.P.; Lu, L.; Zheng, Z.C.; Tang, D.Z.; Cui, H.T. Transcriptome analysis of rice response to blast fungus identified core genes involved in immunity. Plant Cell Environ. 2021, 44, 3103–3121. [Google Scholar] [CrossRef]
- Chen, O.; Deng, L.L.; Ruan, C.Q.; Yi, L.H.; Zeng, K.F. Pichia galeiformis induces resistance in postharvest citrus by activating the phenylpropanoid biosynthesis pathway. J. Agric. Food Chem. 2021, 69, 2619–2631. [Google Scholar] [CrossRef]
- Karpiński, S.; Szechyńska-Hebda, M.; Wituszyńska, W.; Burdiak, P. Light acclimation, retrograde signalling, cell death and immune defences in plants. Plant Cell Environ. 2013, 36, 736–744. [Google Scholar] [CrossRef]
- Ma, L.X.; Zhong, S.F.; Liu, N.; Chen, W.Q.; Liu, T.G.; Li, X.; Zhang, M.; Ren, Z.L.; Yang, J.Z.; Luo, P.G. Gene expression profile and physiological and biochemical characterization of hexaploid wheat inoculated with Blumeria graminis f. Sp. tritici. Physiol. Mol. Plant Pathol. 2015, 90, 39–48. [Google Scholar] [CrossRef]
- Major, I.T.; Nicole, M.C.; Duplessis, S.; Séguin, A. Photosynthetic and respiratory changes in leaves of poplar elicited by rust infection. Photosynth. Res. 2010, 104, 41–48. [Google Scholar] [CrossRef]
- Yang, H.; Luo, P.G. Changes in photosynthesis could provide important insight into the interaction between wheat and fungal pathogens. Int. J. Mol. Sci. 2021, 22, 8865. [Google Scholar] [CrossRef]
- Lu, Y.; Yao, J. Chloroplasts at the crossroad of photosynthesis, pathogen infection and plant defense. Int. J. Mol. Sci. 2018, 19, 3900. [Google Scholar] [CrossRef]
- Nomura, H.; Komori, T.; Uemura, S.; Kanda, Y.; Shimotani, K.; Nakai, K.; Furuichi, T.; Takebayashi, K.; Sugimoto, T.; Sano, S.; et al. Chloroplast-mediated activation of plant immune signalling in Arabidopsis. Nat. Commun. 2012, 3, 926. [Google Scholar] [CrossRef] [PubMed]
- Alazem, M.; Lin, N.S. Interplay between ABA signaling and RNA silencing in plant viral resistance. Curr. Opin. Virol. 2020, 42, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.P.; Yin, D.D.; Wan, W.Y.; Fang, T.T.; Su, X.H.; Cao, X.H. Cloning, characterization, and expression analysis of two MAPKKK genes in Chrysanthemum. Pak. J. Bot. 2022, 54, 143–150. [Google Scholar] [CrossRef]
- Sun, T.J.; Nitta, Y.; Zhang, Q.; Wu, D.; Tian, H.N.; Lee, J.S.; Zhang, Y.L. Antagonistic interactions between two MAP kinase cascades in plant development and immune signaling. EMBO Rep. 2018, 19, e45324. [Google Scholar] [CrossRef]
- Wang, C.; He, X.W.; Wang, X.X.; Zhang, S.X.; Guo, X.Q. Ghr-miR5272a-mediated regulation of GhMKK6 gene transcription contributes to the immune response in cotton. J. Exp. Bot. 2017, 68, 5895–5906. [Google Scholar] [CrossRef]
- Wang, C.; Guo, H.B.; He, X.W.; Zhang, S.X.; Wang, J.Y.; Wang, L.J.; Guo, D.Z.; Guo, X.Q. Scaffold protein GhMORG1 enhances the resistance of cotton to Fusarium oxysporum by facilitating the MKK6-MPK4 cascade. Plant Biotechnol. J. 2020, 18, 1421–1433. [Google Scholar] [CrossRef]
- Guo, D.Z.; Hao, C.H.; Hou, J.B.; Zhao, G.D.; Shan, W.L.; Guo, H.B.; Wang, C.; Guo, X.Q. The protein phosphatase GhAP2C1 interacts together with GhMPK4 to synergistically regulate the immune response to Fusarium oxysporum in cotton. Int. J. Mol. Sci. 2022, 23, 2014. [Google Scholar] [CrossRef]
- Gruner, K.; Griebel, T.; Návarová, H.; Attaran, E.; Zeier, J. Reprogramming of plants during systemic acquired resistance. Front. Plant Sci. 2013, 4, 252. [Google Scholar] [CrossRef]
- Felton, G.W.; Korth, K.L.; Bi, J.L.; Wesley, S.V.; Huhman, D.V.; Mathews, M.C.; Murphy, J.B.; Lamb, C.; Dixon, R.A. Inverse relationship between systemic resistance of plants to microorganisms and to insect herbivory. Curr. Biol. 1999, 9, 317–320. [Google Scholar] [CrossRef]
- Shadle, G.L.; Wesley, S.V.; Korth, K.L.; Chen, F.; Lamb, C.; Dixon, R.A. Phenylpropanoid compounds and disease resistance in transgenic tobacco with altered expression of L-phenylalanine ammonia-lyase. Phytochemistry 2003, 64, 153–161. [Google Scholar] [CrossRef]
- Kim, D.S.; Hwang, B.K. An important role of the pepper phenylalanine ammonia-lyase gene (PAL1) in salicylic acid-dependent signalling of the defence response to microbial pathogens. J. Exp. Bot. 2014, 65, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Chini, A.; Fonseca, S.; Fernández, G.; Adie, B.; Chico, J.M.; Lorenzo, O.; García-Casado, G.; López-Vidriero, I.; Lozano, F.M.; Ponce, M.R.; et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 2007, 448, 666–671. [Google Scholar] [CrossRef]
- Chini, A.; Boter, M.; Solano, R. Plant oxylipins: COI1/JAZs/MYC2 as the core jasmonic acid-signalling module. FEBS J. 2009, 276, 4682–4692. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.X.; Liu, J.; Liu, P.; Ming, D.F.; Sun, J.Q. Overexpression of TaJAZ1 increases powdery mildew resistance through promoting reactive oxygen species accumulation in bread wheat. Sci. Rep. 2019, 9, 5691. [Google Scholar] [CrossRef]
- Kliebenstein, D.J. Secondary metabolites and plant/environment interactions: A view through Arabidopsis thaliana-tinged glasses. Plant Cell Environ. 2004, 27, 675–684. [Google Scholar] [CrossRef]
- Chen, L.G.; Zhang, L.P.; Xiang, S.Y.; Chen, Y.L.; Zhang, H.Y.; Yu, D.Q. The transcription factor WRKY75 positively regulates jasmonate-mediated plant defense to necrotrophic fungal pathogens. J. Exp. Bot. 2021, 4, 1473–1489. [Google Scholar] [CrossRef]
- Piasecka, A.; Jedrzejczak-Rey, N.; Bednarek, P. Secondary metabolites in plant innate immunity: Conserved function of divergent chemicals. New Phytol. 2015, 206, 948–964. [Google Scholar] [CrossRef]
- Vasmatkar, P.; Kaur, K.; Pannu, P.P.S.; Kaur, G.; Kaur, H. Unraveling the metabolite signatures of maize genotypes showing differential response towards southern corn leaf blight by 1H-NMR and FTIR spectroscopy. Physiol. Mol. Plant Pathol. 2019, 108, 101441. [Google Scholar] [CrossRef]
- Christensen, S.A.; Sims, J.; Vaughan, M.M.; Hunter, C.; Block, A.; Willett, D.; Alborn, H.T.; Huffaker, A.; Schmelz, E.A. Commercial hybrids and mutant genotypes reveal complex protective roles for inducible terpenoid defenses in maize. J. Exp. Bot. 2018, 69, 1693–1705. [Google Scholar] [CrossRef]
- Xu, M.J.; Dong, J.F.; Wang, H.Z.; Huang, L.Q. Complementary action of jasmonic acid on salicylic acid in mediating fungal elicitor-induced flavonol glycoside accumulation of ginkgo biloba cells. Plant Cell Environ. 2009, 32, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Jan, R.; Khan, M.A.; Asaf, S.; Lubna; Park, J.R.; Lee, I.J.; Kim, K.M. Flavonone 3-hydroxylase relieves bacterial leaf blight stress in rice via overaccumulation of antioxidant flavonoids and induction of defense genes and hormones. Int. J. Mol. Sci. 2021, 22, 6152. [Google Scholar] [CrossRef]
- Wani, S.H.; Anand, S.; Singh, B.; Bohra, A.; Joshi, R. WRKY transcription factors and plant defense responses: Latest discoveries and future prospects. Plant Cell Rep. 2021, 40, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Cur. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Adachi, H.; Nakano, T.; Miyagawa, N.; Ishihama, N.; Yoshioka, M.; Katou, Y.; Yaeno, T.; Shirasu, K.; Yoshioka, H. WRKY transcription factors phosphorylated by MAPK regulate a plant immune NADPH oxidase in Nicotiana benthamiana. Plant Cell 2015, 27, 2645–2663. [Google Scholar] [CrossRef]
- Zhang, M.F.; Wang, J.Q.; Luo, Q.J.; Yang, C.; Yang, H.B.; Cheng, Y.J. CsMYB96 enhances citrus fruit resistance against fungal pathogen by activating salicylic acid biosynthesis and facilitating defense metabolite accumulation. J. Plant Physiol. 2021, 264, 153472. [Google Scholar] [CrossRef]
- Wei, X.N.; Shan, T.L.; Hong, Y.T.; Xu, H.J.; Liu, X.; Zhang, Z.Y. TaPIMP2, a pathogen-induced MYB protein in wheat, contributes to host resistance to common root rot caused by Bipolaris sorokinian. Sci. Rep. 2017, 7, 1754. [Google Scholar] [CrossRef]
- Li, Y.B.; Xu, R.; Liu, C.; Shen, N.; Han, L.B.; Tang, D. Magnaporthe oryzae fimbrin organizes actin networks in the hyphal tip during polar growth and pathogenesis. PLoS Pathog. 2020, 16, e1008437. [Google Scholar] [CrossRef]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.J.M. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT. Method 2001, 25, 402–408. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | RK13_0d | RK13_1d | RK13_3d | SK39_0d | SK39_1d | SK39_3d |
|---|---|---|---|---|---|---|
| Raw reads (106) | 45.53 | 45.06 | 46.38 | 46.13 | 46.47 | 45.87 |
| Clean reads (106) | 45.30 | 44.84 | 46.17 | 45.88 | 46.24 | 45.65 |
| Error rate (%) | 0.0246 | 0.0246 | 0.0245 | 0.0245 | 0.0248 | 0.0247 |
| Total mapped (%) | 90.22 | 88.56 | 86.21 | 90.28 | 89.13 | 87.56 |
| Multiple mapped (%) | 3.57 | 2.93 | 3.69 | 3.73 | 3.39 | 3.04 |
| Unique mapped (%) | 86.65 | 85.63 | 82.52 | 86.55 | 85.75 | 84.53 |
| Q30 (%) | 94.56 | 94.56 | 94.64 | 94.65 | 94.37 | 94.43 |
| GC content (%) | 56.97 | 55.49 | 55.54 | 56.20 | 55.78 | 55.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, C.; Mo, H.; Fan, J.; Ren, W.; Pei, H.; Zhang, Y.; Ma, Z.; Wang, W.; Huang, J. Physiological and Molecular Characteristics of Southern Leaf Blight Resistance in Sweet Corn Inbred Lines. Int. J. Mol. Sci. 2022, 23, 10236. https://doi.org/10.3390/ijms231810236
Xiong C, Mo H, Fan J, Ren W, Pei H, Zhang Y, Ma Z, Wang W, Huang J. Physiological and Molecular Characteristics of Southern Leaf Blight Resistance in Sweet Corn Inbred Lines. International Journal of Molecular Sciences. 2022; 23(18):10236. https://doi.org/10.3390/ijms231810236
Chicago/Turabian StyleXiong, Caiyun, Haiwei Mo, Jingsheng Fan, Wenchuang Ren, Hu Pei, Yahui Zhang, Ziwei Ma, Wenyi Wang, and Jun Huang. 2022. "Physiological and Molecular Characteristics of Southern Leaf Blight Resistance in Sweet Corn Inbred Lines" International Journal of Molecular Sciences 23, no. 18: 10236. https://doi.org/10.3390/ijms231810236