

Phagocytosis-Regulators-Based Signature to Predict the Prognosis and Chemotherapy Resistance for Breast Cancer Patients

Abstract
:1. Introduction
2. Results
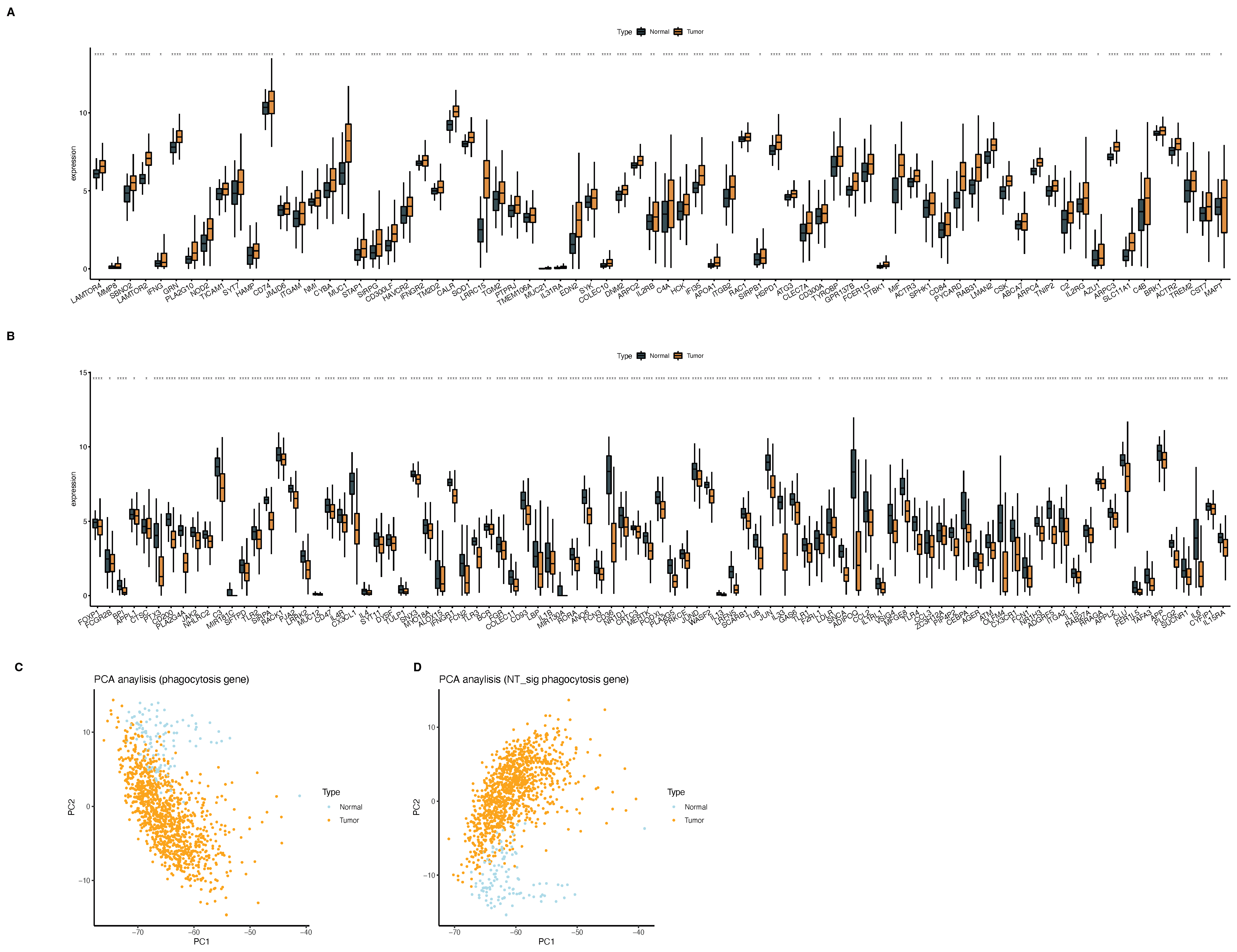
2.1. Phagocytosis Regulators
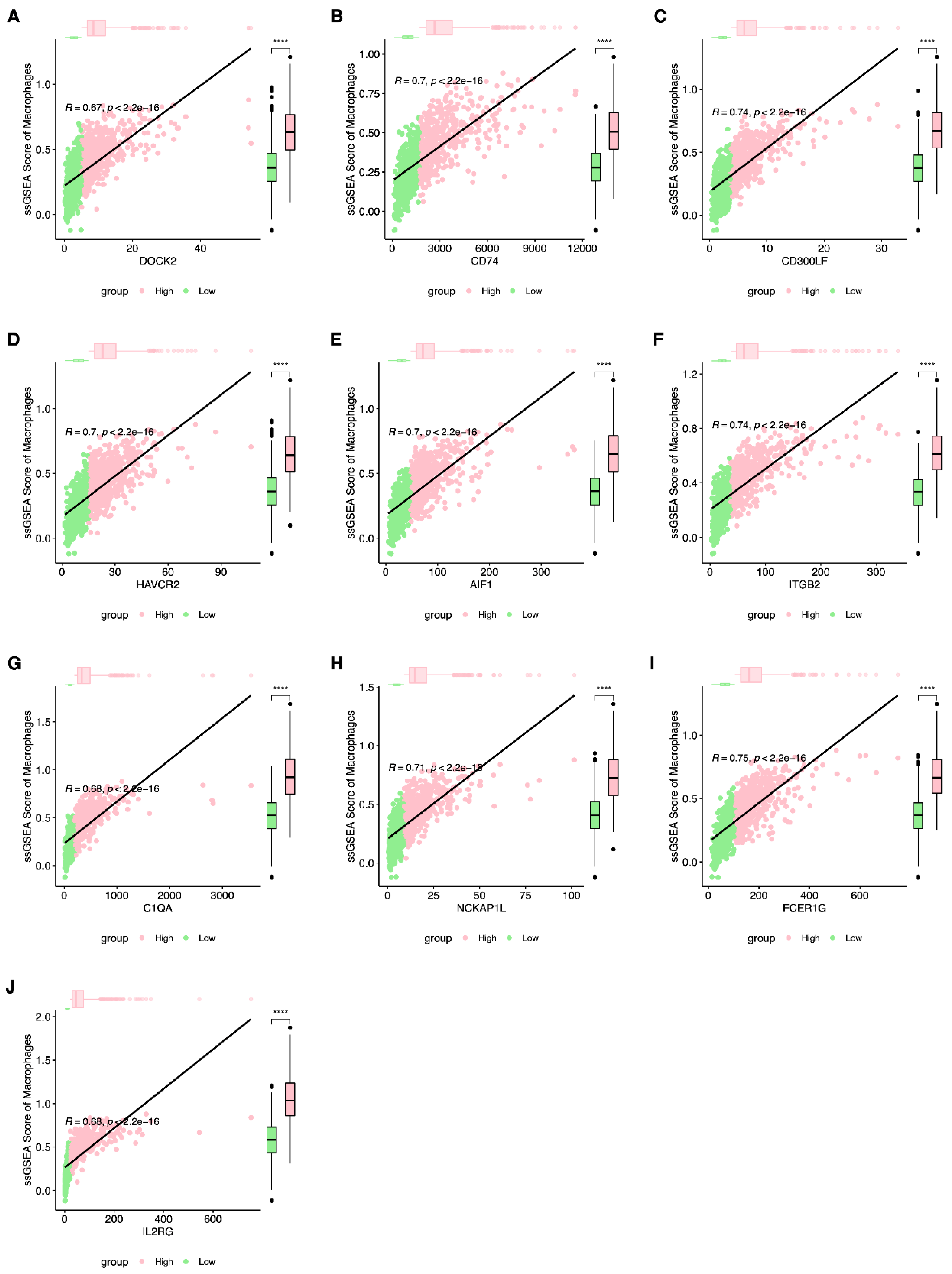
2.2. Phagocytosis Regulators Regulate the Involvement of Macrophages in the Occurrence and Development of Breast Cancer
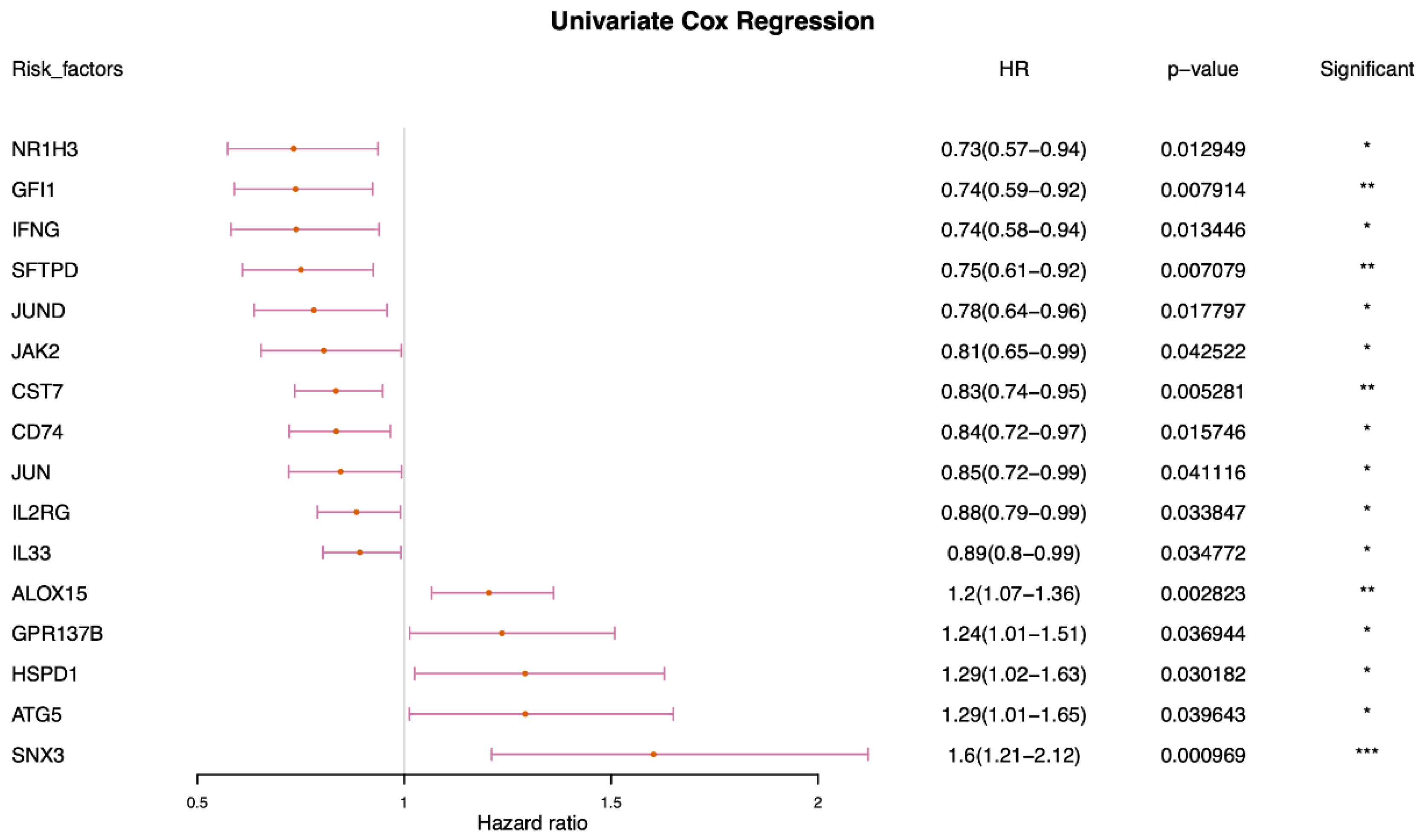
2.3. Identification and Characterization of Prognostic-Related Phagocytosis Regulators
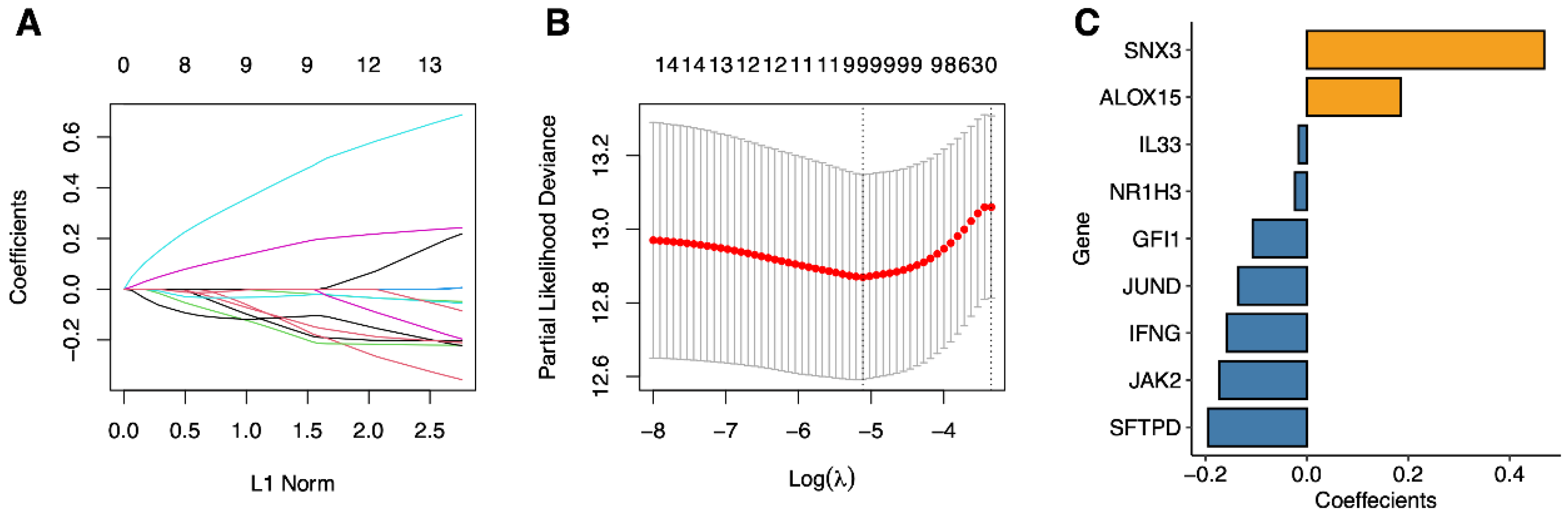
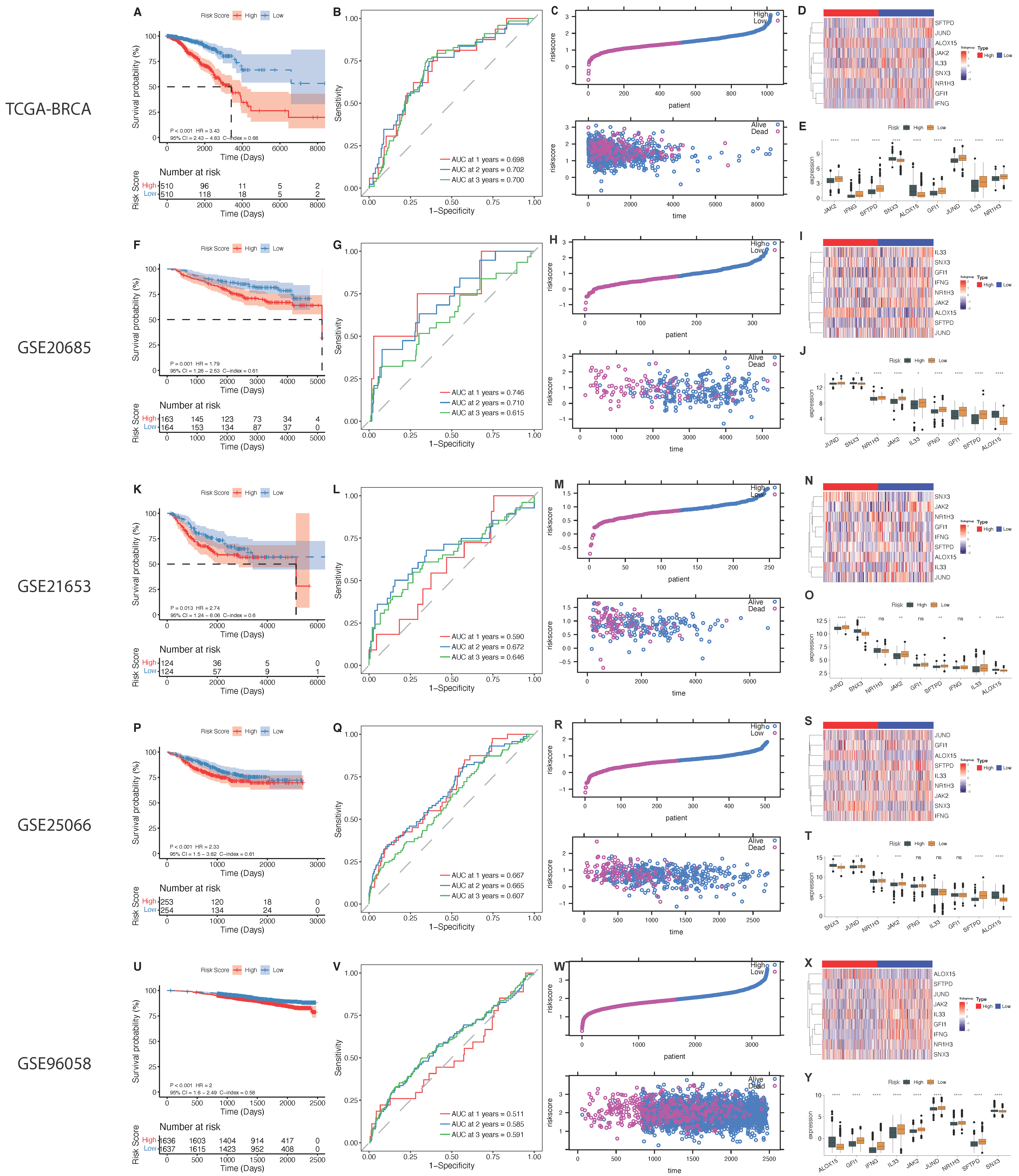
2.4. Signature Construction of Phagocytosis Regulators
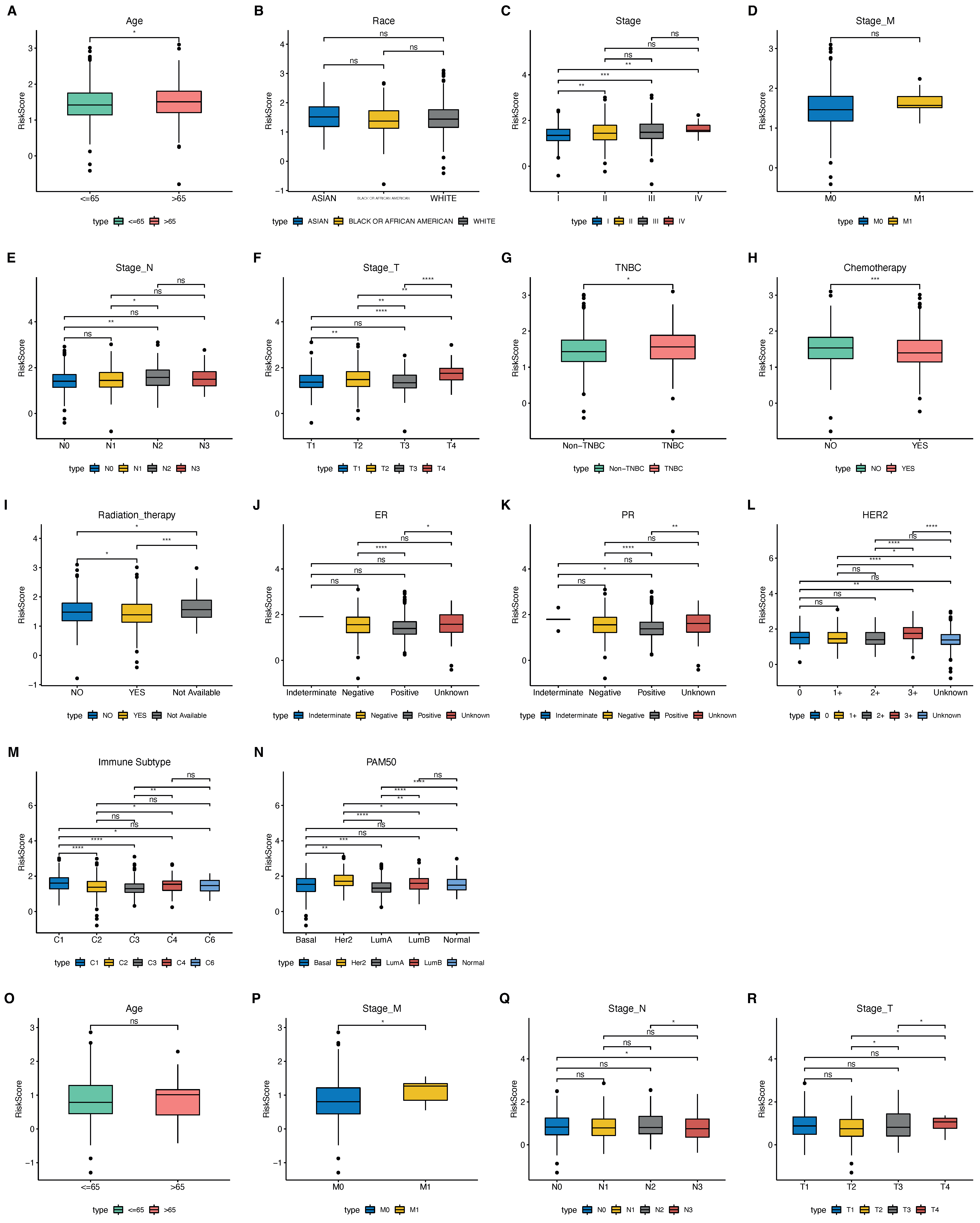
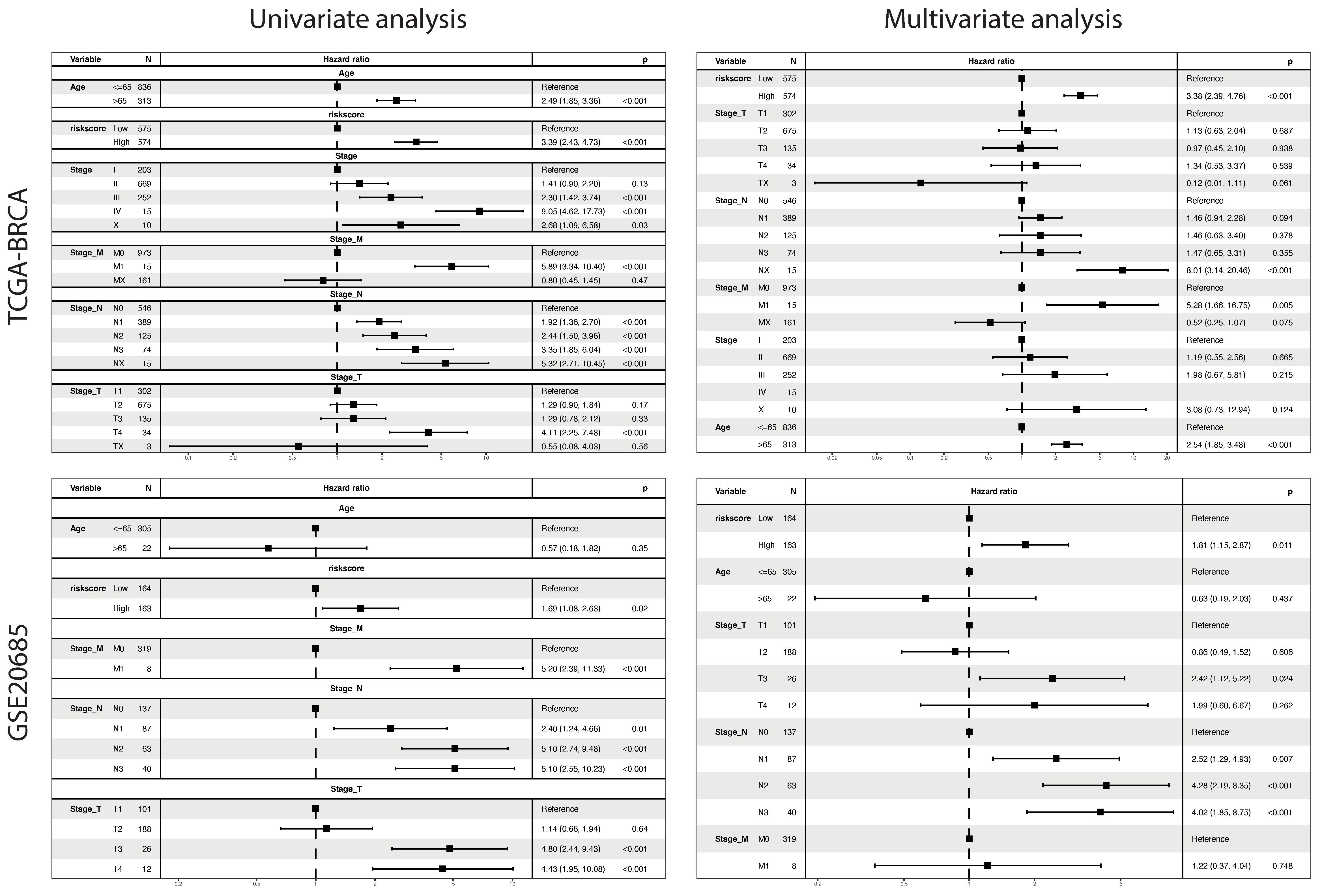
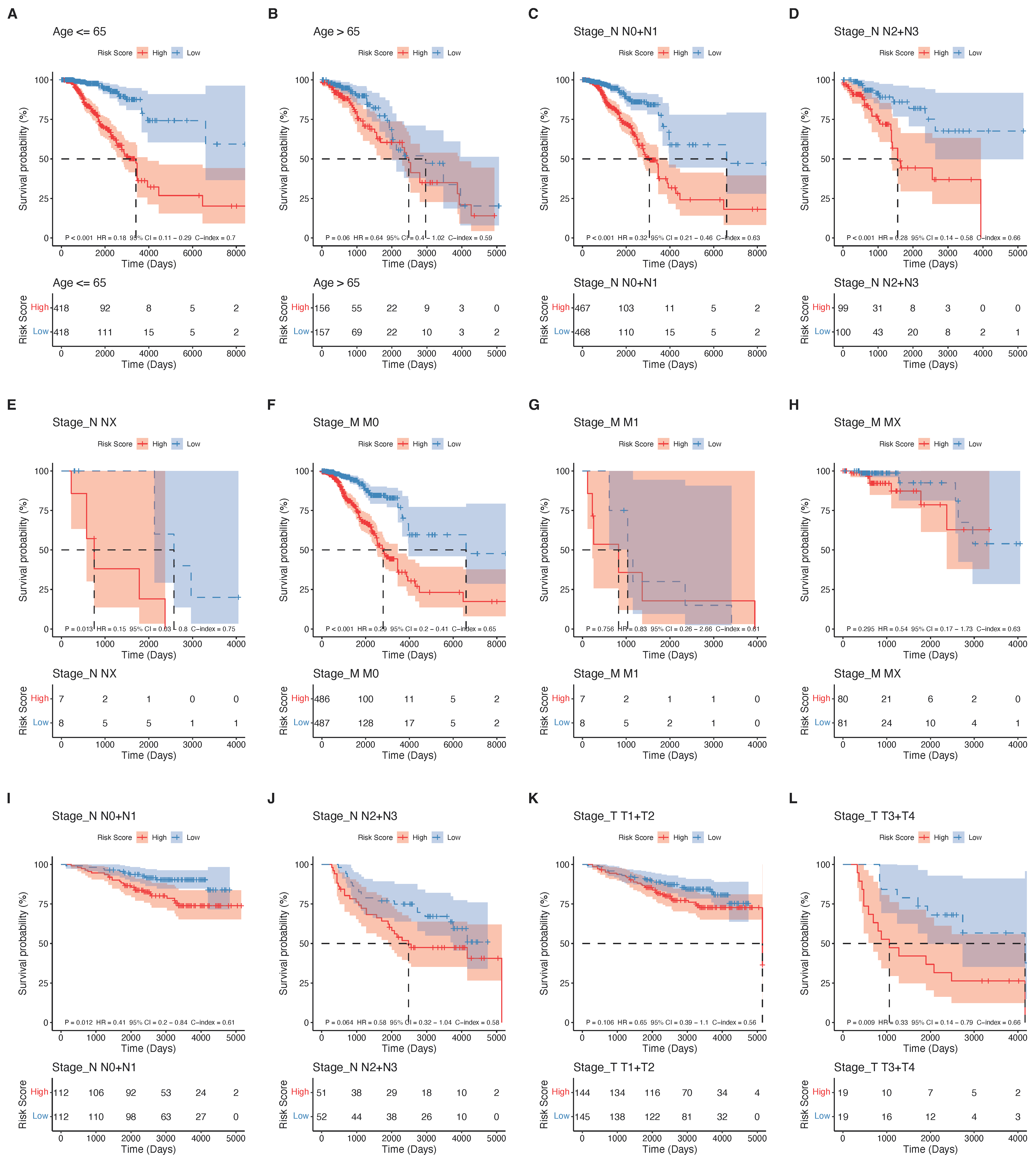
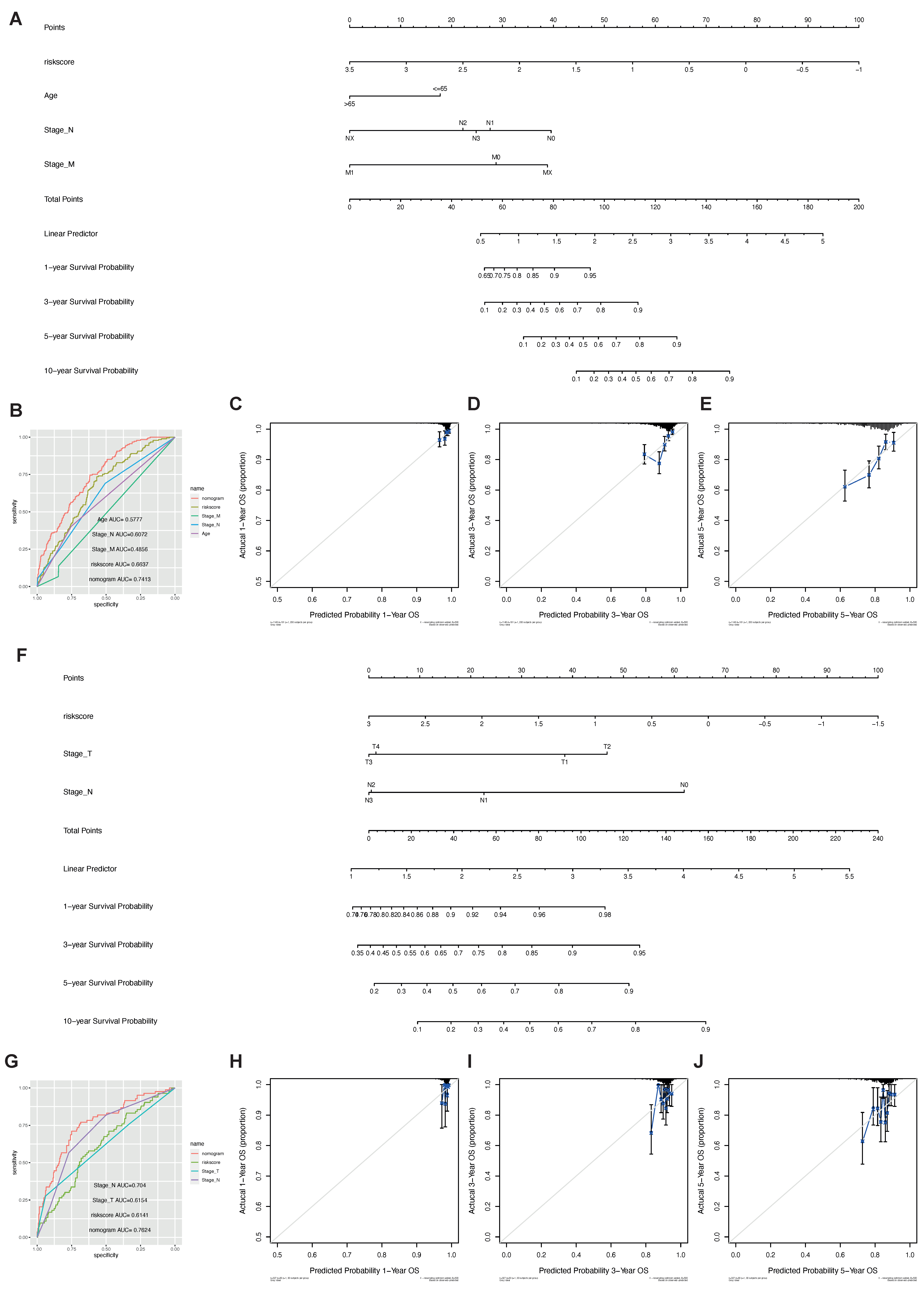
2.5. The Phagocytosis Regulator Signature Is Related to the Prognosis and Clinical Characteristics of Patients
2.6. The Phagocytosis Regulator Signature Is Related to the Patient’s Immune Microenvironment and Immunotherapy
2.7. Genomic Alteration of the Phagocytosis Regulator Signature and Drug Response
3. Discussion
4. Methods and Materials
4.1. Data Download
4.2. Data Overview
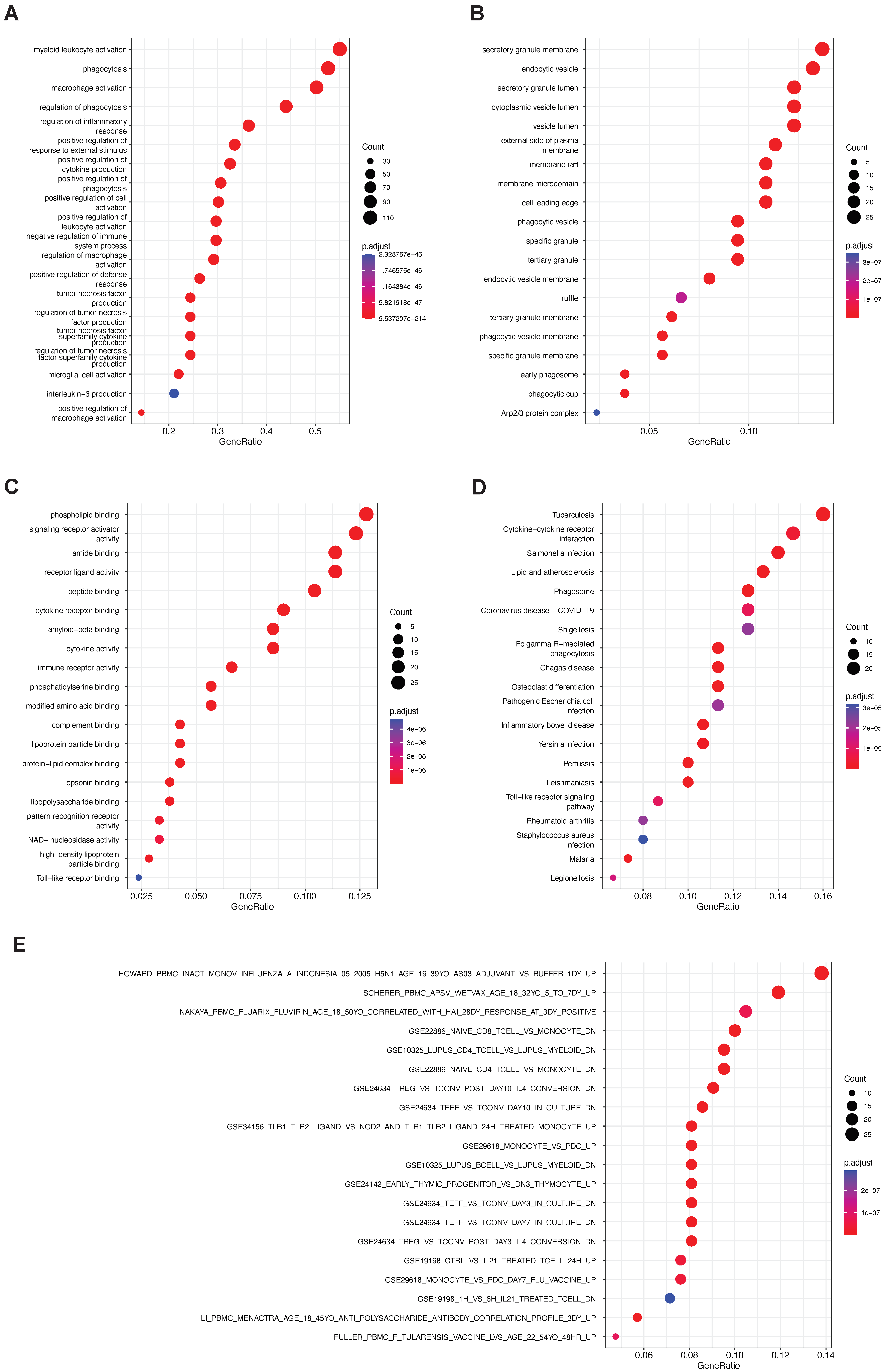
4.3. Pathway Enrichment Analysis
4.4. Analysis of CNV and SNV
4.5. Immune Infiltration Analysis
4.6. Calculation of Tumor Purity, Stromalscore, Immunescore
4.7. Consistent Clustering
4.8. Construction of Immune Infiltrating Cell Marker Scoring System Is Capable of Evaluating Prognosis
4.9. KM Survival Analysis
4.10. Correlation Analysis between Risk Score and Drug Sensitivity
4.11. Principal Component Analysis (PCA) Analysis
4.12. Drawing
4.13. Model Calibration Curve
4.14. Statistical Tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Goldhirsch, A.; Wood, W.C.; Gelber, R.D.; Coates, A.S.; Thürlimann, B.; Senn, H.J. Progress and promise: Highlights of the international expert consensus on the primary therapy of early breast cancer 2007. Ann. Oncol. 2007, 18, 1133–1144. [Google Scholar] [CrossRef]
- Alexander, W. The Checkpoint Immunotherapy Revolution: What Started as a Trickle Has Become a Flood, Despite Some Daunting Adverse Effects; New Drugs, Indications, and Combinations Continue to Emerge. Pharm. Ther. 2016, 41, 185–191. [Google Scholar]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Li, W.; Du, N.; Cui, J. Challenges of evaluating immunotherapy efficacy in solid tumors. Chin. J. Cancer Res. 2019, 31, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Kamber, R.A.; Nishiga, Y.; Morton, B.; Banuelos, A.M.; Barkal, A.A.; Vences-Catalán, F.; Gu, M.; Fernandez, D.; Seoane, J.A.; Yao, D.; et al. Inter-cellular CRISPR screens reveal regulators of cancer cell phagocytosis. Nature 2021, 597, 549–554. [Google Scholar] [CrossRef]
- Haney, M.S.; Bohlen, C.J.; Morgens, D.W.; Ousey, J.A.; Barkal, A.A.; Tsui, C.K.; Ego, B.K.; Levin, R.; Kamber, R.A.; Collins, H.; et al. Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nat. Genet. 2018, 50, 1716–1727. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Sant’ Angelo, M.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Raggi, F.; Pelassa, S.; Pierobon, D.; Penco, F.; Gattorno, M.; Novelli, F.; Eva, A.; Varesio, L.; Giovarelli, M.; Bosco, M.C. Regulation of Human Macrophage M1–M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front. Immunol. 2017, 8, 1097. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef] [PubMed]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Targeting Tumor-Associated Macrophages to Increase the Efficacy of Immune Checkpoint Inhibitors: A Glimpse into Novel Therapeutic Approaches for Metastatic Melanoma. Cancers 2020, 12, 3401. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Palma, A.; Jarrah, A.S.; Tieri, P.; Cesareni, G.; Castiglione, F. Gene Regulatory Network Modeling of Macrophage Differentiation Corroborates the Continuum Hypothesis of Polarization States. Front. Physiol. 2018, 9, 1659. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Choi, S.H. Tumor-associated macrophages in cancer: Recent advancements in cancer nanoimmunotherapies. J. Exp. Clin. Cancer Res. 2022, 41, 68. [Google Scholar] [CrossRef]
- Reis-Sobreiro, M.; Teixeira da Mota, A.; Jardim, C.; Serre, K. Bringing Macrophages to the Frontline against Cancer: Current Immunotherapies Targeting Macrophages. Cells 2021, 10, 2364. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Li, X.; Liu, R.; Su, X.; Pan, Y.; Han, X.; Shao, C.; Shi, Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 177. [Google Scholar] [CrossRef]
- Qiu, S.-Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schröder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic significance of tumor-associated macrophages in breast cancer: A meta-analysis of the literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef] [PubMed]
- Gil Del Alcazar, C.R.; Alečković, M.; Polyak, K. Immune Escape during Breast Tumor Progression. Cancer Immunol. Res. 2020, 8, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef]
- Freeberg, M.A.; Fromont, L.A.; D’Altri, T.; Romero, A.F.; Ciges, J.I.; Jene, A.; Kerry, G.; Moldes, M.; Ariosa, R.; Bahena, S.; et al. The European Genome-phenome Archive in 2021. Nucleic Acids Res. 2021, 50, D980–D987. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Posch, W.; Wilflingseder, D.; Sopper, S.; et al. quanTIseq: Quantifying immune contexture of human tumors. BioRxiv 2017, 223180. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.; Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J. Stat. Softw. 2011, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kattan, M.W. Drawing Nomograms with R: Applications to categorical outcome and survival data. Ann. Transl. Med. 2017, 5, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrell, F.E., Jr.; Harrell, M.F.E., Jr.; Hmisc, D.J.V.U. Package ‘rms’. 2017. Volume 229. Available online: https://github.com/harrelfe/rms (accessed on 16 March 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Rho | p-Value |
|---|---|---|
| FCER1G | 0.74895132 | 7.40 × 10−197 |
| ITGB2 | 0.73940791 | 2.53 × 10−189 |
| CD300LF | 0.73874914 | 8.16 × 10−189 |
| NCKAP1L | 0.71075865 | 1.44 × 10−168 |
| HAVCR2 | 0.7035216 | 1.01 × 10−163 |
| CD74 | 0.69829577 | 2.59 × 10−160 |
| AIF1 | 0.69692323 | 1.98 × 10−159 |
| C1QA | 0.68336001 | 5.75 × 10−151 |
| IL2RG | 0.68100437 | 1.52 × 10−149 |
| DOCK2 | 0.66931193 | 1.13 × 10−142 |
| TYROBP | 0.66309895 | 3.77 × 10−139 |
| CST7 | 0.645747 | 9.63 × 10−130 |
| FGR | 0.64426929 | 5.71 × 10−129 |
| IL15RA | 0.64193309 | 9.33 × 10−128 |
| SIRPG | 0.62837445 | 6.41 × 10−121 |
| GFI1 | 0.61166452 | 6.03 × 10−113 |
| CTSC | 0.60386822 | 2.19 × 10−109 |
| PTPRC | 0.60132084 | 3.03 × 10−108 |
| HCK | 0.59651545 | 4.05 × 10−106 |
| CD300A | 0.58598236 | 1.39 × 10−101 |
| NMI | 0.58263075 | 3.57 × 10−100 |
| IL10 | 0.56451739 | 7.77 × 10−93 |
| FCN1 | 0.55325685 | 1.69 × 10−88 |
| FCGR2B | 0.54762828 | 2.16 × 10−86 |
| SIRPB1 | 0.52573722 | 1.44 × 10−78 |
| C5AR1 | 0.52253377 | 1.80 × 10−77 |
| C3 | 0.51378244 | 1.57 × 10−74 |
| IFNG | 0.5026317 | 6.58 × 10−71 |
| Clinical Traits | p-Value |
|---|---|
| Stage_T | 0.91681495 |
| Stage_N | 0.75402253 |
| Stage_M | 0.82725935 |
| Stage | 0.60040185 |
| Age | 1 |
| ER | 0.77282999 |
| PR | 0.88454944 |
| HER2 | 0.91681495 |
| Symbol | Coefficient |
|---|---|
| JAK2 | −0.1728154 |
| IFNG | −0.1576737 |
| SFTPD | −0.1945068 |
| SNX3 | 0.46774561 |
| ALOX15 | 0.18521293 |
| GFI1 | −0.1071469 |
| JUND | −0.1352052 |
| IL33 | −0.0164065 |
| NR1H3 | −0.0237538 |
| GEO Database | Platform | Platform Access Number | Total Number | Overall Survival |
|---|---|---|---|---|
| TCGA-BRCA | RNA-Seq | - | 1211 | Alive:891 |
| Dead:135 | ||||
| GSE20685 | Affymetrix Human Genome U133 Plus 2.0 Array | GPL570 | 327 | Alive:244 |
| Dead:83 | ||||
| GSE21653 | Affymetrix Human Genome U133 Plus 2.0 Array | GPL570 | 266 | Alive:169 |
| Dead:83 | ||||
| GSE25066 | Affymetrix Human Genome U133A Array | GPL96 | 508 | Alive:397 |
| Dead:111 | ||||
| GSE96058 | Illumina HiSeq 2000 | - | 3409 | Alive:2937 |
| Dead:336 | ||||
| EGAS00001004353 | RNA-Seq | - | 726 | Alive:422 |
| Dead:463 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, J.; Ren, J.; Li, X.; Zhang, X.; Yang, Q.; Wu, Z.; Cui, L.; Liao, L.; Gong, Y.; Cao, D. Phagocytosis-Regulators-Based Signature to Predict the Prognosis and Chemotherapy Resistance for Breast Cancer Patients. Int. J. Mol. Sci. 2022, 23, 10312. https://doi.org/10.3390/ijms231810312
Feng J, Ren J, Li X, Zhang X, Yang Q, Wu Z, Cui L, Liao L, Gong Y, Cao D. Phagocytosis-Regulators-Based Signature to Predict the Prognosis and Chemotherapy Resistance for Breast Cancer Patients. International Journal of Molecular Sciences. 2022; 23(18):10312. https://doi.org/10.3390/ijms231810312
Chicago/Turabian StyleFeng, Juan, Jun Ren, Xiuqi Li, Xue Zhang, Qingfeng Yang, Zankai Wu, Le Cui, Lingxia Liao, Yiping Gong, and Dedong Cao. 2022. "Phagocytosis-Regulators-Based Signature to Predict the Prognosis and Chemotherapy Resistance for Breast Cancer Patients" International Journal of Molecular Sciences 23, no. 18: 10312. https://doi.org/10.3390/ijms231810312
APA StyleFeng, J., Ren, J., Li, X., Zhang, X., Yang, Q., Wu, Z., Cui, L., Liao, L., Gong, Y., & Cao, D. (2022). Phagocytosis-Regulators-Based Signature to Predict the Prognosis and Chemotherapy Resistance for Breast Cancer Patients. International Journal of Molecular Sciences, 23(18), 10312. https://doi.org/10.3390/ijms231810312