2.1. Condensation without Surface Tension
In order to consider cross-linking by molecular association, we refer back to old studies on the condensation phenomena by a short-range attractive force. We start with the similarity between sol–gel transition and gas–liquid transition. In particular, particulate gels formed by the association of colloid particles have a strong similarity to amorphous liquid particles. To construct the analogy between the gelation of functional molecules in a solvent and condensation of gases into dense phases (liquid or solid), we briefly review the statistical theory of condensation by Mayer and his collaborators [
26,
27,
28,
29,
30].
To evaluate the partition function of a gas in which molecules interact with the pairwise additive potential
, Mayer [
26,
30] introduced the function
at a given absolute temperature
T to avoid a divergence due to the repulsive part (hard core) in
, and expanded the partition function in powers of the molecular density. The result of Mayer’s theory can be summarized in terms of the power series [
31]
for
, whose coefficient
is the
l-th cluster integral
The sum is taken over all possible ways of interacting pairs belonging to a single connected cluster. These cluster integrals are formally introduced in the theory to sum up all terms in the density power series. They do not necessarily describe the property of spatially connected molecules (associated molecules), but provide a mathematical tool rather than physical attributes.
The free energy per molecule is
in terms of the activity
z, and the density
of the molecule (
is the volume per particle). The density and the pressure
p are given by
as functions of the activity. Upon eliminating the activity in these coupled equations, we find the equation of state (
p–
isotherm), through which, we can study phase transitions.
The slope of the pressure in this theory is given by
where
is the weight-average number of molecules in the clusters. The nature of the equation of state is therefore determined by the detailed form of the coefficients
. In particular, the radius of the convergence of the functions
and the nature of the singularities on the convergence boundary are governed by the coefficients
for large
l.
To study the degree of discontinuity at the gas–liquid transition point, the second derivative of the pressure was also calculated. It is
where
is the
z-average number of molecules in the clusters.
Using the saddle point method, Mayer et al. [
28] found that, for a usual force potential of van der Waals type, the cluster integrals take the form
for large
l, where
is a certain nonsingular function of
T, and
is the surface tension of the clusters. This asymptotic expression was later derived more rigorously by Born and Fuchs [
32] and Kahn and Uhlenbeck [
33] by applying the method of steepest descent to the grand partition function as a function of the complex activity. From these studies, the radius of the convergence of the power series
is found to be given by
. The nature of the singular behavior of the pressure and the compressibility depends on whether the series Equation (
3)
on the radius of convergence take finite values, or tends to infinity for
. If the surface tension is positive, these series take finite values for any integer
k. Hence, there is a finite value
for the density
at
. Because Equation (
6a) is divergent (
is infinitely large) as soon as
z exceeds
,
is regarded as the density of the condensation point. At this condensation point, the slope of the pressure is finite because
in Equation (
7) is finite.
However, if the surface tension
is zero in a certain temperature range,
and
are finite, but all higher moments are infinite:
(for
). (
is Rieman’s zeta function [
34] of
x.) Therefore, the slope of the pressure is zero at the condensation point.
Mayer assumed the existence of the temperature (below the critical temperature of a gas–liquid transition), at which, the surface tension of the cluster vanishes. For , the condensation takes place with the finite slope of the pressure (first order phase transition). Because there is a sudden break in the shape of the p– curve at , supersaturation of the vapor would be expected. He proved that the existence of the density (the density of the condensed phase) is larger than , at which, the pressure starts to increase again.
For
, the condensation takes place
with zero slope of the pressure, allowing no extrapolation to a higher pressure of the supersaturated vapor. Compression of the system at a constant temperature in this region induces a uniform increase in density throughout the whole system; droplets change into liquid continuously one by one. He also assumed the existence of the density
(volume
) in this region, but it was a logical guess rather than mathematical proof [
29]. The existence of
can only be confirmed after the structure of the condensed phase (liquid, amorphous solid, crystal, etc.) is clarified. In what follows, we show that thermoreversible gelation is a condensation of the latter type: condensation with no surface tension.
2.2. Sponge Phase of Associated Particles
Before moving on to the detailed description of our model of thermoreversible gelation, we next consider the associated particle model of condensing systems proposed by Frenkel [
35,
36] and Band [
37,
38,
39,
40,
41]. Just after Mayer’s work appeared, Band [
37,
38] realized that Mayer’s general results can be derived almost immediately from the theory of associating assemblies discussed by Fowler [
31]. In this model, the field of the molecular interaction force is assumed to be limited to a small region around any molecule, so that it forms an assembly of molecules, referred to as
particles, or
physical clusters, such as dimers, trimers, etc., in order to distinguish itself from Mayer’s mathematical clusters. However, it neglects interactions among associated particles. The system is therefore regarded as an ideal gas of associated particles. Frenkel [
35,
36] also introduced the same model to study heterogeneous fluctuations and pre-transition phenomena in the neighborhood of the phase transition.
Consider a system in a gas phase (A) changing into a condensed phase (B) by forming associated particles. Let
be the number of molecules that remain in the A phase (unimers), and let
be the number of associated particles consisting of
l molecules (referred to as
l-particles) in the B phase. The total number of molecules is given by
The free energy of the system is then found to be
because of the ideality assumption, where
is the chemical potential of the gas phase A, and
is the free energy of a particle of the size
l. Here,
is the chemical potential of a molecule in the B phase, and
is the surface tension of the particles. The ideal mixing entropy is assumed.
By minimization of this free energy under the constraint (
13), we find that the most probable distribution of the particles takes the form
where
is the difference in the chemical potentials, and
is the dimensionless surface tension. The ratio of the number of molecules in the two phases is then given by
The difference from Mayer’s theory is evident. The pre-factor is missing in this treatment. If , the sum is finite at the transition point where holds (and hence holds), so the transition is discontinuous. If , there is no phase transition; all molecules continuously grow to particles of the B phase. The nature of the transition is thus directly related to the convergence of the power series.
Hill [
42] and Stillinger [
43] studied the relation between the Frenkel–Band theory of association equilibrium and Mayer’s cluster integral theory from the viewpoint of intermolecular interaction potential. Stillinger [
43] pointed out the possibility of a
sponge-like structure in physical clusters at temperatures higher than the gas–liquid transition temperature. If the range of the attractive interaction is short enough, thermal agitation would virtually completely overcome the attractive forces. Only the requirement of overlapping particle spheres should hold the clusters together. Such a sponge-like structure can be identified as a gel network, and the transition exactly corresponds to what we will study in the following sections with regard to thermoreversible gelation.
The associated particle model was later refined by Fisher [
44] to apply specifically to the droplet formation in a gas–liquid phase transition. Fisher’s droplet model assumes that the associated particle is a droplet with well-defined surface free energy
, which satisfies the condition
as
, most simply
with
, and neglects interactions between the droplets. The basic equations are the same as Mayer’s Equations (
6a) and (
6b):
where
is the density,
is a constant,
is the exponent of a surface area (
is the space dimension),
z is the activity of the molecule, and
is the parameter due to the closing effect on the surface of the droplet. If
, we have a first-order gas–liquid transition at
. At the critical point [
44,
45], we have the simultaneous conditions
and
. Hence,
and
, where
is the zeta function of
x.
The droplets model was later explored by many researchers in relation to the critical phenomena [
45], nucleation of supersaturated vapor [
46], percolation and spinodal [
47,
48,
49], etc. What we consider for gels in this article is the other possibility, such that
, but
z changes in the region
. We therefore have the coupled equations
2.3. Associated Particle Model of Polymer Solutions
We now apply the associated particle model to the solutions in which primary functional molecules of the molecular weight
n (in terms of the number of statistical repeat units on them) carrying the number
f of functional groups are dissolved in a solvent [
4,
50,
51,
52,
53,
54,
55]. We mainly consider polymers with a high
n value, but can also include low-molecular-weight molecules with small
n, such as low-mass gelators. For simplicity, we assume that the functionality of the primary molecules is monodisperse and that the functional groups form pairwise bonds that can break and recombine by thermal motion. We consider a model incompressible solution consisting of
N primary functional molecules and
solvent molecules. The total volume of the solution is
, where
is the total volume of the solution counted in the unit of the volume
of a solvent molecule, which is assumed to be equal to the volume of a statistical repeat unit on the primary functional molecule.
Our starting free energy is based on the Flory–Huggins theory of polymer solutions [
24,
56,
57,
58], but the molecular association is taken into consideration (referred to as
associating polymer solutions APS). It is given by
where
,
is the number of associated particles (physical clusters) formed with
l primary molecules (referred to as
l-particle),
is their volume fraction,
is the total volume fraction of the primary molecules, and
is the free energy change accompanying the formation of an
l-particle from the separate primary molecules in their standard reference state (superscript circle). The mixing enthalpy (Flory’s
-parameter [
56]) need not be considered here because we are not concerned in this paper with the liquid–liquid phase separation induced by the van der Waals interaction. We focus on a reversible gelation by associative force only.
To incorporate the post-gel regime, we have included the last term: the free energy of the gel network (condensed phase) consisting of a macroscopic number of the primary molecules. The free energy needed to bind a molecule onto the gel part is given by . In general, it depends on the concentration because the structure of the gel changes with the concentration. The specific form of will be discussed in detail in the following section.
By differentiation, we find, for the chemical potentials
an
l-particle, a solvent molecule, and a molecule in the gel, respectively, where
is the total number of particles possessing the translational degree of freedom, and
is the derivative of
. The sum
gives the number density of the finite particles in the solution. For the gel part,
is the number density of the primary molecules in the gel, and
is the volume fraction of the gel. The gel fraction, defined by the weight fraction of the gel relative to the total weight of the primary molecules, is given by
. The volume fraction of the sol part is then
.
In thermal equilibrium, the solution has a distribution of connected particles with a population distribution fixed by the equilibrium condition
for association and dissociation. Then, we find that the volume fraction of the
l-particles is given by
where
is the volume fraction of the primary molecules that remain free from association (referred to as
unimers), and
is the equilibrium constant.
Because the volume fraction
of the unimers plays the role of the activity, let us rewrite it as
. The number
and the volume fraction
are then given by
where the coefficient
has been introduced. The volume fraction
of the sol part and the total number of associated particles
are then given by
where functions
for
have been introduced. In terms of the gel fraction
, the relation
is transformed into the equation for the volume fraction of the sol part
In the post-gel regime where the gel exists, there is an additional condition regarding the equilibrium between sol and gel. It is
which leads to the relation
Hence, the binding free energy is uniquely related to the activity z of the functional molecule.
The osmotic pressure of the solution is related to the chemical potential of the solvent by the thermodynamic relation
. Explicitly, we have
In the pre-gel regime, the pressure is basically proportional to the total number
of associated particles, since all molecules with a translational degree of freedom equally contribute to the pressure within the ideal gas approximation. In the post-gel regime, there is a contribution from the gel given by the last term. If the binding energy is independent of the concentration, however, the osmotic pressure is independent of the gel fraction. By solving Equation (
30) with respect to
z, and substituting the result into Equation (
33), we obtain the osmotic pressure as a function of the temperature and volume fraction of the primary molecules.
The total free energy per primary molecule is
This is analogous to Mayer’s formula (
5), except the last term (mixing entropy of the solvent).
We next calculate the slope of the osmotic pressure , and find the osmotic compressibility defined by as a function of the temperature and the volume fraction. If the pressure slope becomes zero, the compressibility is divergent. Thus, it indicates a phase transition.
By taking the concentration derivative of Equation (
33), we find
for the slope of the osmotic pressure. Here, the new factor
is defined by
in the pre-gel regime, where
is the weight-average degree of polymerization of cross-linked polymers. The activity
z is a function of
and temperature
T through the relation (
27a). Since we have, alternatively,
this result is the same as Mayer’s one (
7), except the last term
in the slope (
35), which comes from the mixing entropy of solvent molecules. (In the case of gas–liquid transition, vacancy plays the role of the solvent and has no mixing entropy.) This term does not cause any singularity. It simply gives the increment of the pressure in the high concentration region of the primary molecules due to the finiteness of the molecular volume. If we use the approximation
in the last term of Equation (
34) by assuming a low concentration, and put
for low-molecular-weight molecules, Mayer’s formula is exactly recovered.
In the post-gel regime, the function
in the slope has an additional term due to the finite gel fraction
The singularity in the osmotic pressure originates in this function: the translational entropy of associated particles.
At this stage, we introduce a new function
by the definition
for later convenience. The osmotic compressibility is described by
, so that the spinodal condition is simply
If we included the enthalpy of mixing (van der Waals interaction) in terms of Flory’s
-parameter in our starting free energy of the APS model, we would have obtained
for the spinodal condition.
2.4. Application of the Classical Gelation Theory
We now consider specific models for association [
4]. We first split the free energy of association into three parts:
In order to find the combinatorial part
, all particles are assumed to take tree forms. The cycle formation within a particle is neglected. We consider the entropy change in combining
l identical
f-functional molecules into a single Cayley tree. The classical tree statistics [
22,
23] (see also Chapter XII in Flory’s textbook [
24]) gives
for the entropy of combination, where
is Stockmayer’s combinatorial factor. The free energy is given by
.
For the conformational free energy
, we employ the lattice theoretical entropy of disorientation [
24,
56]
for a chain consisting of
n statistical units, where
is the lattice coordination number, and
the symmetry number of the chain. This entropy is produced when a polymer chain carrying the number
n of the statistical units is brought from the hypothetical crystalline state to an amorphous one. The first bond starting from the chain end can be placed in any direction among the nearest-neighboring
cells. The following bonds have only
possible ways of placement because one of the nearest-neighboring cells is already occupied by the preceding monomer. We thus have the factor
. The remaining factor
is the artifact of the lattice theory. We then find
Finally, the free energy of bond formation
is given by
because there are
bonds in a tree of
l molecules (
is the free energy change on forming one bond).
Combining all of the results together, we find
for the equilibrium constant, where
is the
association constant, which provides a measure of the strength of a physical bond.
The total volume fraction and the total number of particles in the solution are then given by using Equations (
27a) and (
27b) as
The parameter z is here defined by . ( being the volume fraction of the unimers.)
2.4.1. Pre-Gel Regime
We first consider the pre-gel regime where all particles are finite. From the fundamental two relations (
48a) and (
48b) given above with
, the function
is given by
so that it is identified to be the reciprocal of the weight-average aggregation number
of particles. In what follows, we show that
is continuous at the gel point concentration, but its derivative
has a discontinuity.
It is now clear that the moments of the Stockmayer’s distribution defined by
play exactly the same roles of the functions
in the theories of Mayer and Frenkel–Band. The number-average and weight-average of the particle distribution are given by
The osmotic pressure is
where the activity
z is related to the volume fraction
by
The slope of the pressure is
The moments
can be exactly calculated by introducing a new parameter
, which is the positive root of the equation
for a given value of
z. The first three moments of Stockmayer’s distribution are explicitly calculated to be [
22]
In order to see the physical meaning of
, let us calculate the extent of the reaction, i.e., the probability for a randomly chosen functional group to be associated. Since an
l-particle includes the total of
functional groups, among which,
are associated, the extent of the reaction is given by
Thus, it turns out that
, introduced by the formal relation (
55), actually gives the extent of the reaction.
By using
, the average particle sizes are given by
Therefore, it is obvious that the gel point is identified to be
, where
because the weight-average particle size becomes infinite at this point. The activity at the gel point is then given by
The volume fraction at the gel point is therefore
All moments are monotonically increasing functions of z, and have a common radius of convergence . For , all moments diverge. Exactly on the radius of convergence , and take finite values, but all moments with are infinite. The number average also diverges at , but, since , we have to study its post-gel behavior on the basis of the proposed treatment of the post-gel regime.
In terms of the reactivity
, we find the osmotic pressure as
The slope of the pressure is
In the pre-gel regime (
), the volume fraction
occupied by the molecules belonging to the sol must always be equal the total polymer volume fraction
. Thus, from Equation (
48a), the total volume fraction
and the extent of association
satisfy the relation
where
(the total number concentration of the functional groups) is used instead of the volume fraction
.
We can solve this equation for
, and find
At the sol–gel transition point, the binding free energy, the free energy for a primary molecule to be bound to the gel network, is
from Equation (
32). This critical value gives
which agrees with Equation (
59). The concentration of polymers at the gel point is then given by Equation (
60), or
This condition gives the sol–gel transition line on the temperature-concentration plane.
To study the singularity at the gel point, we examine how the weight-average diverges. A simple calculation gives
and hence
with the amplitude
The analogy between vapor condensation and gelation in the random polymerization of polyfunctional molecules was originally noticed by Stockmayer [
22] in his statistical–mechanical analysis of the gelation reaction and molecular weight distribution function. Later, the analogy was explored by introducing physical clusters rather than Mayer’s mathematical clusters [
42,
43]. On the basis of Hill’s criterion for a bond formation, an attempt to derive Stockmayer distribution within the theoretical framework of Mayer was made under the condition of no ring-closure [
59,
60]. Gibbs et al. [
61] applied the tree approximation to the Mayer’s cluster integrals and derived very flat
p–
v isotherms, the horizontal portion of which represents gas–liquid coexistence. The form that they assumed for the cluster integrals was
so that it is very close to the asymptotic form of Equation (
10), but leads to slightly different behaviors of
near the transition point.
2.4.2. Similarity to Bose–Einstein Condensation
At this stage, we readily realize that Equations (
48a) and (
48b) are mathematically parallel to those we encountered in the study of the Bose–Einstein condensation (BEC) of ideal Bose gases [
30,
62,
63,
64]. The number density
and the pressure
p of an ideal Bose gas consisting of
N molecules confined in the volume
V is given by
where
z is the activity of the molecule, and
is the thermal de Broglie wave length. The coefficient of the infinite series on the right hand side is replaced from Stockmayer’s combinatorial factor
to
, but other parts are completely analogous. The infinite summations on the right hand side of these equations are known as Truesdell’s function [
65] of order 3/2 and 5/2. Their singularity appearing at the convergence radius
was studied in detail [
65]. Since the internal energy of the Bose gas is related to the pressure as
, the singularity in the compressibility and that in the specific heat have the same nature and reveal discontinuity in their derivatives [
62,
63,
64]. (See also Chapter 14 in Mayer’s textbook [
30].) Hence, the transition (condensation of macroscopic number of molecules into a single quantum state of zero momentum) turns out to be a third-order phase transition.
We now show that a similar picture holds for our gelling solution; a finite fraction of the total number of primary molecules condenses into a single state (gel network) with no center of mass translational degree of freedom (no momentum), although we have no quantum effect. The gel network extends to the entire system, and hence loses its translational degree of freedom.
The analogy of BEC can be seen more clearly if we replace Stockmayer’s combinatorial factor
by its asymptotic form
for large
l, where
is the critical value (
59). This form can be derived by applying Stirling’s formula
for a large
N to Equation (
42). We find that
Thus, we can see that the singularity at
is identical to those in Truesdell’s functions at
. In our previous study [
54], we referred to this important similarity in the more general case of multiple association. Later, the nature of the singularity was clarified [
66] in relation to the coexisting phase separation.
Comparing with the asymptotic form of Equation (
10) of the cluster integral, we can readily see that there is no term corresponding to the surface tension in
for the tree particles. The reason why particles of the tree form have no surface tension is easily understood as follows. The surface free energy of a particle is given by
in a space of dimensions
d. Because the dimensions of a tree are infinite [
67], the surface free energy is proportional to the size
l of the tree, so that it can be adsorbed into the factor
.
Physically, a primary molecule on the surface of an associated particle of tree form has the same, or with negligible difference, contact number with solvent molecules as that of a free primary molecule because of the geometrical characteristics of a tree form. Therefore, no significant difference in the interaction energy is produced when a primary molecule is attached on a tree particle, thus leading to no surface tension.
We can push this analogy further still by considering loops (rings) instead of trees. To overcome the severe limitation in accounting for cyclic structures in tree statistics, Jacobson and Stockmayer [
68,
69] studied the linear polycondensation (
) in which open chains and closed loops coexist. If we apply their idea to reversible loop formation, we can treat it within the theoretical framework of Mayer, Frenkel–Band, or APS. The cluster integrals for a loop of size
l is exactly given by
because the probability of closing the ends of an open chain constructed with the number
l of constituent Gaussian chains is proportional to
(including the symmetry number). If the primary chains are not Gaussian, but obey the scaling law due to the excluded volume effect, the ring closure probability is proportional to
, where
. (
is the space dimensions,
is the Flory’s exponent of the radius of gyration of a chain, and
is the exponent of the total number of self-avoiding random walks [
70].) The exponent
changes from
to
, but the nature of the functions
(
are finite whereas
are infinite at
) remains the same, so the singular behavior of the osmotic pressure remains the same [
71].
A similar factor of the loop entropy appeared in the fusion of DNA double helices [
72,
73,
74]. When a double helix melts partially, a loop made up of the combination of the complementary strands is created, and the entropy of loop formation appears. Thus, the BEC of loops is exactly reproduced within the classical statistical mechanics [
71,
72,
73,
74].
2.4.3. Analyses of the Singularity
In the post-gel regime where
(
), we have an additional equilibrium condition (
32). The activity
z of the solute molecule is related to its binding free energy of the gel.
Since the reactivity of a functional (associative) group in the sol can, in general, be different from that in the gel, let us write the former as
and the latter as
. The average reactivity
of the system as a whole is given by
where
w is the weight fraction of the gel.
The volume fraction
of polymers belonging to the sol is consequently given by
in the post-gel regime, so it is different from the total
that is given by
. The total number of finite clusters must also be replaced by
This gives the number of particles that have a translational degree of freedom. The gel network covers the entire solution and has no translational degree of freedom. If we use the total reactivity
in this equation instead of
in the post-gel regime, we have an unphysical result such that
becomes negative for
because of Equation (
56a).
The problem regarding how the reaction inside the gel proceeds in the post-gel regime depends on the structure of the gel. To find as a function of the concentration of the primary functional molecules, several models are possible.
2.4.4. Stockmayer’s Treatment
Because all particles are assumed to take a tree form, Stockmayer [
22,
23] proposed that the gel must also retain a tree form. Hence, in the post-gel regime, the extent of the reaction in the gel should take the value
, where
is the reactivity of an infinite tree structure without cycles. He also assumed that the extent of the reaction of functional groups in the finite particles remains at the critical value
throughout the post-gel regime. The osmotic pressure is therefore given by
From the definition (
76), the gel fraction
w takes the form
where
(
is the extent of the reaction of the entire system, including all functional groups. It is a linear function of
, and reaches unity (all molecules belong to the gel) at
,
before the reaction is completed. The volume fraction of the sol remains constant at
. The number-average particle size remains constant at
, whereas the weight-average is divergent
in the post-gel regime.
In
Figure 1, the osmotic pressure
as functions of the volume fraction of primary molecules is shown. For simplicity, the primary molecules are assumed to be trifunctional
and of low molecular weight (
). The association constant
is changed from curve to curve. The gel point is indicated by a circle for each value of
. The pressure is continuous at the gel point.
In
Figure 2, the compressibility for the same systems is shown. At the gel point, it is continuous, but has a cusp whose slope is discontinuous. This slope discontinuity is enhanced by the increase in the association constant
. The line connecting the top of the cusps forms a sol–gel transition line.
We thus find that the discontinuity in the slope of the function
is given by
This leads to a discontinuity in the osmotic compressibility of the form
where
is a constant depending only on the temperature, functionality, and the number of statistical units on a chain. For large-molecular-weight primary molecules, the amplitude
B is small. This is the main reason why the experimental detection of the singularity has so far been difficult.
The binding free energy is constant at the value of Equation (
65). From Equation (
48a), which is now equivalent to
we find
The result is schematically shown in
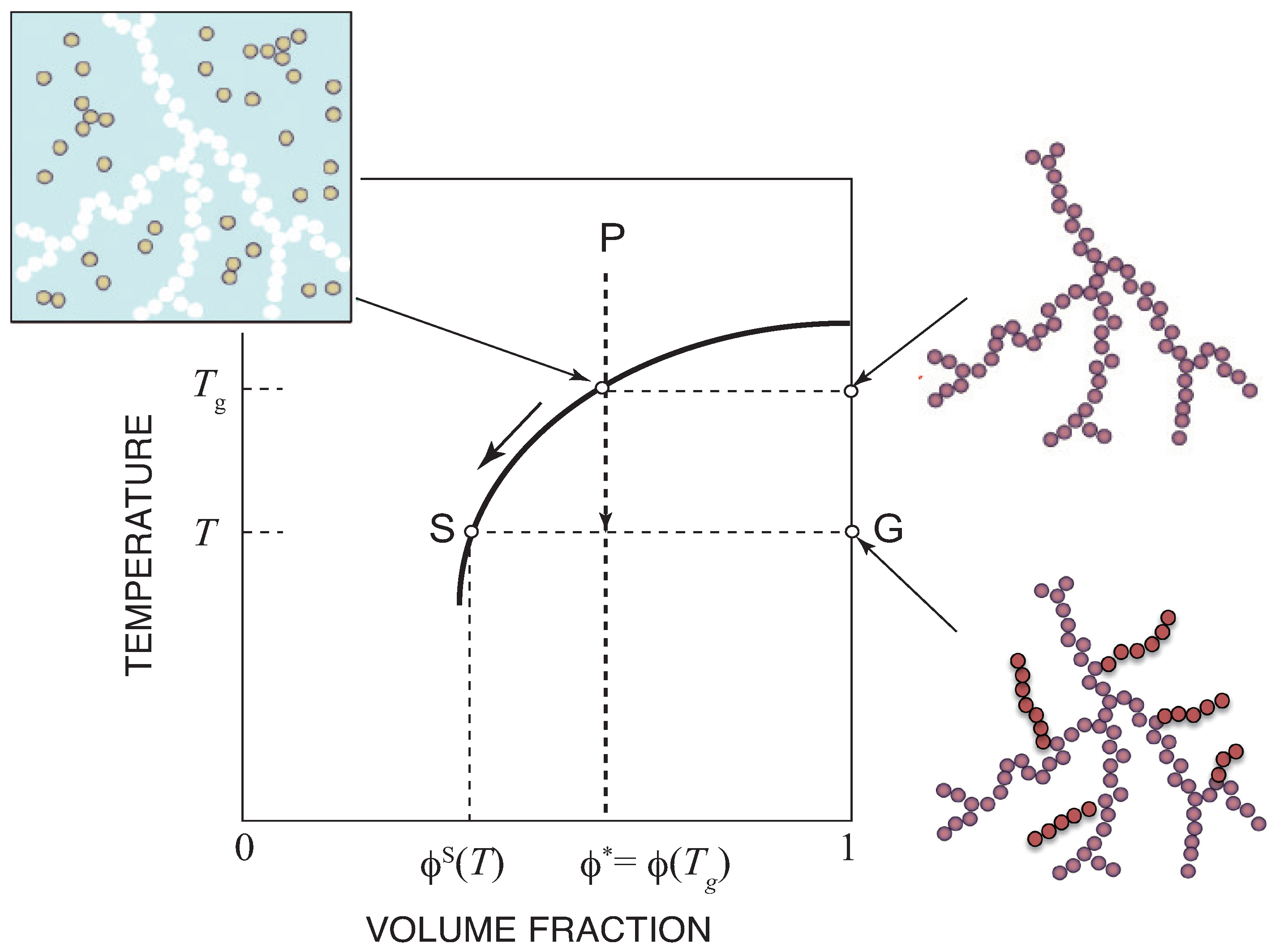
Figure 3 on the temperature–concentration phase plane. If we cool the solution from P at a constant concentration, we hit the sol–gel transition line at temperature
, where the gel network starts to appear. The gel extends to the entire system without phase separation in the coordination space. The molecules are however separated into two phases in the momentum space—molecules with finite momentum (sol) and with zero momentum (gel)—because the gel is macroscopic and has no degree of freedom for the center of mass translational motion. The concentration of the sol remains at the critical value, and therefore the solution moves along the sol–gel transition line by further cooling. The gel grows, but retains a tree structure.
2.4.5. Flory’s Treatment
Theoretically, Stockmayer’s picture [
22,
23] is not the only consistent way to treat the post-gel regime, but other pictures are possible without breaching the fundamental laws of thermodynamics. In fact, Flory proposed a different picture in his work on the gelation reaction of trifunctional molecules [
19,
20,
21]. In his treatment, molecules in the sol part react with those in the gel part with an increase in the concentration, and, as a result, the formation of
cyclic linkages within the gel part is allowed.
Using the definition (
55) for
, the activity
z takes a maximum value
at
. Therefore, two values of
can be found for a given value of
z in the post-gel regime, where
holds. For a given
, the value of
z is fixed by the relation (
55). There is another root
(shadow root) lying below
of the equation for a given value of
z. Flory postulated that
gives the extent of the reaction in the sol. Hence, we have
For the total volume fraction, the relation
remains valid. The volume fraction of the sol part in Equation (
77) is changed to
leading to the sol fraction
. Equation (
78) is also changed to
because the number of particles can be counted only for finite particles In the literature [
75], there is a statement that “In fact one can easily check that the free energy, eqn (2.15), and all its derivatives are perfectly analytical at the gel point”. The error in this paper resides in the treatment of the reaction in the postgel regime. It claims that the study is based on Flory’s postgel picture, but in fact it simply missed the extent of reaction
of the sol, and hence it failed to find the gel fraction. (Superscript S indicates the sol part.)
The function
that appeared in the compressibility is
where
refers to the weight-average particle size in the sol part in the post-gel regime. Thus, from the post-gel form of Equation (
38) of
, we also find a discontinuity
in the slope of the osmotic compressibility in Flory’s treatment, although the sign of the discontinuity becomes negative. The gel fraction is given by
The gel fraction reaches unity only at the limit of complete reaction
. The extent of association
in the gel can be obtained by the definition of the total reactivity (
76). Explicitly, it gives
This value is obviously larger than
(infinite limit of the tree) so that, in Flory’s picture,
cycle formation is allowed within the gel network. Its cycle rank is given by
The free energy
for binding a primary molecule onto the gel network turns out to be
It is a monotonically decreasing function of the concentration. With an increase in the concentration, the network structure becomes tighter, so the binding of a polymer chain becomes stronger. Since the average number of bonds per molecule is , the binding free energy per bond is given by . This is not a constant, but changes as the reaction proceeds.
The result is schematically shown in
Figure 4. If we cool the solution from P at a constant concentration, we hit the sol–gel transition line at temperature
, where the gel network starts to appear. By further cooling, the solution moves along the new line, which lies above the sol–gel transition line because the concentration of the sol decreases below the critical value. The gel network grows and forms cycles within it.
2.4.6. Other Post-Gel Treatments
The difference in the above two treatments was later examined from a kinetic point of view. For an irreversible cross-linking reaction, Ziff and Stell [
76] clarified the reaction mechanism (sol–gel interaction) in these two treatments after the gel point is passed. They found that, in Stockmayer’s treatment, reactive groups in the sol do not interact with those in the gel, and the gel grows only through a
cascade process from the sol to the gel, whereas, in Flory’s treatment, all functional groups are allowed to react. On the basis of their kinetic study, they proposed a third model that takes the reaction between the sol and gel into account as in Flory’s picture, while the cycle formation in the gel is forbidden as in Stockmayer’s one.
In classical tree statistics, the number of the functional groups on the surface of a tree-like cluster is of the same order as that of the groups inside the cluster, so that a simple thermodynamic limit without a surface term is impossible to take. The equilibrium statistical mechanics for the polycondensation was later refined by Yan [
77] by taking the surface correction into tree-like systems. He found the same result as Ziff and Stell.
Later, in order to ensure the equilibrium distribution, additional terms describing the reversible reaction (fragmentation) were introduced to the kinetic equation by van Dongen and Ernst [
78]. Since their study was limited only to Flory’s and Stockmayer’s model, the possibility of other new treatments within the classical tree statistics for reversible gelation remains an open question. From the mathematical analysis given in this paper, however, it is highly probable that a new thermodynamically consistent treatment, even if it exists, leads to the third-order singularity lying somewhere between Stockmayer’s one and Flory’s one.
Since the appearance of APS, there have been studies accumulated on thermoreversible gelation by computer simulation. They are mainly concerned with the percolation of the clusters, and there have only been a few reports that seriously check the osmotic pressure. Kumar and Panagiotopoulos [
79] used a chain model carrying strongly associative stickers regularly placed along the chain, and studied the nature of the transition by a grand canonical Monte Carlo simulation on a lattice. They showed that the osmotic pressure exhibits a cusp-like behavior at a low temperature, just like the form shown in
Figure 1 of the present paper. They attributed the behavior to the critical micelle concentration (cmc) because it was independent of the system size, and reached the conclusion that it would not grow to a singularity. Since the third order singularity is expected to be very weak and difficult to detect experimentally, further careful studies on the pressure by computer simulation are eagerly hoped for.
2.5. Percolation Models for Gelation
In a quite different way from the classical theory of gelation, the percolation model of gelation focuses on the geometrical structure and connectivity of the system. We describe the percolation model with an attempt to apply it to the gelation problem [
70,
80,
81,
82].
Percolation models are roughly classified into percolation on regular lattices and percolation in continuum space. Both of them study the scaling laws for the anomaly of geometrical and physical properties near the percolation threshold on the basis of the self-similarity of the connected objects. In this section, we consider percolation on regular lattices. Percolation in continuum space will be discussed in the following section.
There are two types of lattice percolation problems: site percolation and bond percolation.
2.5.1. Site Percolation
First, we focus on the site percolation. In a site percolation, molecules are randomly distributed on the lattice sites. Neighboring pairs of molecules are regarded as connected. Let be the total number of the lattice sites, and N be the number of molecules placed on them. The percolation probability p defined by is identical to the volume fraction discussed above. When p exceeds a certain threshold value , connected particles (clusters) of infinite size appear. This critical value depends on the space dimensions d and the lattice structure.
The cluster distribution function
is defined by
wher
(
) is the number of connected clusters consisting of
l molecules (referred to as
l-mers). The number density
of
l-mers is defined by
In the region
after the percolation threshold is passed, the infinite cluster coexists with finite clusters. Let
be the number of molecules in the infinite cluster. The total molecules are decomposed into two parts
Dividing by
, we find the relation
where
is the volume fraction of the infinite cluster. The gel fraction
is then given by
In the critical region near the percolation threshold, the structure of the clusters are self-similar; the structure observed in a certain length scale looks similar to a part of it when the part is magnified properly, and, hence, they are superimposable to each other. Thus, it is known that the cluster distribution function obeys the scaling law
where
is a power index referred to as the Fisher index, and
is the reference size of the clusters [
70,
80,
81]. The size
is shown to be the
z-average cluster size
. Practically, it is the size of the largest cluster. Since it diverges at
, the index
is introduced by the scaling law
The index
and
are two fundamental structural indices of the percolation theory. The function
is a smooth scaling function that decays sufficiently quickly. On the basis of these two power indices
and
, scaling laws of various averages and the gel fraction can be derived [
80,
81].
The defect of the site percolation model is that all clusters are fixed on the lattice. There is no translational motion of their mass centers, so the pressure and temperature effect cannot be studied. If we assume that clusters obeying the distribution function (
99) move freely with no inter-cluster interaction as in the Frenkel–Band model of associated particles, the pressure is proportional to the total number
of finite clusters. Its slope is therefore
and we go back to Mayer’s Equation (
7). The scaling law with the index
leads to a singularity of the compressibility.
Another serious deficiency of the site percolation model resides in the effect of temperature. At high percolation probability p (volume fraction of the particles), in particular, at the limit of , the system always remains percolated no matter how high the temperature is. In other words, there is no temperature-dependent transition.
2.5.2. Site–Bond Correlated Percolation
To overcome such deficiencies of the site percolation model, Coniglio et al. [
83,
84] introduced random bond formation between the nearest neighboring molecules in the standard lattice gas model. Solvent (A) and solute (B) molecules placed on a lattice interact with each other in two ways: the usual van der Waals interaction
(
A,B) and reversible bond formation with the bond energy
. In what follows, the model is referred to as
site–bond percolation (SBP). In this paper, we focus only on the bond formation and neglect the van der Waals interaction, as in the preceding sections. Hence, we fix
, and
with probability
for non-bonded solute pairs, and
with probability
for bonded pairs. The Hamiltonian is
where
is the variable for the lattice gas solute molecules,
is the variable of the bond formation for the pair
, and
is the chemical potential of the particle. The grand partition function is then calculated by
Summing up with respect to all
(annealed average), we find that
where
and
. By introducing the Ising variables
through the relation
, the grand partition function can be related to the canonical partition function
for the Ising magnet by the equation
where
and
(
f is the number of the nearest neighboring sites). The volume fraction of the solute molecules is given by
where
is the maginitization of the Ising magnet, and
is the free energy of the Ising magnet. The pressure is given by
Therefore, if we knew the free energy of an Ising ferromagnet as a function of the temperature and the magnetic field, we can study the p–v curve of the lattice gas.
The slope of the pressure in the isotherm is then obtained by
Using the definition of the magnetization, we find
so that
where
is the activity. This relation is in agreement with the relation (
7) in Mayer’s theory, and also Equation (
35) in APS, except the last term. The last term of Equation (
35) comes from the difference between the chemical potential of the solvent in a solution and that of a vacancy of a gas. If we have vacancies instead of solvent molecules, we have, for the activity of the solute molecules,
Hence, the two relations are identical., so that it is a model-independent general relation.
In the ideal case where there is no bond formation, we have
, so that
. The pressure and concentration are given by
and
By eliminating
H, we find
and hence
This agrees with the APS Equationn (
54), with
and
.
2.5.3. Exact Solution on the Bethe Lattice
We next consider the exact solution of SBP on the Bethe lattice presented by Coniglio et al. [
83,
84]. It is well known that the Ising model of a ferromagnet on a Bethe lattice can be solved exactly. The result is summarized for instance in Baxter [
67]. The solution was applied to the site percolation problem of the Ising lattice gas by Odagaki [
85] and Coniglio [
86]. We first review these works on the site percolation problem, and then go back to the SBP problem.
The solution of the Ising ferromagnet on a Bethe lattice can be described as follows. Let
,
, and
be the three fundamental parameters. These parameters are written as
,
and
in Ref. [
67]. Since we use the letter
z for the activity and
for the chemical potential, we have changed these letters. The coordination number
q is replaced by
f to compare with the Flory–Stockmayer theory. The parameter
x is defined by
and is related to the magnetization by
In terms of the activity
, we find
because
. By eliminating
and
, we find
The concentration of the solute molecules is
and the pressure can be found from the exact solution of the Ising free energy [
67] as
We then eliminate the activity
z by using Equations (
125) and (
126), and find
x in terms of the concentration and temperature as [
85]
The activity as a function of the concentration is found by substituting this into Equation (
125). With all of these results, we can find the pressure explicitly as a function of the concentration
We focus on a solute molecule and find the probability
q for one of its nearest neighboring cites to be occupied by a solute molecule. It is given by the correlation function
and calculated to be [
85,
86]
Substituting the above
x, we find
In this result, we consider the limit of strong bond energy
with a fixed
. The factor
in the square root can be replaced by 1 in this limit, so the probability
q agrees with the reactivity
in Equation (
64) for APS. The association constant
corresponds to the factor
of the Ising model. Because
, the relation is summarized as
In the limit of strong bond energy, 1 is neglected in the second factor. The relation reduces to
Therefore, we see that, if we take the strong bond limit
with finite
, the site percolation model reduces to APS with association constant (
133).
The volume fraction of the particles can be written as
in terms of the probability
q. This equation reduces to APS Equation (
63) in the limit of the strong bonds (
).
The weight-average molecular weight of the connected clusters in the site percolation problem is given by [
85,
86]
so that the percolation point is decided by the equation
or
. The concentration at the percolation point is therefore
where
, etc., have been used.
We now see the trouble with this site percolation model. In the limit of high temperature
, the percolation line has a finite limiting value
. In other word, the system remains percolated no matter how high the temperature is if the concentration is higher than the critical value
. There is no percolation transition by the temperature change. This unphysical result originates from the assumption that a nearest neighboring pair is always regarded as connected. In fact,
(connectivity 1) always trivially holds at
(no solvent) as is seen from Equation (
131).
To remedy this defect of site percolation, Coniglio et al. [
83,
84] introduced the SBP model, in which, the nearest neighboring pair is either bonded (with probability
) or unbonded (with probability
). The connectivity probability
q is replaced by
in all of the above relations. In particular, the weight-average molecular weight and the percolation threshold is given by
as in Equation (
57b), but now
.
This relation is the same both in APS (the limit of strong bond) and SBP (bond formation introduced for the nearest neighboring pairs). Both models have a sol–gel transition at full volume fraction . However, the thermodynamic nature of the transition is different.
In order to see the difference, we study the slope (
118) of the pressure. First, by taking the derivative of Equation (
125) with respect to the concentration, we find
Substituting into the relation
obtained from the derivative of Equation (
126), we find
where
The spinodal condition is therefore given by
, or, explicitly,
This condition for the spinodal of the Ising model is different from the percolation condition (
136), and hence the percolation line is not accompanied by any singularity. However, if we take the strong bond limit
in this spinodal condition (with finite
), we can see that the spinodal condition becomes identical to the percolation condition. In other words, the slope of the pressure vanishes on the percolation line.
As for the treatment of the post-gel regime, the percolation model on a Bethe lattice has a serious problem. A Bethe lattice with a finite number N of cites has the total of the free functional groups on the surface if we fill the lattice with functional molecules. Hence, in the thermodynamic limit of , the reactivity of the nearest neighboring molecular pairs has an upper limit . Therefore, Flory’s treatment allowing for cycle formation is impossible. This is one of the most important differences between the classical tree statistics of the polyfunctional molecules in a three-dimensional off-lattice free space and the percolation model on a Bethe lattice. For the latter, Stockmayer’s treatment is the only possible treatment of the post-gel regime.
2.6. Adhesive Hard Sphere Model
Let us move onto percolation in a continuum space. In the study of the gas–liquid phase transition of spherical particles, Baxter introduced a model in which hard sphere particles interact with each other by the attractive force potential of a rectangular well shape (referred to as an adhesive hard sphere model AHS; see
Figure 5a,b) [
87,
88]. In the limit of the surface adhesion, i.e., narrow force range and strong attractive force, he found the analytic solution of the thermodynamic problem within Percus–Yevick approximation. Since then, AHS is often used as a model system for the study of gelation phenomena in globular proteins, colloid dispersions, silica aerogels, and other particulate gels.
Consider
N spherical particles of radius
in a container of volume
V. The volume fraction
is used for the concentration. The attractive potential
has a depth
and width
. Baxter [
87] introduced the reduced temperature
by the definition
The second virial coefficient of AHS is
where
is the Mayer’s function (
2) for AHS. It can be normalized as
by using the hard sphere system
as the reference system. In the Baxter limit,
. If we apply Mayer’s theory of condensation, the coefficients (cluster integrals)
for AHS are constructed by this special form of
.
In the limit of short range
and strong force
, the spheres form
branched sponge-like clusters in which they are connected to each other by surface adhesion [
87]. Above a certain volume fraction, the clusters percolate over the entire container [
89,
90,
91,
92].
AHS systems show interesting phase diagrams in which gas–liquid phase transition (thermal problem) coexists with percolation transition (connectivity problem). The percolated cluster can be regarded as a porous gel comprising spherical particles. There have been theoretical attempts to construct phase diagrams on the
-
plane [
90,
91]. Within Percus–Yevick approximation, Chiew and Glandt [
90] found that the percolation line is given by In the dilute region, this approximation is poor. In particular, the gelation temperature must decrease to
in the limit of
, while Equation (
148) gives a finite
.
Because the analytical solution of the problem is difficult to find, molecular simulations are used to construct the phase diagram [
93,
94,
95,
96,
97].
From a thermodynamic point of view on the sol–gel transition, AHS retains the same deficiency as the site percolation; in the limit of high density, the system remains percolated at any high temperature. To refine AHS, there have been several attempt to replace the Baxter potential by patchy sticky hard spheres [
98,
99,
100], which carry a varied number
f of attractive patches on the surface (
Figure 5c). Two spheres interact via a sticky Baxter potential if the line joining the centers of the two spheres intersects a patch on each sphere, and via a hard sphere potential otherwise. The area and distribution of attractive patches on the sphere surface are changed to study how the percolation line and phase separation (gas–liquid) line shift. The analytical study and Monte Carlo simulation [
98] on the systems with
with a varied patch area showed that the percolation line shifts to a higher concentration and lower temperature region as the patch area decreases. However, because the analytical study of the pressure was based on the virial expansion up to the third order of the density, or on the integral equation approximation for closure, it was impossible to find any singularity across the percolation line. If the number
f is increased, with a smaller patch area, the cluster integrals of such a patchy AHS take the form similar to
in tree statistics with the functionality
f (
Figure 5d) for a large cluster size
l. There is a strong tendency to form tree-type clusters rather than spherical droplets. As a result, the percolation transition splits substantially away from the gas–liquid transition line, as was pointed out by Stillinger [
43] for the association model. We can expect that patchy AHS with
reveals a temperature-controlled transition even in the limit of a high density.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




