
Clinical Implications of the Genetic Background in Pediatric Pulmonary Arterial Hypertension: Data from the Spanish REHIPED Registry
 , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Methods
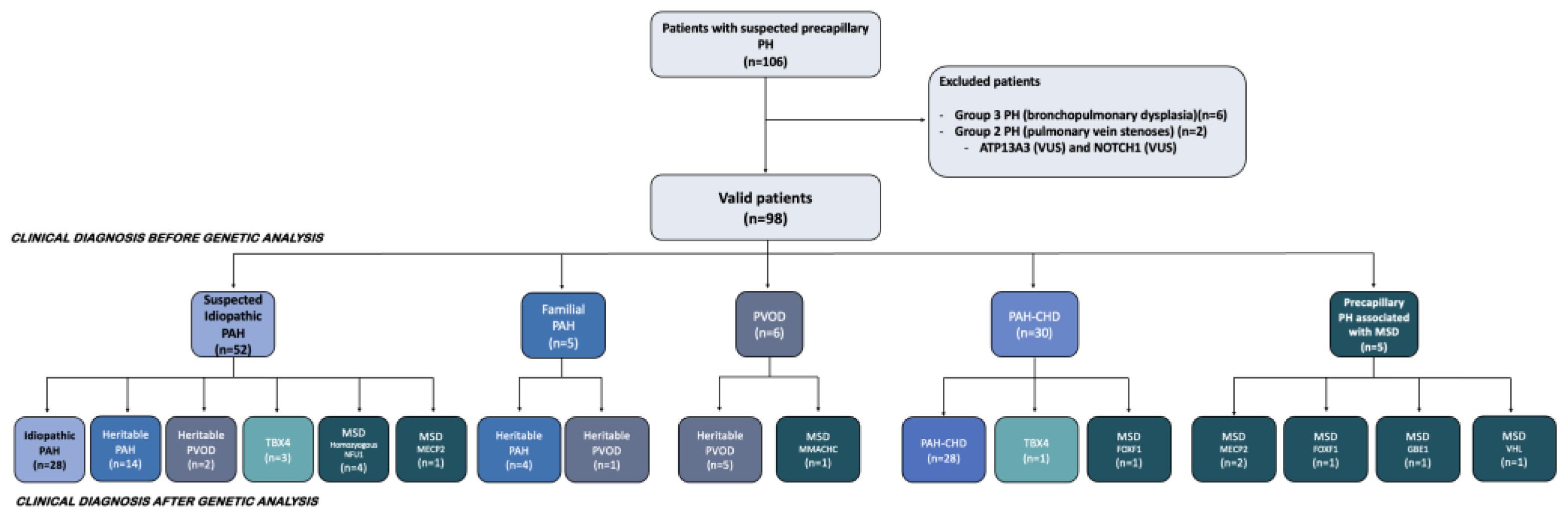
2.1. Study Population
2.2. Clinical Classification of Included Patients
2.3. Genetic Analyses
2.4. Outcomes
2.5. Statistics
3. Results
3.1. Genetic Findings in Idiopathic, Heritable PAH and TBX4 Forms
3.2. Findings in the PAH-CHD Population
3.3. Genetic Findings in the Pediatric PVOD Population
3.4. Genetic Analysis in Patients with Pulmonary Hypertension Associated with a Multisystemic Disorder
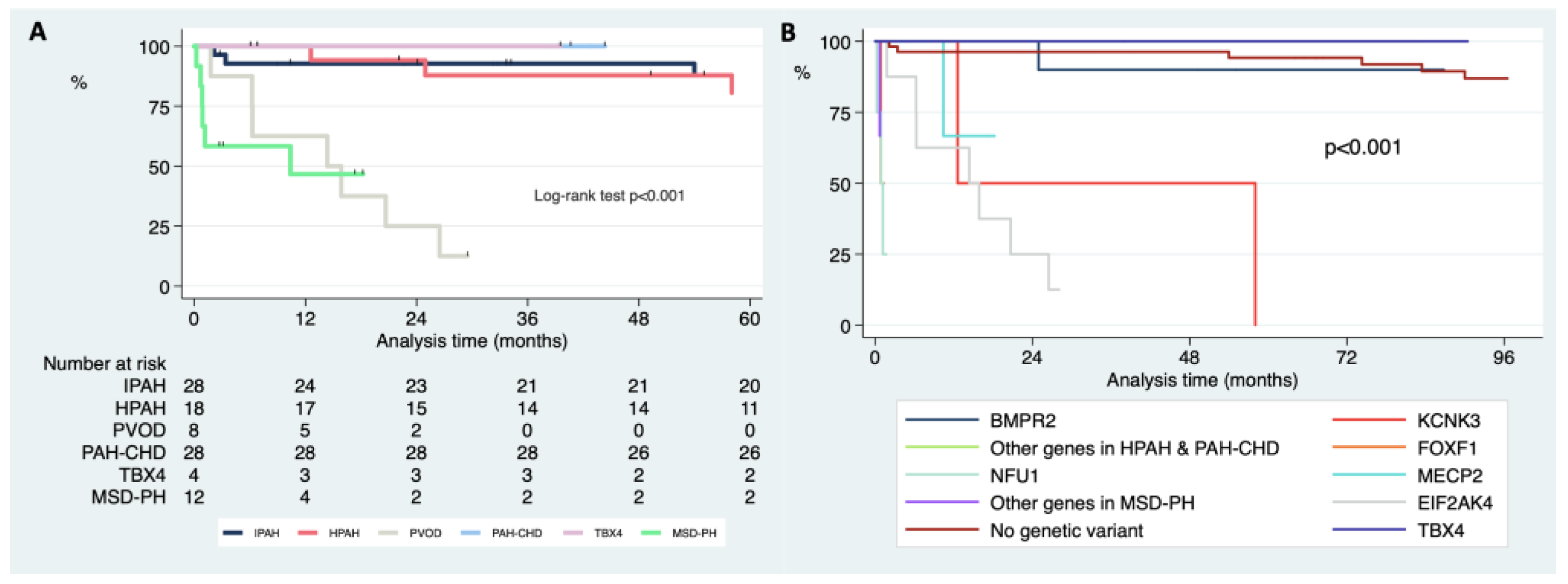
3.5. Differences in Survival Free of Death or Lung Transplantation according to the Clinical Classification and Genetics after Genetic Testing
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, R.L.; Roofthooft, M.T.R.; van Osch-Gevers, M.; Delhaas, T.; Strengers, j.M.; Blom, N.A.; Backx, A.; Berger, R.M.F. Clinical characterization of pediatric pulmonary hypertension: Complex presentation and diagnosis. J. Pediatr. 2009, 155, 176–182.e1. [Google Scholar] [CrossRef] [PubMed]
- Cerro, M.J.; Abman, S.; Diaz, G.; Freudenthal, A.H.; Freudenthal, F.; Harikrishnan, S.; Haworth, S.G.; Ivy, D.; Lopes, A.A.; Raj, J.U.; et al. A consensus approach tothe classification of pediatric pulmonary hypertensive vascular disease: Report from the PVRI Pediatric Taskforce, Panama 2011. Pulm. Circ. 2011, 1, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Moledina, S.; Hislop, A.A.; Foster, H.; Schulze-Neick, I.; Haworth, S.G. Childhood idiopathic pulmonary arterial hypertension: A national cohort study. Heart 2010, 96, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H.; Mullen, M.P.; Sleeper, L.A.; Austin, E.D.; Rosenzweig, E.B.; Kinsella, J.P.; Ivy, D.; Hopper, R.K.; Raj, J.U.; Fineman, J.; et al. Characterisation of paediatric pulmonary hypertensive vascular disease from the PPHNet Registry. Eur. Respir. J. 2022, 59, 2003337. [Google Scholar] [CrossRef]
- Marín, M.D.C.; Rotés, A.S.; Ogando, A.R.; Soto, A.M.; Jiménez, M.Q.; Camacho, J.G.; Sonnenfeld, I.R.; Bonora, A.M.; Brotons, D.A.; Galdó, A.M. Assessing pulmonary hypertensive vascular disease in childhood. Data from the Spanish registry. Am. J. Respir. Crit. Care Med. 2014, 190, 1421–1429. [Google Scholar] [CrossRef]
- Welch, C.L.; Austin, E.D.; Chung, W.K. Genes that drive the pathobiology of pediatric pulmonary arterial hypertension. Pediatr. Pulmonol. 2021, 56, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Gonzaga-Jauregui, C.; Welch, C.L.; Ma, L.; Qi, H.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; Austin, E.D.; et al. Exome Sequencing in Children With Pulmonary Arterial Hypertension Demonstrates Differences Compared With Adults. Circ. Genomic. Precis. Med. 2018, 11, e001887. [Google Scholar] [CrossRef]
- Southgate, L.; Machado, R.D.; Gräf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2020, 17, 85–95. [Google Scholar] [CrossRef]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Subias, P.E.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. USA 2021, 384, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, E.B.; Abman, S.H.; Adatia, I.; Beghetti, M.; Bonnet, D.; Haworth, S.; Ivy, D.D.; Berger, R.M.F. Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur. Respir. J. 2019, 53, 1801916. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, H.; De Backer, J.; Babu-Narayan, S.V.; Budts, W.; Chessa, M.; Diller, G.P.; Lung, B.; Kluin, J.; Lang, I.M.; Meijboom, F.; et al. 2020 ESC Guidelines for the management of adult congenital heart disease: The Task Force for the management of adult congenital heart disease of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 42, 563–645. [Google Scholar] [CrossRef] [PubMed]
- Castaño, J.A.T.; Hernández-Gonzalez, I.; Gallego, N.; Pérez-Olivares, C.; Ochoa Parra, N.; Arias, P.; Granda, E.; Acebo, G.G.; Lago-Docampo, M.; Palomino-Doza, J.; et al. Customized Massive Parallel Sequencing Panel for Diagnosis of Pulmonary Arterial Hypertension. Genes 2020, 11, 1158. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Zazo, N.; Cruz-Utrilla, A.; del Cerro, M.J.; Parra, N.O.; Blanco, J.N.; Arias, P.; Lapunzina, P.; Escribano-Subias, P.; Tenorio-Castaño, J. Description of Two New Cases of AQP1 Related Pulmonary Arterial Hypertension and Review of the Literature. Genes 2022, 13, 927. [Google Scholar] [CrossRef] [PubMed]
- van Gent, M.W.; Velthuis, S.; Post, M.C.; Snijder, R.J.; Westermann, C.J.; Letteboer, T.G.; Mager, J.J. Hereditary hemorrhagic telangiectasia: How accurate are the clinical criteria? Am. J. Med. Genet. A 2013, 161A, 461–466. [Google Scholar] [CrossRef]
- Chida, A.; Shintani, M.; Yagi, H.; Fujiwara, M.; Kojima, Y.; Sato, H.; Imamura, S.; Yokozawa, M.; Onodera, N.; Horigome, H.; et al. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am. J. Cardiol. 2012, 110, 586–593. [Google Scholar] [CrossRef]
- Levy, M.; Eyries, M.; Szezepanski, I.; Ladouceur, M.; Nadaud, S.; Bonnet, D.; Soubrier, F. Genetic analyses in a cohort of children with pulmonary hypertension. Eur. Respir. J. 2016, 48, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Haarman, M.G.; Kerstjens-Frederikse, W.S.; Vissia-Kazemier, T.R.; Breeman, K.T.; Timens, W.; Vos, Y.J.; Roofthooft, M.T.; Hillege, H.L.; Berger, R.M. The Genetic Epidemiology of Pediatric Pulmonary Arterial Hypertension. J. Pediatr. 2020, 225, 65–73.e5. [Google Scholar] [CrossRef]
- Tenorio, J.; Navas, P.; Barrios, E.; Fernández, L.; Nevado, J.; Quezada, C.A.; López-Meseguer, M.; Arias, P.; Mena, R.; Lobo, J.L.; et al. A founder EIF2AK4 mutation causes an aggressive form of pulmonary arterial hypertension in Iberian Gypsies. Clin. Genet. 2015, 88, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Hadinnapola, C.; Bleda, M.; Haimel, M.; Screaton, N.; Swift, A.; Dorfmüller, P.; Preston, S.D.; Southwood, M.; Hernandez-Sanchez, J.; Martin, J.; et al. Phenotypic Characterization of EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically With Pulmonary Arterial Hypertension. Circulation 2017, 136, 2022–2033. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Girerd, B.; Jaïs, X.; Laveneziana, P.; Lau, E.M.; Bouchachi, A.; Hascoët, S.; Günther, S.; Godinas, L.; Parent, F.; et al. Screening for pulmonary arterial hypertension in adults carrying a BMPR2 mutation. Eur. Respir. J. 2021, 58, 2004229. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.E.; McElroy, J.J.; Wong, W.P.K.; Yen, E.; Widlitz, A.; Barst, R.J.; Knowles, J.A.; Morse, J.H. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur. Respir. J. 2004, 24, 371–374. [Google Scholar] [CrossRef]
- Liu, D.; Liu, Q.-Q.; Guan, L.-H.; Jiang, X.; Zhou, D.-X.; Beghetti, M.; Qu, J.-M.; Jing, Z.-C. BMPR2 mutation is a potential predisposing genetic risk factor for congenital heart disease associated pulmonary vascular disease. Int. J. Cardiol. 2016, 211, 132–136. [Google Scholar] [CrossRef]
- Montani, D.; Lechartier, B.; Girerd, B.; Eyries, M.; Ghigna, M.-R.; Savale, L.; Jaïs, X.; Seferian, A.; Jevnikar, M.; Boucly, A.; et al. An emerging phenotype of pulmonary arterial hypertension patients carrying SOX17 variants. Eur. Respir. J. 2022, 2200656. [Google Scholar] [CrossRef]
- Cruz-Utrilla, A.; Gallego, N.; Segura de la Cal, T.; Tenorio-Castaño, J.; Arribas-Ynsaurriaga, F.; Escribano Subias, P. The role of genetics in pulmonary arterial hypertension associated with congenital heart disease. Rev. Esp. Cardiol. (Engl. Ed.) 2021, 74, 884–886. [Google Scholar] [CrossRef]
- Navarro-Sastre, A.; Tort, F.; Stehling, O.; Uzarska, M.A.; Arranz, J.A.; del Toro, M.; Labayru, M.T.; Landa, J.; Font, A.; Garcia-Villoria, J.; et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am. J. Hum. Genet. 2011, 89, 656–667. [Google Scholar] [CrossRef]
- Menéndez Suso, J.J.; Del Cerro Marín, M.J.; Dorao Martínez-Romillo, P.; Labrandero de Lera, C.; Fernández García-Moya, L.; Rodríguez González, J.I. Nonketotic hyperglycinemia presenting as pulmonary hypertensive vascular disease and fatal pulmonary edema in response to pulmonary vasodilator therapy. J. Pediatr. 2012, 161, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Ahting, U.; Mayr, J.A.; Vanlander, A.V.; Hardy, S.A.; Santra, S.; Makowski, C.; Alston, C.L.; Zimmermann, F.A.; Abela, L.; Plecko, B.; et al. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Front. Genet. 2015, 6, 123. [Google Scholar] [CrossRef] [Green Version]
- Bourque, D.K.; Fonseca, I.C.; Staines, A.; Teitelbaum, R.; Axford, M.M.; Jobling, R.; Chiasson, D.; Chitayat, D. Alveolar capillary dysplasia with misalignment of the pulmonary veins and hypoplastic left heart sequence caused by an in frame deletion within FOXF1. Am. J. Med. Genet. A 2019, 179, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Kömhoff, M.; Roofthooft, M.T.; Westra, D.; Teertstra, T.K.; Losito, A.; van de Kar, N.C.; Berger, R.M. Combined pulmonary hypertension and renal thrombotic microangiopathy in cobalamin C deficiency. Pediatrics 2013, 132, e540–e544. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Labrune, P.; Sitbon, O.; Le Gall, C.; Callebert, J.; Herve, P.; Samuel, D.; Machado, R.; Trembath, R.; Drouet, L.; et al. Pulmonary arterial hypertension and type-I glycogen-storage disease: The serotonin hypothesis. Eur. Respir. J. 2002, 20, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-P.; Xie, W.-M.; Huang, X.; Lu, X.; Zhai, Z.-G.; Zhan, Q.-Y.; Wang, C. Pulmonary Hypertension in Glycogen Storage Disease Type II. Chin. Med. J. (Engl.) 2018, 131, 1375–1376. [Google Scholar] [CrossRef] [PubMed]
- Giudice-Nairn, P.; Downs, J.; Wong, K.; Wilson, D.; Ta, D.; Gattas, M.; Amor, D.; Thompson, E.; Kirrali-Borri, C.; Ellaway, C.; et al. The incidence, prevalence and clinical features of MECP2 duplication syndrome in Australian children. J. Paediatr. Child Health 2019, 55, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- De Felice, C.; Guazzi, G.; Rossi, M.; Ciccoli, L.; Signorini, C.; Leoncini, S.; Tonni, G.; Latini, G.; Valacchi, G.; Hayek, J. Unrecognized lung disease in classic Rett syndrome: A physiologic and high-resolution CT imaging study. Chest 2010, 138, 386–392. [Google Scholar] [CrossRef]
- Caravita, S.; Deboeck, G.; Vachiery, J.-L.; Naeije, R. Pulmonary arterial hypertension associated with a von Hippel-Lindau gene mutation. J. Hear Lung Transplant Off. Publ. Int. Soc. Hear Transplant 2016, 35, 1138–1139. [Google Scholar] [CrossRef]
- Chomette, L.; Migeotte, I.; Dewachter, C.; Vachiery, J.-L.; Smits, G.; Bondue, A. Early-onset and severe pulmonary arterial hypertension due to a novel compound heterozygous association of rare VHL mutations: A case report and review of existing data. Pulm. Circ. 2022, 12, e12052. [Google Scholar] [CrossRef]
- Austin, E.D.; Elliott, C.G. TBX4 syndrome: A systemic disease highlighted by pulmonary arterial hypertension in its most severe form. Eur. Respir. J. 2020, 55, 2000585. [Google Scholar] [CrossRef]
- Hernandez-Gonzalez, I.; Tenorio, J.A.; Palomino-Doza, J.; Meñaca, A.M.; Ruiz, R.M.; Lago-Docampo, M.; Gomez, M.V.; Roman, J.G.; Valls, A.B.E.; Perez-Olivares, C.; et al. Clinical heterogeneity of Pulmonary Arterial Hypertension associated with variants in TBX4. PLoS ONE 2020, 15, e0232216. [Google Scholar] [CrossRef]
- Pienkos, S.; Gallego, N.; Condon, D.F.; Cruz-Utrilla, A.; Ochoa, N.; Nevado, J.; Arias, P.; Agarwal, S.; Patel, H.; Chakraborty, A.; et al. Novel TNIP2 and TRAF2 Variants Are Implicated in the Pathogenesis of Pulmonary Arterial Hypertension. Front. Med. 2021, 8, 625763. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Cohort (n = 98) | Idiopathic (n = 28) | Heritable PAH (n = 18) | PVOD (n = 8) | PAH-CHD (n = 28) | TBX4 (n = 4) | PH-MSD (n = 12) | p Value | |
|---|---|---|---|---|---|---|---|---|
| Age—years (median—IQr) | 7.1 (1.5–14.7) | 6.3 (3.6–12.6) | 10.2 (5.6–17.7) | 15.7 (12.4–17.3) | 6.3 (2.1–10.5) | 0.4 (0.2–8.1) | 0.1 (0.1–0.4) | <0.001 |
| Gender—male (n—%) | 43 (43.9) | 13 (46.4) | 6 (33.3) | 6 (75.0) | 10 (35.7) | 2 (50.0) | 6 (50.0) | 0.412 |
| Descendance (n—%) | <0.001 * | |||||||
| Arabic | 2 (2.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (16.7) | |
| Asiatic | 1 (1.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Caucasian | 80 (81.6) | 27 (96.4) | 13 (72.2) | 2 (25.0) | 25 (89.3) | 4 (100.0) | 9 (75.0) | |
| Romani | 8 (8.2) | 0 (0.0) | 2 (11.1) | 6 (75.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Hispanic | 6 (6.1) | 1 (3.6) | 2 (11.1) | 0 (0.0) | 2 (7.1) | 0 (0.0) | 1 (8.3) | |
| Black | 1 (1.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (3.6) | 0 (0.0) | 0 (0.0) | |
| Mode of diagnosis—Index case vs. family screening (n—%) | 91 (92.9) | 28 (100.0) | 14 (77.8) | 5 (62.5) | 28 (100.0) | 4 (100.0) | 12 (100.0) | 0.001 * |
| Baseline | ||||||||
| hemodynamics | N = 82 | N = 27 | N = 16 | N = 7 | N= 21 | N = 3 | N = 8 | |
| mPAP—mmHg (median—IQr) | 53.0 (40.0–63.0) | 54.0 (39.0–70.0) | 55.0 (42.5–68.5) | 44.0 (22.0–56.0) | 61.0 (46.0–65.0) | 39.0 (27.0–61.0) | 41.0 (32.0–49.5) | 0.081 |
| RAP—mmHg (median—IQr) | 7.0 (5.0–10.0) | 7.0 (6.0–10.0) | 5.5 (2.0–11.5) | 7.0 (4.0–10.0) | 7.5 (6.0–10.0) | 11.0 (8.0–12.0) | 6.5 (5.0–12.0) | 0.814 |
| iPVR—WU * m2 (median—IQr) | 11.3 (7.0–17.4) | 11.7 (7.9–16.9) | 15.6 (7.0–20.4) | 7.2 (3.2–13.7) | 11.8 (5.4–18.3) | 10.8 (3.8–16.2) | 8.6 (7.1–14.3) | 0.500 |
| CI—L/m/m2 | 3.1 (2.0–4.0) | 3.1 (1.9–3.9) | 2.6 (2.0–3.2) | 2.3 (1.8–2.7) | 4.0 (3.1–4.8) | 6.4 (4.8–8.0) | 2.2 (1.6–3.1) | 0.007 |
| (Median—IQr) | ||||||||
| iPVR/ iSVR (median—IQr) | 0.9 (0.6–1.0) | 0.8 (0.6–0.9) | 0.8 (0.6–0.9) | N/A | 1.0 (0.6–1.3) | 0.8 (0.8–2.2) | 0.9 (0.8–1.0) | 0.178 |
| Evaluated Genes | Number of Patients | PH Groups (IPAH, HPAH, PVOD, CHD, Others) | Diagnostic Yield of Genetic Analysis * | Results in IPAH/HPAH | Results in PVOD | Results in CHD-PAH | Age at Diagnosis of Mutation Carriers (Mean) | |
|---|---|---|---|---|---|---|---|---|
| Chida et al., 2012 [18] | BMPR2, ACVRL1, SMAD8, BMPR1B | 57 | 46/10/0/0/0 | 25/54 (46.3%) | BMPR2 n = 13/57 (33%) ALK1 n = 6/57 (13%) SMAD8 n = 1/57 (1.7%) BMPR1B n = 2 (3.5%) | NA | NA | 9.2 |
| Levy et al., 2016 [19] | BMPR2, ACVRL1, TBX4, KCNK3, EIF2AK4 | 42 | 35/5/3/23 | 14/42 (33.3%) | BMPR2 n = 5/40 (12.5%) ACVRL1 n = 4/40 (10%) TBX4 n = 3/40 (7.5%) | EIF2AK4 2/3 (67.7%) | 0/23 (0%) | 8.3 |
| Zhu et al., 2018 [9] | BMPR2, TBX4, ACVRL1, BMPR1B, CAV1, EIF2AK4, ENG, KCNK3, SMAD4, SMAD9 | 155 | 130/25 | 48/155 (31.0%) | BMPR2 n = 27/155 (17.7%) TBX4 n = 12/155 (7.4%) ACVRL1 n = 3/155 (1.9%), KCNK3 n = 2/155 (1.3%), SMAD9 n = 1/155 (0.6%) | N/A | N/A | |
| Haarman et al., 2020 [20] | BMPR2, ACVRL1, CAV1, ENG, KCNK3, SMAD9, TBX4, EIF2AK4, VHL, MMACHC, CBLC, ACTA2 | 70 | 19/16/5/20/10 | 19/70 (27.1%) | BMPR2 n = 7/35 (20%) TBX4 n = 7/35 (20%) ACVRL1 n = 1/35 (2.8%), PTPN11 n = 2/35 (5.7%) KCNK3 n = 1/35 (2.8%) | EIF2AK4 2/5 (40.0%) | ACTA2 1/20 (5%) | 2.8 years TBX4 and14.0 years in BMPR2 carriers |
| PPHNet Registry. 2021 [6] | BMPR2, ACVRL1, SMAD9, CAV1, KCNK3, TBX4, GDF2 | 40 FPAH | 0/40/0/0/0 | 36/40 heritable cases (90.0%) ** | BMPR2 n = 17/40 (43%) TBX4 n = 6/40 (15%) ALK/ENG n = 5/40 (5%) GDF2 n = 2 /40 (5%) CAV1 n = 1/40 (3%) KCNK3 n = 1 (3%) | N/A | N/A | N/A |
| REHIPED registry (this study) | ABCC8, ACVRL1, BMPR1B, BMPR2, CAV1, CBLN2, CPS1, EIF2AK4, ENG, GDF2, KCNA5, KCNK3, MMACHC, NOTCH3, SARS2, SMAD1, SMAD4, SMAD5, SMAD9, TBX4, and TOPBP1 | 98 | 28/18/8/28/16 | 44/98 (44.9%) | BMPR2 n = 12/50 (24.0%) TBX4 n = 4/50 (8.0%) KCNK3 n = 2/50 (4.0%) ACVRL1 n = 1 (2.0%) BMPR1B n = 1 (2.0%) GDF2 n = 1 (2.0%) | EIF2AK4N = 8/8 (100.0%) | N = 7/28 (25.0%) - BMPR2 n = 3 - ENG n = 1 - CPS1 n = 1 - ABCC8/SMAD1 n = 1 - SOX17 n = 1 | 8.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Utrilla, A.; Gallego-Zazo, N.; Tenorio-Castaño, J.A.; Guillén, I.; Torrent-Vernetta, A.; Moya-Bonora, A.; Labrandero, C.; Rodríguez-Monte, M.E.G.-L.; Rodríguez-Ogando, A.; Rey, M.d.M.R.V.D.; et al. Clinical Implications of the Genetic Background in Pediatric Pulmonary Arterial Hypertension: Data from the Spanish REHIPED Registry. Int. J. Mol. Sci. 2022, 23, 10433. https://doi.org/10.3390/ijms231810433
Cruz-Utrilla A, Gallego-Zazo N, Tenorio-Castaño JA, Guillén I, Torrent-Vernetta A, Moya-Bonora A, Labrandero C, Rodríguez-Monte MEG-L, Rodríguez-Ogando A, Rey MdMRVD, et al. Clinical Implications of the Genetic Background in Pediatric Pulmonary Arterial Hypertension: Data from the Spanish REHIPED Registry. International Journal of Molecular Sciences. 2022; 23(18):10433. https://doi.org/10.3390/ijms231810433
Chicago/Turabian StyleCruz-Utrilla, Alejandro, Natalia Gallego-Zazo, Jair Antonio Tenorio-Castaño, Inmaculada Guillén, Alba Torrent-Vernetta, Amparo Moya-Bonora, Carlos Labrandero, María Elvira Garrido-Lestache Rodríguez-Monte, Alejandro Rodríguez-Ogando, María del Mar Rodríguez Vázquez Del Rey, and et al. 2022. "Clinical Implications of the Genetic Background in Pediatric Pulmonary Arterial Hypertension: Data from the Spanish REHIPED Registry" International Journal of Molecular Sciences 23, no. 18: 10433. https://doi.org/10.3390/ijms231810433