
Lysophosphatidic Acid Promotes Epithelial–Mesenchymal Transition in Kidney Epithelial Cells via the LPAR1/MAPK-AKT/KLF5 Signaling Pathway in Diabetic Nephropathy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
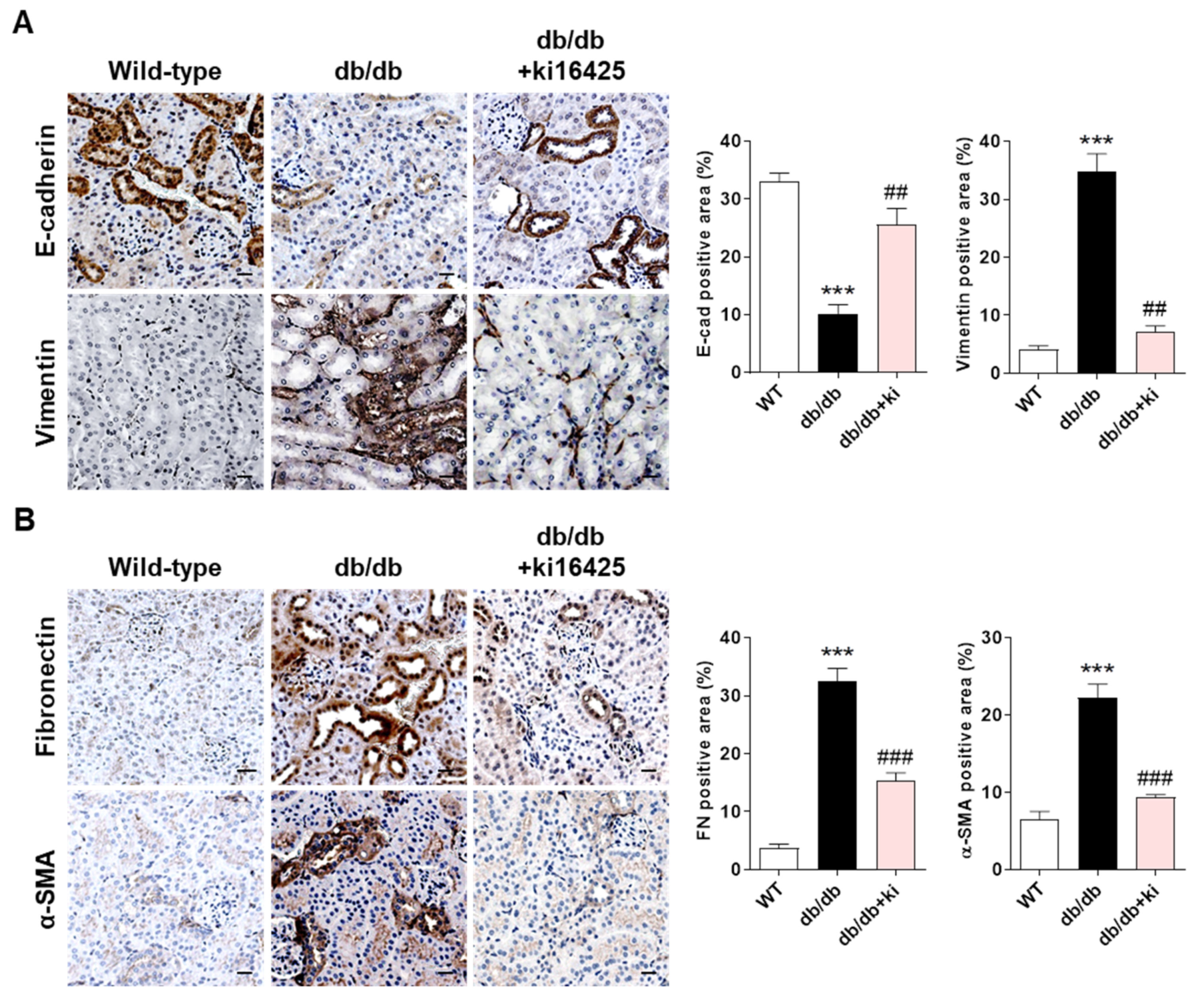
2.1. Treatment with the LPAR1/3 Antagonist, ki16425, Inhibits the Change in the Expression Levels of EMT Markers and Fibrotic Factors in Renal Tubular Epithelial Cells of db/db Mice
2.2. LPAR1/3 or LPAR1 Inhibitor Treatment Inhibits LPA−Induced Changes in the Expression Levels of EMT Markers and Fibrotic Factors
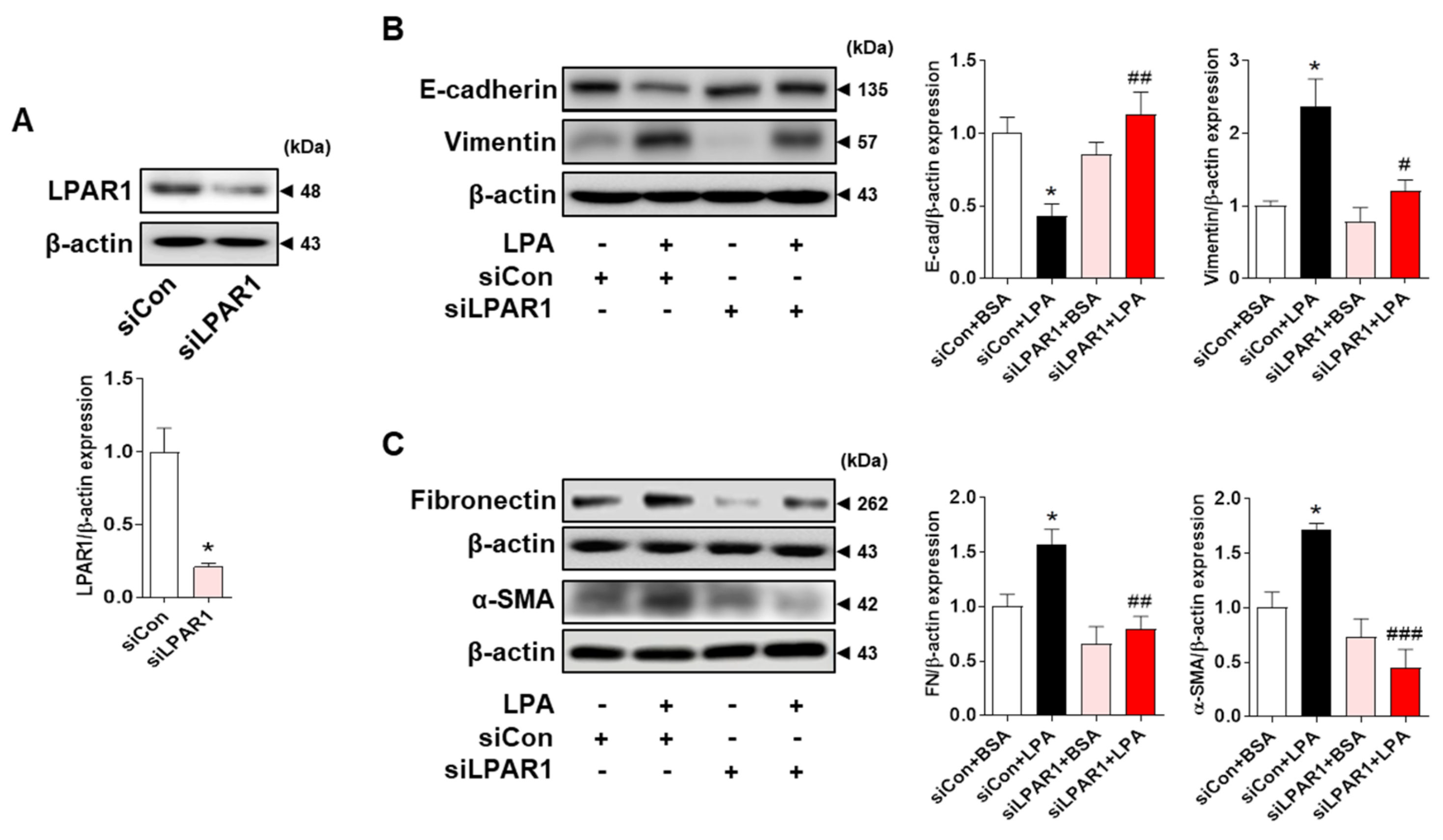
2.3. LPA Induces the Expression of EMT Markers and Fibrotic Factors through LPAR1 in HK-2 Cells
2.4. LPA/LPAR1 Signaling Induces KLF5 Expression
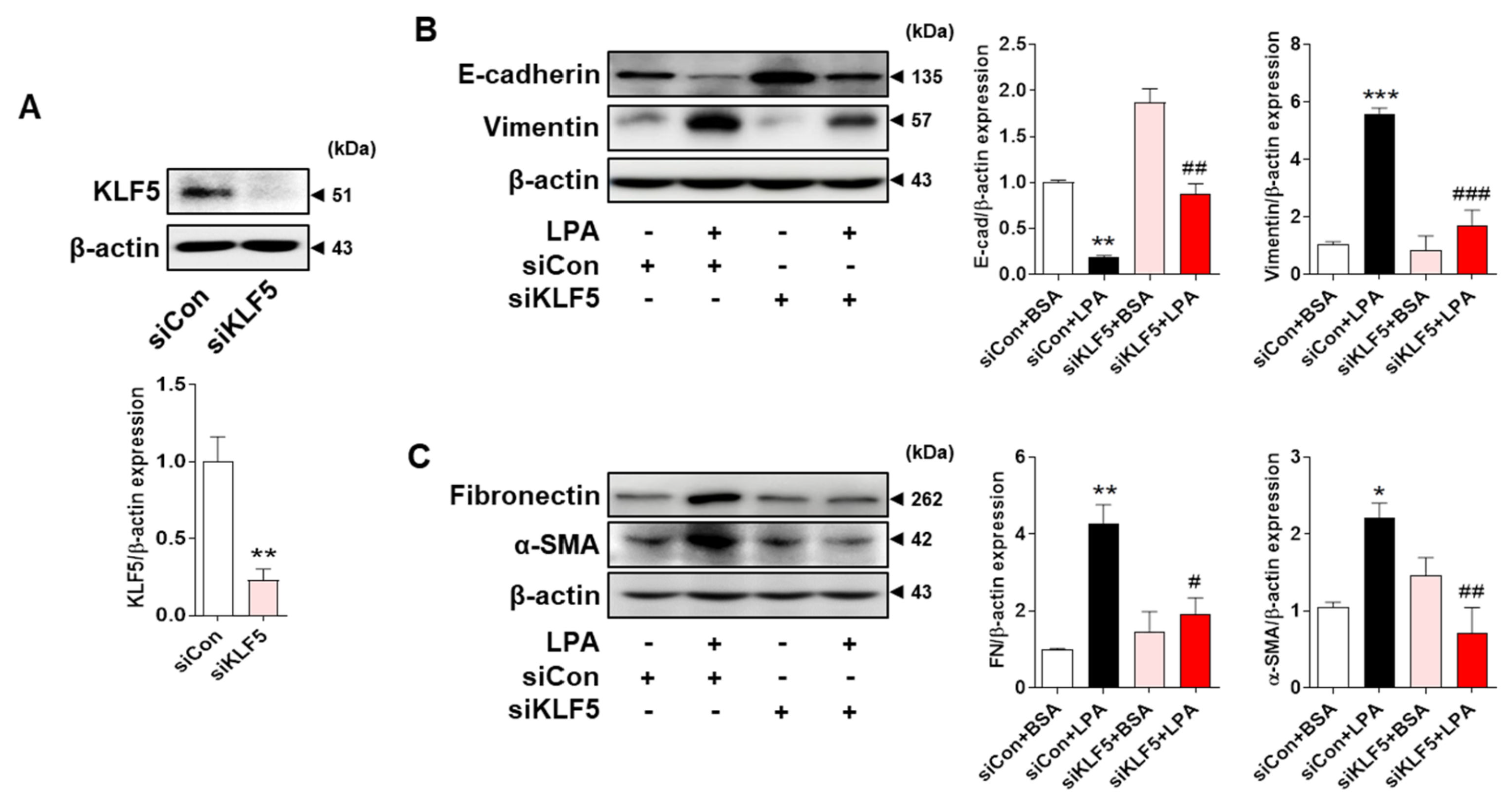
2.5. LPA−Induced Expression of EMT Markers and Fibrotic Factors Is Mediated by KLF5 in HK-2 Cells
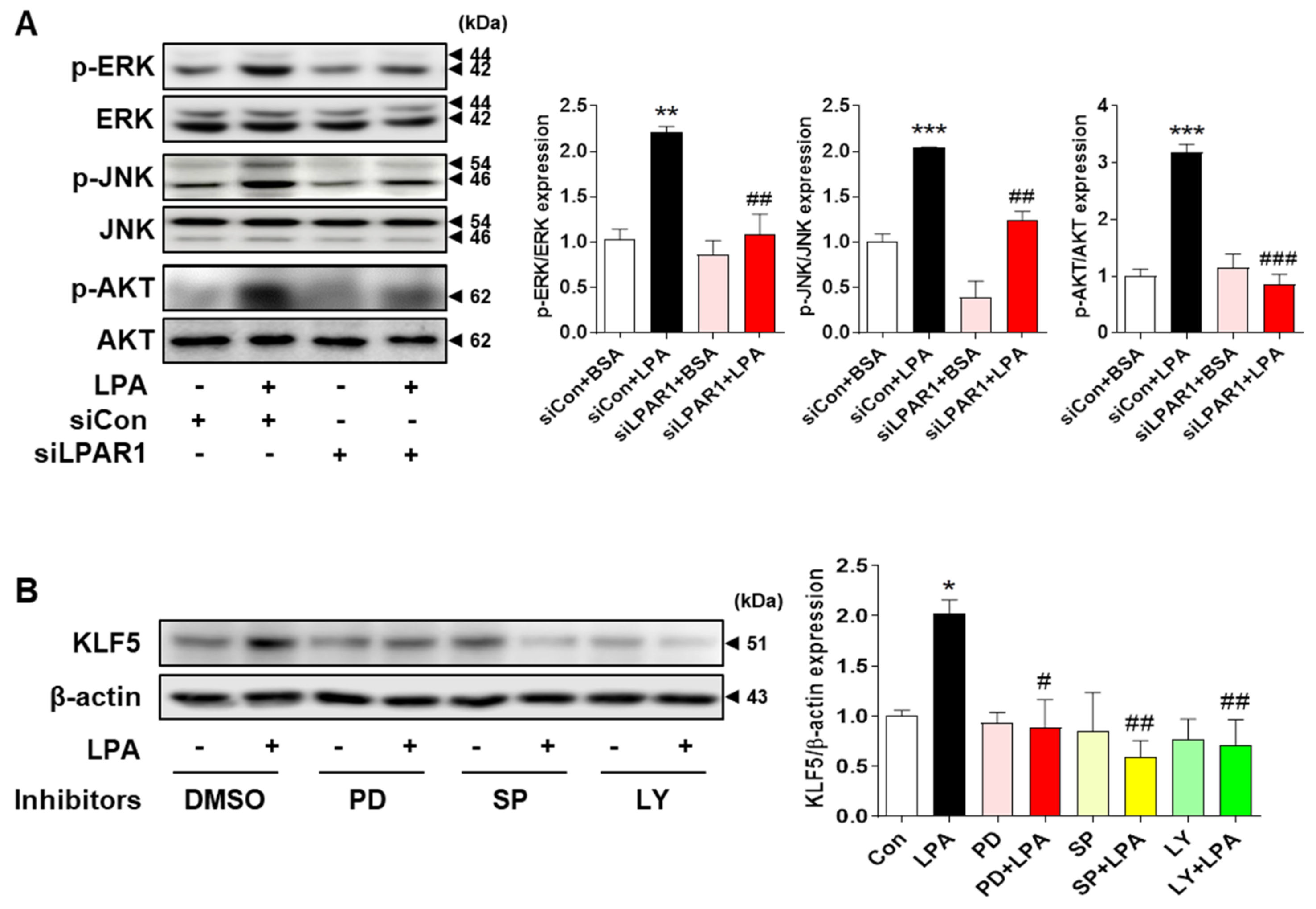
2.6. LPA−Induced KLF5 Expression Is Regulated by MAPK and AKT Signaling Pathways in HK-2 Cells
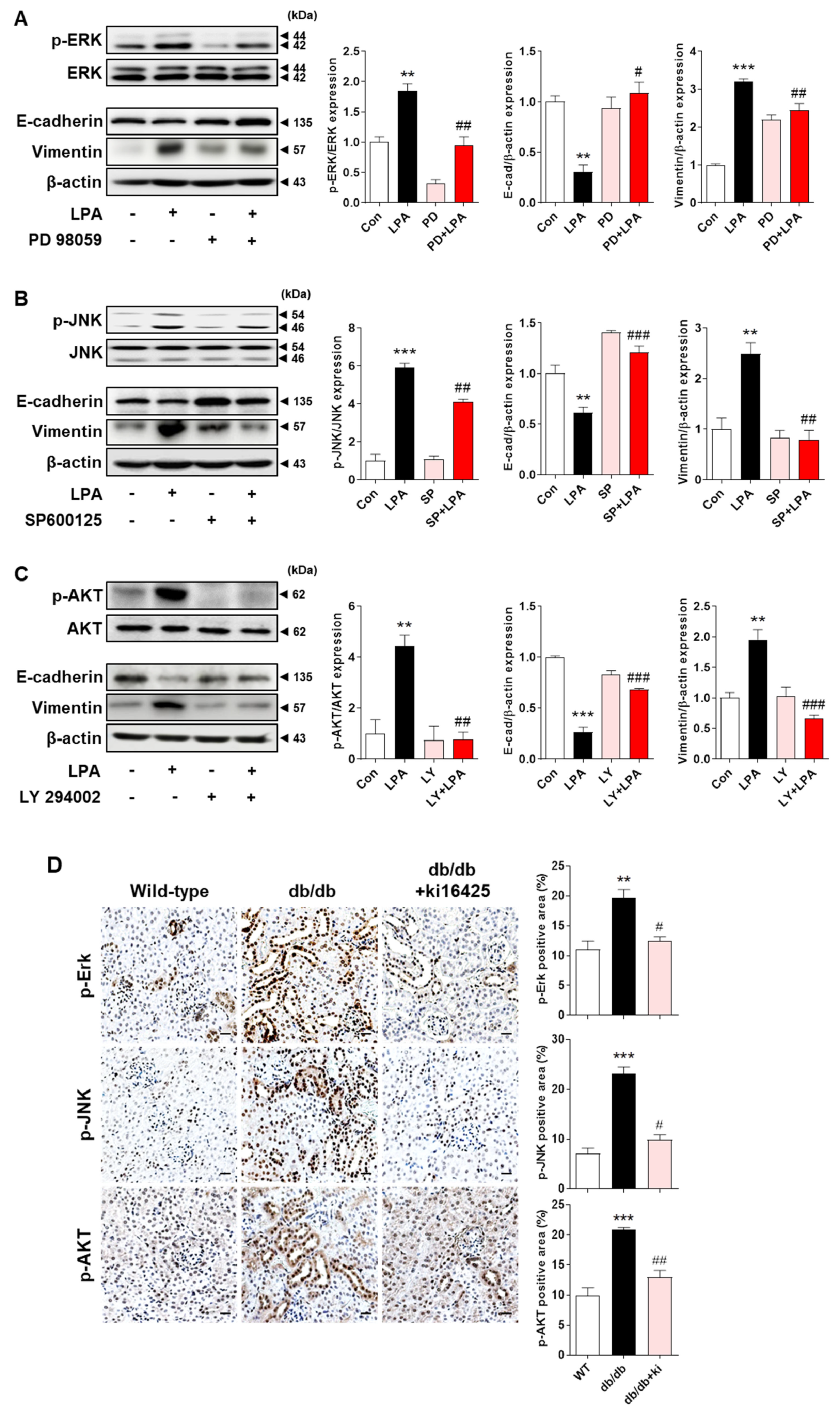
2.7. LPA−Induced EMT Marker Expression Is Mediated via the MAPK and AKT Signaling Pathways
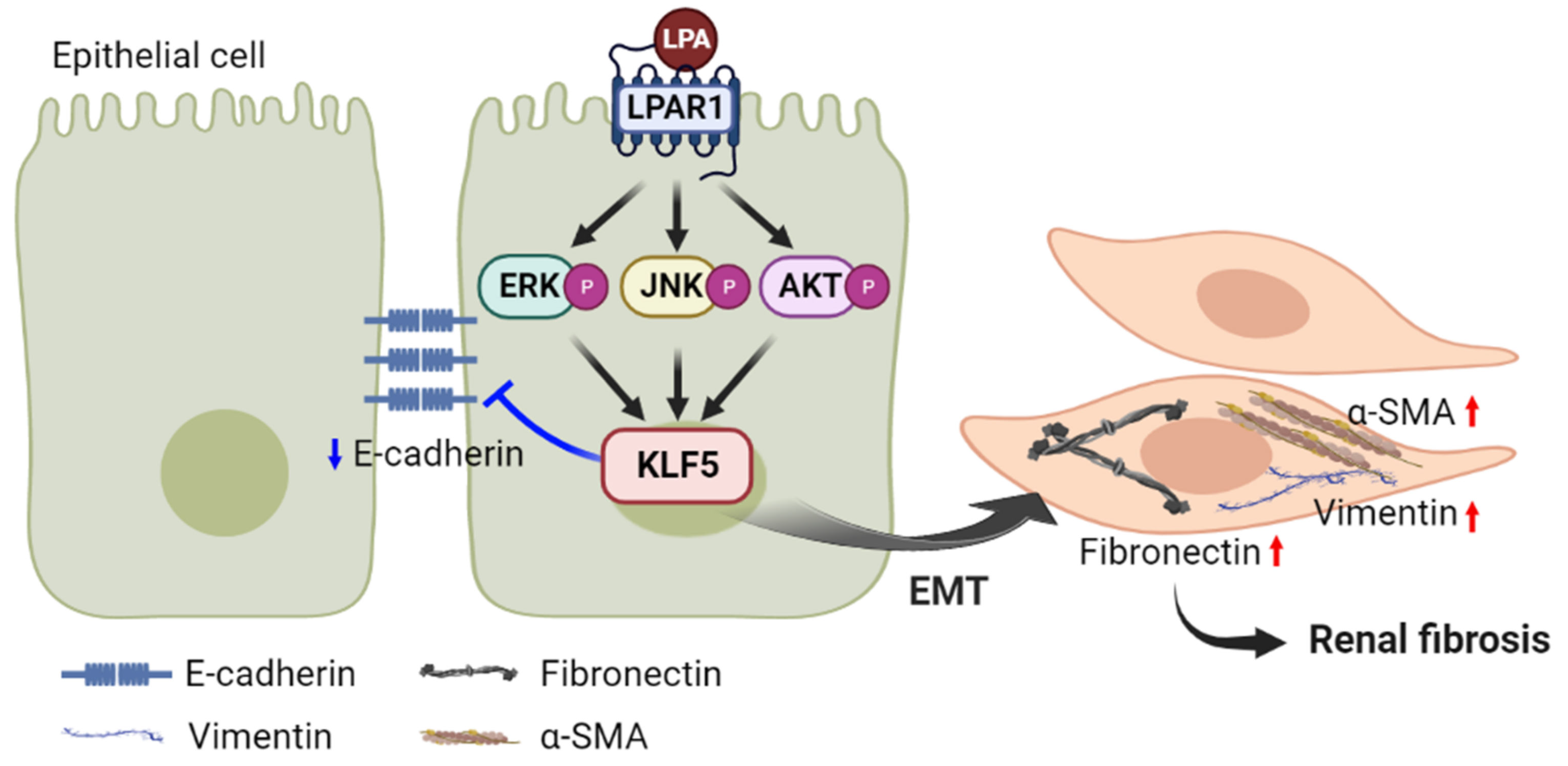
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Immunohistochemical Staining
4.3. Cell Culture and Treatment
4.4. Transfection
4.5. Western Blotting
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arora, M.K.; Singh, U.K. Molecular mechanisms in the pathogenesis of diabetic nephropathy: An update. Vasc. Pharm. 2013, 58, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Xinaris, C.; Macconi, D. Diabetic Nephropathy: Novel Molecular Mechanisms and Therapeutic Targets. Front. Pharm. 2020, 11, 586892. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Cooper, M.E. The tubulointerstitium in progressive diabetic kidney disease: More than an aftermath of glomerular injury? Kidney Int. 1999, 56, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am. J. Kidney Dis. 1992, 20, 1–17. [Google Scholar] [CrossRef]
- Tonolo, G.; Cherchi, S. Tubulointerstitial disease in diabetic nephropathy. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 107–115. [Google Scholar] [CrossRef]
- Hu, N.; Duan, J.; Li, H.; Wang, Y.; Wang, F.; Chu, J.; Sun, J.; Liu, M.; Wang, C.; Lu, C.; et al. Hydroxysafflor Yellow A Ameliorates Renal Fibrosis by Suppressing TGF-β1-Induced Epithelial-to-Mesenchymal Transition. PLoS ONE 2016, 11, e0153409. [Google Scholar] [CrossRef]
- Inoue, T.; Umezawa, A.; Takenaka, T.; Suzuki, H.; Okada, H. The contribution of epithelial-mesenchymal transition to renal fibrosis differs among kidney disease models. Kidney Int. 2015, 87, 233–238. [Google Scholar] [CrossRef]
- Yang, Y.L.; Ju, H.Z.; Liu, S.F.; Lee, T.C.; Shih, Y.W.; Chuang, L.Y.; Guh, J.Y.; Yang, Y.Y.; Liao, T.N.; Hung, T.J.; et al. BMP-2 suppresses renal interstitial fibrosis by regulating epithelial-mesenchymal transition. J. Cell Biochem. 2011, 112, 2558–2565. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; Zeisberg, M.; Kalluri, R. Partial Epithelial-to-Mesenchymal Transition and Other New Mechanisms of Kidney Fibrosis. Trends Endocrinol. Metab. 2016, 27, 681–695. [Google Scholar] [CrossRef]
- Zhao, Y.; Yin, Z.; Li, H.; Fan, J.; Yang, S.; Chen, C.; Wang, D.W. MiR-30c protects diabetic nephropathy by suppressing epithelial-to-mesenchymal transition in db/db mice. Aging Cell 2017, 16, 387–400. [Google Scholar] [CrossRef]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N. Lysophosphatidic Acid Signaling in Cancer. Cancers 2020, 12, 3791. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.Z. Lysophosphatidic acid effects on atherosclerosis and thrombosis. Clin. Lipidol. 2011, 6, 413–426. [Google Scholar] [PubMed]
- Moll, S.; Chaykovska, L.; Meier, M.; Budd, D.C.; Formentini, I.; Pomposiello, S.; Prunotto, M. Targeting the epithelial cells in fibrosis: A new concept for an old disease. Drug Discov. Today 2013, 18, 582–591. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, D.; Oh, Y.S.; Jun, H.S. Lysophosphatidic Acid Signaling in Diabetic Nephropathy. Int. J. Mol. Sci. 2019, 20, 2850. [Google Scholar] [CrossRef]
- Rancoule, C.; Attané, C.; Grès, S.; Fournel, A.; Dusaulcy, R.; Bertrand, C.; Vinel, C.; Tréguer, K.; Prentki, M.; Valet, P.; et al. Lysophosphatidic acid impairs glucose homeostasis and inhibits insulin secretion in high-fat diet obese mice. Diabetologia 2013, 56, 1394–1402. [Google Scholar] [CrossRef]
- Tomizawa, S. Analysis of data in square contingency tables with ordered categories using the conditional symmetry model and its decomposed models. Environ. Health Perspect. 1985, 63, 235–239. [Google Scholar] [CrossRef]
- Saulnier-Blache, J.S.; Feigerlova, E.; Halimi, J.M.; Gourdy, P.; Roussel, R.; Guerci, B.; Dupuy, A.; Bertrand-Michel, J.; Bascands, J.L.; Hadjadj, S.; et al. Urinary lysophopholipids are increased in diabetic patients with nephropathy. J. Diabetes Complicat. 2017, 31, 1103–1108. [Google Scholar] [CrossRef]
- Li, H.Y.; Oh, Y.S.; Choi, J.W.; Jung, J.Y.; Jun, H.S. Blocking lysophosphatidic acid receptor 1 signaling inhibits diabetic nephropathy in db/db mice. Kidney Int. 2017, 91, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Sarker, M.K.; Choi, H.; Shin, D.; Kim, D.; Jun, H.S. Lysophosphatidic acid receptor 1 inhibitor, AM095, attenuates diabetic nephropathy in mice by downregulation of TLR4/NF-kappaB signaling and NADPH oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.T.; Chen, C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol. Life Sci. 2009, 66, 2691–2706. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, L.; Zhou, W.Q.; Cai, L.; Xu, Z.G.; Rane, M.J. Roles of Kruppel-like factor 5 in kidney disease. J. Cell Mol. Med. 2021, 25, 2342–2355. [Google Scholar] [CrossRef]
- Fujiu, K.; Manabe, I.; Nagai, R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J. Clin. Invest. 2011, 121, 3425–3441. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Lv, L.L.; Wang, B.; Tang, T.T.; Feng, Y.; Cao, J.Y.; Jiang, L.Q.; Sun, Y.B.; Liu, H.; Zhang, X.L.; et al. The profibrotic effects of MK-8617 on tubulointerstitial fibrosis mediated by the KLF5 regulating pathway. FASEB J. 2019, 33, 12630–12643. [Google Scholar] [CrossRef]
- Du, L.; Qian, X.; Li, Y.; Li, X.Z.; He, L.L.; Xu, L.; Liu, Y.Q.; Li, C.C.; Ma, P.; Shu, F.L.; et al. Sirt1 inhibits renal tubular cell epithelial-mesenchymal transition through YY1 deacetylation in diabetic nephropathy. Acta Pharmacol. Sin. 2021, 42, 242–251. [Google Scholar] [CrossRef]
- Zhang, X.; Guan, T.; Yang, B.; Gu, H.F.; Chi, Z. Effects of ZnT8 on epithelial-to-mesenchymal transition and tubulointerstitial fibrosis in diabetic kidney disease. Cell Death Dis. 2020, 11, 544. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J. Fibrosis: Targeting EMT to reverse renal fibrosis. Nat. Rev. Nephrol. 2015, 11, 565. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Mao, X.; Miao, S.; He, H.; Miao, S.; Pei, R.; Yang, X.; Xiu, C.; Song, K.; Zhang, J.; Sun, J.; et al. Krüppel-like factor 5: A novel biomarker for lymph node metastasis and recurrence in supraglottic squamous cell laryngeal carcinoma. Tumour Biol. 2014, 35, 623–629. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.B.; Ghaleb, A.M.; Nandan, M.O.; Yang, V.W. The diverse functions of Krüppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays 2007, 29, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Fukuda, N.; Matsuda, H.; Endo, M.; Wang, X.; Saito, K.; Ueno, T.; Matsumoto, T.; Matsumoto, K.; Soma, M.; et al. Complement 3 activates the renal renin-angiotensin system by induction of epithelial-to-mesenchymal transition of the nephrotubulus in mice. Am. J. Physiol. Ren. Physiol. 2013, 305, F957–F967. [Google Scholar] [CrossRef] [PubMed]
- Ayasolla, K.R.; Rai, P.; Rahimipour, S.; Hussain, M.; Malhotra, A.; Singhal, P.C. Tubular cell phenotype in HIV-associated nephropathy: Role of phospholipid lysophosphatidic acid. Exp. Mol. Pathol. 2015, 99, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Lan, R.; Liu, Y.; Chen, W.; Wu, M.; Saikumar, P.; Weinberg, J.M.; Venkatachalam, M.A. Proximal tubule LPA1 and LPA2 receptors use divergent signaling pathways to additively increase profibrotic cytokine secretion. Am. J. Physiol. Ren. Physiol. 2021, 320, F359–F374. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Sun, L.; Xiao, L.; Han, Y.; Fu, X.; Xiong, X.; Xu, X.; Liu, Y.; Yang, S.; Liu, F.; et al. Insights into the Mechanisms Involved in the Expression and Regulation of Extracellular Matrix Proteins in Diabetic Nephropathy. Curr. Med. Chem. 2015, 22, 2858–2870. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [PubMed]
- Morcos, M.; Sayed, A.A.; Bierhaus, A.; Yard, B.; Waldherr, R.; Merz, W.; Kloeting, I.; Schleicher, E.; Mentz, S.; Abd el Baki, R.F.; et al. Activation of tubular epithelial cells in diabetic nephropathy. Diabetes 2002, 51, 3532–3544. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, T.; Suzuki, K.; Shiota, T.; Kondo, T.; Okita, M. The significance of plasma lysophospholipids in patients with renal failure on hemodialysis. J. Nutr. Sci. Vitaminol. 1998, 44, 809–818. [Google Scholar] [CrossRef]
- Shimizu, M.; Furuichi, K.; Toyama, T.; Yamahana, J.; Ohkawa, R.; Igarashi, K.; Aoki, J.; Kaneko, S.; Yatomi, Y.; Wada, T. Serum Autotaxin Levels Are Associated with Proteinuria and Kidney Lesions in Japanese Type 2 Diabetic Patients with Biopsy-proven Diabetic Nephropathy. Intern. Med. 2016, 55, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Z.; Wang, X.; Yang, H.; Fogo, A.B.; Murphy, B.J.; Kaltenbach, R.; Cheng, P.; Zinker, B.; Harris, R.C. Lysophosphatidic Acid Receptor Antagonism Protects against Diabetic Nephropathy in a Type 2 Diabetic Model. J. Am. Soc. Nephrol. 2017, 28, 3300–3311. [Google Scholar] [CrossRef]
- Kim, D.; Li, H.Y.; Lee, J.H.; Oh, Y.S.; Jun, H.S. Lysophosphatidic acid increases mesangial cell proliferation in models of diabetic nephropathy via Rac1/MAPK/KLF5 signaling. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nam, G.Y.; Seo, E.; Jun, H.S. Inhibition of ChREBP ubiquitination via the ROS/Akt-dependent downregulation of Smurf2 contributes to lysophosphatidic acid-induced fibrosis in renal mesangial cells. J. Biomed. Sci. 2022, 29, 31. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Ray, U.; Roy, S.S.; Chowdhury, S.R. Lysophosphatidic Acid Promotes Epithelial to Mesenchymal Transition in Ovarian Cancer Cells by Repressing SIRT1. Cell Physiol. Biochem. 2017, 41, 795–805. [Google Scholar] [CrossRef]
- Ohta, H.; Sato, K.; Murata, N.; Damirin, A.; Malchinkhuu, E.; Kon, J.; Kimura, T.; Tobo, M.; Yamazaki, Y.; Watanabe, T.; et al. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol. Pharmacol. 2003, 64, 994–1005. [Google Scholar] [CrossRef]
- Sakai, N.; Chun, J.; Duffield, J.S.; Lagares, D.; Wada, T.; Luster, A.D.; Tager, A.M. Lysophosphatidic acid signaling through its receptor initiates profibrotic epithelial cell fibroblast communication mediated by epithelial cell derived connective tissue growth factor. Kidney Int. 2017, 91, 628–641. [Google Scholar] [CrossRef]
- Chen, W.C.; Lin, H.H.; Tang, M.J. Matrix-Stiffness-Regulated Inverse Expression of Krüppel-Like Factor 5 and Krüppel-Like Factor 4 in the Pathogenesis of Renal Fibrosis. Am. J. Pathol. 2015, 185, 2468–2481. [Google Scholar] [CrossRef]
- Zhang, H.; Bialkowska, A.; Rusovici, R.; Chanchevalap, S.; Shim, H.; Katz, J.P.; Yang, V.W.; Yun, C.C. Lysophosphatidic acid facilitates proliferation of colon cancer cells via induction of Krüppel-like factor 5. J. Biol. Chem. 2007, 282, 15541–15549. [Google Scholar] [CrossRef]
- Liu, R.; Shi, P.; Zhou, Z.; Zhang, H.; Li, W.; Zhang, H.; Chen, C. Krüpple-like factor 5 is essential for mammary gland development and tumorigenesis. J. Pathol. 2018, 246, 497–507. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, Q.; Liu, F.; Ma, X.; Wu, L.; Guo, F.; Zhao, S.; Huang, F.; Qin, G. KLF5 promotes the tumorigenesis and metastatic potential of thyroid cancer cells through the NF-κB signaling pathway. Oncol. Rep. 2018, 40, 2608–2618. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xu, J.; Zhang, C.; Wen, Y. Aberrant histone deacetylase 1 expression upregulates vimentin expression via an NF-κB-dependent pathway in hepatocellular carcinoma. Oncol. Lett. 2019, 18, 339–347. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Guan, X.; Wang, S.; Xiao, T.; Yang, K.; Xu, X.; Wang, J.; Zhao, J. Resveratrol prevents high glucose-induced epithelial-mesenchymal transition in renal tubular epithelial cells by inhibiting NADPH oxidase/ROS/ERK pathway. Mol. Cell Endocrinol. 2015, 402, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.W.; Tsai, J.P.; Lin, S.H.; Lee, C.H.; Hsieh, Y.H.; Chang, H.R. Pentraxin 3 Activates JNK Signaling and Regulates the Epithelial-To-Mesenchymal Transition in Renal Fibrosis. Cell Physiol. Biochem. 2016, 40, 1029–1038. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, W.W.; Zhang, M.Z.; Ma, Z.X.; Qiu, X.R.; Shen, M.; Yin, X.X. ROS induces epithelial-mesenchymal transition via the TGF-β1/PI3K/Akt/mTOR pathway in diabetic nephropathy. Exp. Ther. Med. 2019, 17, 835–846. [Google Scholar] [CrossRef]
- Zhang, L.; Shen, Z.Y.; Wang, K.; Li, W.; Shi, J.M.; Osoro, E.K.; Ullah, N.; Zhou, Y.; Ji, S.R. C-reactive protein exacerbates epithelial-mesenchymal transition through Wnt/β-catenin and ERK signaling in streptozocin-induced diabetic nephropathy. FASEB J. 2019, 33, 6551–6563. [Google Scholar] [CrossRef] [PubMed]
- Annaldas, S.; Saifi, M.A.; Khurana, A.; Godugu, C. Nimbolide ameliorates unilateral ureteral obstruction-induced renal fibrosis by inhibition of TGF-beta and EMT/Slug signalling. Mol. Immunol. 2019, 112, 247–255. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, D.; Yoon, H.; Choi, C.S.; Oh, Y.S.; Jun, H.S. Prevention of Oxidative Stress-Induced Pancreatic Beta Cell Damage by Broussonetia Kazinoki Siebold Fruit Extract Via the ERK-Nox4 Pathway. Antioxidants 2020, 9, 406. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, G.-H.; Cheon, J.; Kim, D.; Jun, H.-S. Lysophosphatidic Acid Promotes Epithelial–Mesenchymal Transition in Kidney Epithelial Cells via the LPAR1/MAPK-AKT/KLF5 Signaling Pathway in Diabetic Nephropathy. Int. J. Mol. Sci. 2022, 23, 10497. https://doi.org/10.3390/ijms231810497
Lee G-H, Cheon J, Kim D, Jun H-S. Lysophosphatidic Acid Promotes Epithelial–Mesenchymal Transition in Kidney Epithelial Cells via the LPAR1/MAPK-AKT/KLF5 Signaling Pathway in Diabetic Nephropathy. International Journal of Molecular Sciences. 2022; 23(18):10497. https://doi.org/10.3390/ijms231810497
Chicago/Turabian StyleLee, Geon-Ho, Jayeon Cheon, Donghee Kim, and Hee-Sook Jun. 2022. "Lysophosphatidic Acid Promotes Epithelial–Mesenchymal Transition in Kidney Epithelial Cells via the LPAR1/MAPK-AKT/KLF5 Signaling Pathway in Diabetic Nephropathy" International Journal of Molecular Sciences 23, no. 18: 10497. https://doi.org/10.3390/ijms231810497