
Cardio-Oncology: Mechanisms, Drug Combinations, and Reverse Cardio-Oncology

Abstract
:1. Introduction
2. Cardiovascular Toxicity of Antitumor Therapy and Mechanisms
2.1. ANTs
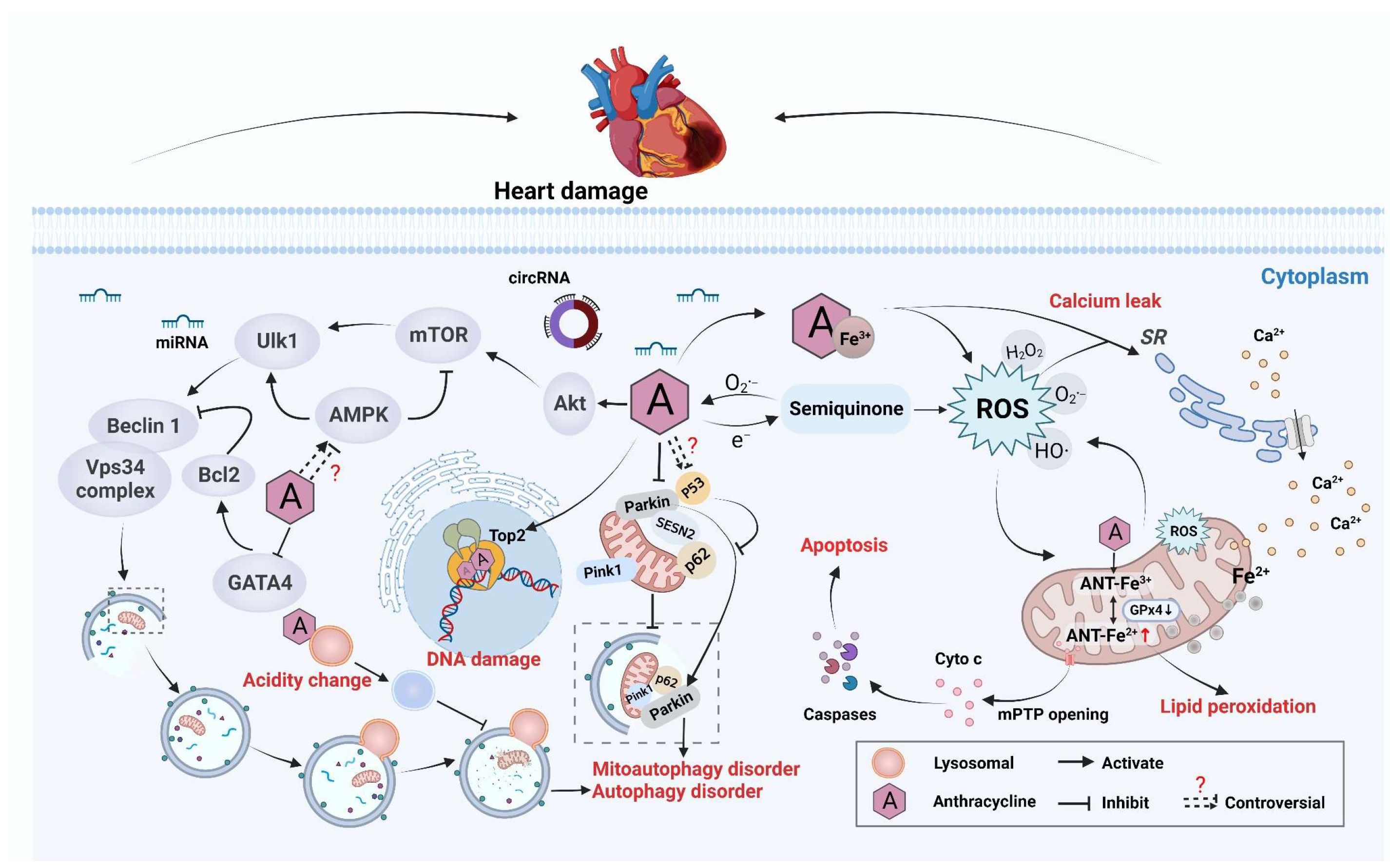
2.1.1. Antitumor Mechanism and Cardiotoxicity
2.1.2. Imbalance in Redox Reactions
2.1.3. Disruption of Mitochondrial Function
2.1.4. Autophagy Abnormality
2.1.5. Targeting Top2
2.1.6. Dysregulation of Ca2+ Homeostasis
2.1.7. Changes in Epigenetic Modification
2.2. Non-ANT-Based Chemotherapy
2.2.1. Platinum
2.2.2. Alkylating Agents
2.2.3. Antimetabolites
2.3. Radiotherapy
2.4. Targeted Therapy
2.5. Immunotherapy
3. Combination of Drugs to Achieve Cardiac Detoxification
3.1. Common Clinical Drugs
3.2. Natural Products/TCM Formulations
3.2.1. Phenolic Acids
3.2.2. Polysaccharides
3.2.3. Flavonoids
3.2.4. Saponins
3.2.5. Alkaloids
3.2.6. TCM Formulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Compound/Herb Name | Source | Chemotherapy Drug | Experimental Model | Mechanism of Action | Ref. |
|---|---|---|---|---|---|---|
| Phenol acid | Salvianolic acid | Salvia miltiorrhiza | DOX | Male KM mice | Increased absorbance of oxygen radicals, decreased levels of creatine kinase and malondialdehyde. | [255] |
| Curcumin | Turmeric | DOX | Kunming mice, primary neonatal rat cardiomyocytes | Regulated the 14-3-3γ/BAD/Bcl-2 pathway and mPTP. | [37] | |
| Resveratrol | Grapes and peanuts | DOX | Male Sprague-Dawley rats, rat embryonic cardiomyoblast-derived cells, H9c2 cells, male FBN rats | Regulated VEGF-B/Akt/GSK-3β signaling pathway; regulated mTOR, AMPK, and SIRT1 pathways. | [258] | |
| Polysaccharides | Ganoderma lucidum polysaccharides | Ganoderma lucidum | DOX | H9c2, MCF-7, HepG2 cells | Regulated polyubiquitin modification, inhibited the NF-κB signaling pathway. | [262] |
| Astragalus polysaccharide | Astragalus membranaceus | DOX | Neonatal rat ventricular myocytes, male C57BL/6 mice | Activated the PI3K/Akt pathway, inhibited the p38MAPK pathway, reduced levels of caspase 3 and caspase 9. | [263] | |
| Ganoderma atrum polysaccharide (PSG-1) | Ganoderma atrum | DOX | Kunming mice, primary myocardial cells | Improved antioxidant capacity, reduced cytochrome-c release, increased mitochondrial membrane potential, inhibited mPTP opening. | [264] | |
| Flavonoids | Chrysin | Mushrooms, bee propolis, et al. | DOX | Male Sprague-Dawley rats | Regulated oxidative stress, as well as VEGF/PI3K/Akt, p53, p38, JNK, and NF-κB signaling pathways. | [271] |
| Cardamonin | Alpinia katsumadai, Ginkgo biloba, and Carya cathayensis Sarg | DOX | Male C57BL/6 J mice, rat-myocardium-derived cardiomyoblast H9C2 and mouse cardiomyocyte HL-1 | Regulated Nrf2, NF-κB, and caspase-3 pathways. | [272] | |
| Calycosin | Radix astragali | DOX | Rat cardiomyocyte line H9c2, male Kunming mice | Inhibited Bax; increased Bcl-2; activated PI3K/Akt pathway; reduced NLRP3, TXNIP, IL-1β, and caspase-1. | [273] | |
| Saponin | Ginsenoside Rg3 | Ginseng | DOX | Adult male Sprague-Dawley rats, cardiac microvascular endothelial cells from neonatal rats | Increased EF and FS, improved LV outflow, activated Nrf2-ARE and Akt pathways. | [280] |
| Astragaloside IV | Astragalus membranaceus | DOX | C57BL/6 mice, NRCMs | Alleviated myocardial fibrosis and left-ventricular systolic dysfunction, inhibited NOX2 and NOX4. | [281] | |
| Saikosaponin D | Radix bupleuri | DOX | Male mice | Inhibited excessive oxidative stress via the p38MAPK signaling pathway. | [282] | |
| Alkaloids | Neferine | Nelumbo nucifera Gaertn | DOX | Rat cardiomyoblasts, H9c2 | Activated IGF-1R, PI3K/Akt/mTOR pathway, increased expression of SOD-1 and HO-1. | [294] |
| Berberine | Rhizoma coptidis | DOX | Sprague-Dawley rats, rat cardiac H9c2 cell line | Improved ST, QRS complexes, and QT; regulated SIRT1-p66shc pathway. | [295] | |
| Cyclovirobuxine D | Buxus microphylla | DOX | C57BL mice injected (i.p.) with DOX | Ameliorated cardiac oxidative damage, prevented impairment of mitochondrial biogenesis. | [304] | |
| Single herb | - | Platycodon grandiflorum | DOX | Clinical trial | Ongoing | [299] |
| - | Hypericum hircinum | DOX | Wistar rats | Increased antioxidant defenses, reversed ECG changes, and prevented reductions in heart weight. | [300] | |
| - | Lycium barbarum | DOX | Male Sprague-Dawley rats | Prevented loss of myofibrils and improved heart function, normalized antioxidative activity. | [301] | |
| TCM formulation | Modified ZGCT | Radix Glycyrrhirae preparata, Radix Ginseng, Ramulus Cinnamomi, Colla Corii Asini, Radix Rehmanniae, Radix Ophiopogonis, et al. | DOX | 18-year-old male adolescent with refractory acute lymphoblastic leukemia | Chest radiograph showed great improvements in pulmonary edema and cardiomegaly. | [302] |
| Antarth | Boswellia serrata, Commiphora mukul, Withania somnifera, Smilax china, Tribulus terrestris, Curcuma longa, et al. | DOX | 12-week-old male Swiss albino mice | Inhibited DOX-induced decline in antioxidant status. | [303] | |
| Tongmai Yangxin pills | Radix Rehmanniae, Radix Glycyrrhizae, Ramulus Cinnamomi, Fructus Schisandrae, Radix Ophiopogonis, Radix Polygoni multiflori preparata, et al. | Cisplatin | Male Wistar rats | Improved anti-oxidative stress and reduced apoptosis through regulating Nrf2/HO-1 and p38MAPK pathways. | [298] |
4. Advances in Reverse Cardiac Oncology
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-FU | 5-Fluorouracil | LDH | Lactate dehydrogenase |
| ACEI | Angiotensin-converting enzyme inhibitor | LPs | Lipid peroxides |
| AMPK | Adenosine 5‘-monophosphate -activated protein kinase | LSD1 | Lysine-specific demethylase 1 |
| ANT | Anthracycline | MAPK | p38-mitogen-activated protein kinase |
| ARE | Anti-oxidant reactive elements | MDA | Malondialdehyde |
| Bak | BCL2 antagonist/killer | MDM2 | Murine double minute2 |
| Bax | BCL2 associated X | MMTV | Mouse mammary tumor virus |
| Bcl-2 | B-cell lymphoma-2 | mPTP | Mitochondrial permeability transition pore |
| Ca2+ | Calcium ions | mTOR | Mammalian target of rapamycin |
| [Ca2+]i | Intracellular Ca2+ concentration | MTX | Methotrexate |
| CaMKII | Calcium/calmodulin-dependent protein kinase II | NADH | Nicotinamide adenine dinucleotide |
| CAR T-cell | Chimeric antigen receptor T cell | ncRNA | Non-coding RNA |
| CAT | Catalase | NF-κB | Nuclear factor-kappa B |
| CD68 | Cluster of differentiation 68 | NOX2 | Nicotinamide adenine dinucleotide phosphate oxidase 2 |
| CircITCH | CircRNA generated from several exons of itchy E3 ubiquitin protein ligase | NRF-1 | Nuclear respiratory factor-1 |
| CK | Creatine kinase | Nrf2 | NF-E2-related factor 2 |
| CK-MB | Creatine kinase-MB | NT-proBNP | Natriuretic peptide |
| CoQ10 | Coenzyme Q10 | PD-1 | Programmed death protein-1 |
| CP | Cyclophosphamide | PD-L1 | Programmed death-ligand 1 |
| CTLA-4 | Cytotoxic T lymphocyte antigen-4 | Pgc1 | Peroxisome proliferator activated receptor gamma coactivator 1 |
| Cul3 | Cullin 3 | PI3Kγ/Akt | Phosphoinositide 3-kinase gamma/protein kinase B |
| Cyto c | Cytochrome c | PINK1 | PTEN-induced kinase 1 |
| DDR | DNA damage response | PTEN | Phosphatase and tensin homolog |
| DOX | Doxorubicin | PyMT | Polyoma middle T-antigen |
| eNOS | Endothelial nitric oxide synthase | Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| ER | Endoplasmic reticulum | Raf/MEK | MAP kinse-ERK kinase |
| ERK | Extracellular signal-regulated kinase | RE-LY | Randomized Evaluation Long-Term Anticoagulant Therapy Study |
| FBAL | α-Fluoro-β-alanine | ROS | Reactive oxygen species |
| FoxO3a | Forkhead box O3a | RSV | Resveratrol |
| GOT | Glutamate oxaloacetate transaminase | RyR2 | Ryanodin receptor-2 |
| GPT | Glutamic-pyruvic transaminase | S6K1 | Ribosomal protein S6 kinase 1 |
| GPx4 | Glutathione peroxidase 4 | SESN2 | Sestrin2 |
| GR | Glutathione reductase | SMAD2/3 | Suppressor of mothers against decapentaplegic 2/3 |
| GSH | Glutathione | SOD | Superoxide dismutase |
| GSK-3β | Glycogen synthase kinase-3 beta | SR | Sarcoplasmic reticulum |
| GSTP1 | Glutathione S-transferase pi 1 | ST18 | Suppression of tumorigenicity 18 |
| HDAC2 | Histone deacetylase 2 | TAC | Transverse aortic constriction |
| HER2 | Human epidermal growth factor receptor 2 | TCA cycle | Tricarboxylic acid cycle |
| HF | Heart failure | TCM | Traditional Chinese medicine |
| HO-1 | Heme oxygenase-1 | TGF-β | Transforming growth factor-beta |
| hs-CRP | High-sensitivity C-reactive protein | TKIs | Tyrosine kinase inhibitors |
| ICIs | Immune-checkpoint inhibitors | TLR9 | Toll-like receptor-9 |
| IGF-1R | Insulin-like growth factor 1 receptor | TMYXP | Tongmai Yangxin pill |
| IL-6 | Interleukin-6 | TNF-α | Tumor necrosis factor alpha |
| ING5 | The inhibitor of growth 5 | Top2 | Topoisomerase type 2 |
| JNK | Janus kinase | TP53 | Tumor protein p53 |
| K48 | Lysine 48 | Ulk-1 | Unc-51-like autophagy activating kinase 1 |
| KDM3A | Lysine-specific histone demethylase 3A | VEGF-A | vascular endothelial growth factor A |
| Keap1 | Kelch-like ECH-associated protein 1 | VigiBase | Global Individual Case Safety Report database of the World Health Organization |
| LC3 | Light chain 3 | ZGCT | Zhigancaotang |
References
- Stoltzfus, K.C.; Zhang, Y.; Sturgeon, K.; Sinoway, L.I.; Trifiletti, D.M.; Chinchilli, V.M.; Zaorsky, N.G. Fatal heart disease among cancer patients. Nat. Commun. 2020, 11, 2011. [Google Scholar] [CrossRef] [PubMed]
- Okwuosa, T.M.; Anzevino, S.; Rao, R. Cardiovascular disease in cancer survivors. Postgrad. Med. J. 2017, 93, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.B.; Ramin, C.; Barac, A.; de Gonzalez, A.B.; Veiga, L. Trends in heart disease mortality among breast cancer survivors in the US, 1975–2017. Breast Cancer Res. Treat. 2022, 192, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Dent, S.; Liu, P.; Brezden-Masley, C.; Lenihan, D. Cancer and Cardiovascular Disease: The Complex Labyrinth. J. Oncol. 2015, 2015, 516450. [Google Scholar] [CrossRef]
- Coufal, N.; Farnaes, L. Anthracyclines and Anthracenediones. In Cancer Management in Man: Chemotherapy, Biological Therapy, Hyperthermia and Supporting Measures; Minev, B.R., Ed.; Springer: Dordrecht, The Netherlands, 2011; pp. 87–102. [Google Scholar] [CrossRef]
- Mott, M.G. Anthracycline cardiotoxicity and its prevention. Ann. N. Y. Acad. Sci. 1997, 824, 221–228. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L., Jr.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-induced congestive heart failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Rozencweig, M.; Layard, M.; Slavik, M.; Muggia, F.M. Daunomycin-induced cardiotoxicity in children and adults. A review of 110 cases. Am. J. Med. 1977, 62, 200–208. [Google Scholar] [CrossRef]
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol. 2015, 12, 547–558. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Ma, Q.; Zhang, S.L.; Liu, H.Y.; Zhao, B.Q.; Du, B.; Wang, W.; Lin, P.; Zhang, Z.; Zhong, Y.X.; et al. Digoxin Enhances the Anticancer Effect on Non-Small Cell Lung Cancer While Reducing the Cardiotoxicity of Adriamycin. Front. Pharmacol. 2020, 11, 186. [Google Scholar] [CrossRef]
- Aboumsallem, J.P.; Moslehi, J.; de Boer, R.A. Reverse Cardio-Oncology: Cancer Development in Patients With Cardiovascular Disease. J. Am. Heart Assoc. 2020, 9, e013754. [Google Scholar] [CrossRef] [PubMed]
- Seretis, A.; Cividini, S.; Markozannes, G.; Tseretopoulou, X.; Lopez, D.S.; Ntzani, E.E.; Tsilidis, K.K. Association between blood pressure and risk of cancer development: A systematic review and meta-analysis of observational studies. Sci. Rep. 2019, 9, 8565. [Google Scholar] [CrossRef] [PubMed]
- MacLellan, D.G.; Richardson, A.; Stoodley, M.A. Venous thromboembolism and cancer. ANZ J. Surg. 2012, 82, 294–298. [Google Scholar] [CrossRef]
- Conen, D.; Wong, J.A.; Sandhu, R.K.; Cook, N.R.; Lee, I.M.; Buring, J.E.; Albert, C.M. Risk of Malignant Cancer Among Women With New-Onset Atrial Fibrillation. JAMA Cardiol. 2016, 1, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Meijers, W.C.; Maglione, M.; Bakker, S.J.L.; Oberhuber, R.; Kieneker, L.M.; de Jong, S.; Haubner, B.J.; Nagengast, W.B.; Lyon, A.R.; van der Vegt, B.; et al. Heart Failure Stimulates Tumor Growth by Circulating Factors. Circulation 2018, 138, 678–691. [Google Scholar] [CrossRef]
- Totzeck, M.; Schuler, M.; Stuschke, M.; Heusch, G.; Rassaf, T. Cardio-oncology-strategies for management of cancer-therapy related cardiovascular disease. Int. J. Cardiol. 2019, 280, 163–175. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Y.; Liu, W. Precision cardio-oncology: Understanding the cardiotoxicity of cancer therapy. NPJ Precis. Oncol. 2017, 1, 31. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Yu, X.Y.; Wang, Z.; Wu, P.; Huang, J. Anthracyclines potentiate anti-tumor immunity: A new opportunity for chemoimmunotherapy. Cancer Lett. 2015, 369, 331–335. [Google Scholar] [CrossRef]
- Yeh, E.T.; Chang, H.M. Oncocardiology-Past, Present, and Future: A Review. JAMA Cardiol. 2016, 1, 1066–1072. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, P.; Kasi, A. Anthracyclines; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Roca-Alonso, L.; Pellegrino, L.; Castellano, L.; Stebbing, J. Breast Cancer Treatment and Adverse Cardiac Events: What Are the Molecular Mechanisms? Cardiology 2012, 122, 253–259. [Google Scholar] [CrossRef]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Sala, V.; Della Sala, A.; Hirsch, E.; Ghigo, A. Signaling Pathways Underlying Anthracycline Cardiotoxicity. Antioxid. Redox Signal. 2020, 32, 1098–1114. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.J.; Jorden, M. Pathology of radiation and anthracycline cardiotoxicity. Pediatr. Blood Cancer 2005, 44, 630–637. [Google Scholar] [CrossRef]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Kong, C.Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.P.; Teng, T.; Yan, L.; Tang, Q.Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef]
- Menna, P.; Salvatorelli, E. Primary Prevention Strategies for Anthracycline Cardiotoxicity: A Brief Overview. Chemotherapy 2017, 62, 159–168. [Google Scholar] [CrossRef]
- Fang, X.X.; Wang, H.; Han, D.; Xie, E.J.; Yang, X.; Wei, J.Y.; Gu, S.S.; Gao, F.; Zhu, N.L.; Yin, X.J.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Doroshow, J.H.; Davies, K.J. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J. Biol. Chem. 1986, 261, 3068–3074. [Google Scholar] [CrossRef]
- Wouters, K.A.; Kremer, L.C.M.; Miller, T.L.; Herman, E.H.; Lipshultz, S.E. Protecting against anthracycline-induced myocardial damage: A review of the most promising strategies. Br. J. Haematol. 2005, 131, 561–578. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.B.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, E.; Huart, P.; Praet, M.; Brasseur, R.; Ruysschaert, J.M. Structure of the adriamycin-cardiolipin complex: Role in mitochondrial toxicity. Biophys. Chem. 1990, 35, 247–257. [Google Scholar] [CrossRef]
- Murabito, A.; Hirsch, E.; Ghigo, A. Mechanisms of Anthracycline-Induced Cardiotoxicity: Is Mitochondrial Dysfunction the Answer? Front. Cardiovasc. Med. 2020, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Baccouch, R.; Modine, T.; Preau, S.; Zannis, K.; Marchetti, P.; Lancel, S.; Neviere, R. Stabilization of mitochondrial membrane potential prevents doxorubicin-induced cardiotoxicity in isolated rat heart. Toxicol. Appl. Pharmacol. 2010, 244, 300–307. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Luo, Y.; Qiao, Y.; Zhang, Z.; Yin, D.; Yao, J.; You, J.; He, M. Curcumin attenuates doxorubicin-induced cardiotoxicity via suppressing oxidative stress and preventing mitochondrial dysfunction mediated by 14-3-3gamma. Food Funct. 2018, 9, 4404–4418. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.N.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019, 9, 2002. [Google Scholar] [CrossRef]
- Wang, P.; Wang, L.; Lu, J.; Hu, Y.; Wang, Q.; Li, Z.; Cai, S.; Liang, L.; Guo, K.; Xie, J.; et al. SESN2 protects against doxorubicin-induced cardiomyopathy via rescuing mitophagy and improving mitochondrial function. J. Mol. Cell. Cardiol. 2019, 133, 125–137. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef] [Green Version]
- Wallace, K.B.; Sardao, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Yin, J.; Guo, J.B.; Zhang, Q.; Cui, L.; Zhang, L.; Zhang, T.F.; Zhao, J.; Li, J.; Middleton, A.; Carmichael, P.L.; et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol In Vitro 2018, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef] [PubMed]
- Nazarko, V.Y.; Zhong, Q. ULK1 targets Beclin-1 in autophagy. Nat. Cell Biol. 2013, 15, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 2016, 473, 2251–2271. [Google Scholar] [CrossRef]
- Feng, Y.C.; He, D.; Yao, Z.Y.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.L.; Chen, H.L.; Wu, D.; Chen, J.X.; Wang, X.X.; Li, R.L.; He, J.H.; Mo, L.; Cen, X.B.; et al. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through AMPK and p38-MAPK. Biochem. Pharmacol. 2014, 88, 334–350. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Meng, C.; Zhang, X.M.; Yuan, C.H.; Wen, M.D.; Chen, Z.; Dong, D.C.; Gao, Y.H.; Liu, C.; Zhang, Z. Ophiopogonin D Attenuates Doxorubicin-Induced Autophagic Cell Death by Relieving Mitochondrial Damage In Vitro and In Vivo. J. Pharmacol. Exp. Ther. 2015, 352, 166–174. [Google Scholar] [CrossRef]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Volden, P.; Timm, D.; Mao, K.; Xu, X.; Liang, Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 2010, 285, 793–804. [Google Scholar] [CrossRef]
- Li, M.; Sala, V.; De Santis, M.C.; Cimino, J.; Cappello, P.; Pianca, N.; Di Bona, A.; Margaria, J.P.; Martini, M.; Lazzarini, E.; et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects From Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation 2018, 138, 696–711. [Google Scholar] [CrossRef]
- Li, S.; Wang, W.; Niu, T.; Wang, H.; Li, B.; Shao, L.; Lai, Y.; Li, H.; Janicki, J.S.; Wang, X.L.; et al. Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid. Med. Cell. Longev. 2014, 2014, 748524. [Google Scholar] [CrossRef]
- Bartlett, J.J.; Trivedi, P.C.; Yeung, P.; Kienesberger, P.C.; Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 2016, 473, 3769–3789. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Russo, M.; Pirozzi, F.; Tocchetti, C.G.; Ghigo, A. Autophagy and cancer therapy cardiotoxicity: From molecular mechanisms to therapeutic opportunities. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118493. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.J.; Trivedi, P.C.; Pulinilkunnil, T. Autophagic dysregulation in doxorubicin cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 104, 1–8. [Google Scholar] [CrossRef]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.M.; Gamblin, S.J.; Harrison, S.C.; Wang, J.C. Structure and mechanism of DNA topoisomerase II. Nature 1996, 379, 225–232. [Google Scholar] [CrossRef]
- Moro, S.; Beretta, G.L.; Dal Ben, D.; Nitiss, J.; Palumbo, M.; Capranico, G. Interaction model for anthracycline activity against DNA topoisomerase II. Biochemistry 2004, 43, 7503–7513. [Google Scholar] [CrossRef]
- Swift, L.P.; Cutts, S.M.; Nudelman, A.; Levovich, I.; Rephaeli, A.; Phillips, D.R. The cardio-protecting agent and topoisomerase II catalytic inhibitor sobuzoxane enhances doxorubicin-DNA adduct mediated cytotoxicity. Cancer Chemother. Pharmacol. 2008, 61, 739–749. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Drake, F.H.; Zimmerman, J.P.; McCabe, F.L.; Bartus, H.F.; Per, S.R.; Sullivan, D.M.; Ross, W.E.; Mattern, M.R.; Johnson, R.K.; Crooke, S.T.; et al. Purification of topoisomerase II from amsacrine-resistant P388 leukemia cells. Evidence for two forms of the enzyme. J. Biol. Chem. 1987, 262, 16739–16747. [Google Scholar] [CrossRef]
- Capranico, G.; Tinelli, S.; Austin, C.A.; Fisher, M.L.; Zunino, F. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim. Biophys. Acta 1992, 1132, 43–48. [Google Scholar] [CrossRef]
- Lyu, Y.L.; Kerrigan, J.E.; Lin, C.P.; Azarova, A.M.; Tsai, Y.C.; Ban, Y.; Liu, L.F. Topoisomerase IIbeta mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007, 67, 8839–8846. [Google Scholar] [CrossRef] [PubMed]
- Vejpongsa, P.; Yeh, E.T. Topoisomerase 2beta: A promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin. Pharmacol. Ther. 2014, 95, 45–52. [Google Scholar] [CrossRef]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1α and ERRα target gene downregulation is a signature of the failing human heart. J. Mol. Cell. Cardiol. 2009, 46, 201–212. [Google Scholar] [CrossRef]
- Sebastiani, M.; Giordano, C.; Nediani, C.; Travaglini, C.; Borchi, E.; Zani, M.; Feccia, M.; Mancini, M.; Petrozza, V.; Cossarizza, A.; et al. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J. Am. Coll. Cardiol. 2007, 50, 1362–1369. [Google Scholar] [CrossRef]
- Jirkovsky, E.; Popelova, O.; Krivakova-Stankova, P.; Vavrova, A.; Hroch, M.; Haskova, P.; Brcakova-Dolezelova, E.; Micuda, S.; Adamcova, M.; Simunek, T.; et al. Chronic Anthracycline Cardiotoxicity: Molecular and Functional Analysis with Focus on Nuclear Factor Erythroid 2-Related Factor 2 and Mitochondrial Biogenesis Pathways. J. Pharmacol. Exp. Ther. 2012, 343, 468–478. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Huelsenbeck, S.C.; Schorr, A.; Roos, W.P.; Huelsenbeck, J.; Henninger, C.; Kaina, B.; Fritz, G. Rac1 Protein Signaling Is Required for DNA Damage Response Stimulated by Topoisomerase II Poisons. J. Biol. Chem. 2012, 287, 38590–38599. [Google Scholar] [CrossRef] [Green Version]
- Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. The effects of doxorubicin on cardiac calcium homeostasis and contractile function. J. Cardiol. 2022, 80, 125–132. [Google Scholar] [CrossRef]
- Arai, M.; Tomaru, K.; Takizawa, T.; Sekiguchi, K.; Yokoyama, T.; Suzuki, T.; Nagai, R. Sarcoplasmic reticulum genes are selectively down-regulated in cardiomyopathy produced by doxorubicin in rabbits. J. Mol. Cell. Cardiol. 1998, 30, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.E.; Olson, R.D.; Cusack, B.J.; Gambliel, H.A.; Dillmann, W.H. Anthracycline cardiotoxicity in transgenic mice overexpressing SR Ca2+-ATPase. Biochem. Biophys. Res. Commun. 2003, 303, 504–507. [Google Scholar] [CrossRef]
- Takahashi, S.-s.; Denvir, M.A.; Harder, L.; Miller, D.J.; Cobbe, S.M.; Kawakami, M.; MacFarlane, N.G.; Okabe, E. Effects of In Vitro and In Vivo Exposure to Doxorubicin (Adriamycin) on Caffeine-Induced Ca2+ Release from Sarcoplasmic Reticulum and Contractile Protein Function in ‘Chemically-Skinned’ Rabbit Ventricular Trabeculae. Jpn. J. Pharmacol. 1998, 76, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.C.; Toll, L.; Ellis, M.; Jensen, R.A. L-type cardiac calcium channels in doxorubicin cardiomyopathy in rats morphological, biochemical, and functional correlations. J. Clin. Investig. 1991, 87, 2108–2113. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Miller, S.C.; Billingham, M.E.; Akimoto, H.; Torti, S.V.; Wade, R.; Gahlmann, R.; Lyons, G.; Kedes, L.; Torti, F.M. Doxorubicin selectively inhibits muscle gene expression in cardiac muscle cells in vivo and in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 4275–4279. [Google Scholar] [CrossRef]
- Hanna, A.D.; Lam, A.; Tham, S.; Dulhunty, A.F.; Beard, N.A. Adverse Effects of Doxorubicin and Its Metabolic Product on Cardiac RyR2 and SERCA2A. Mol. Pharmacol. 2014, 86, 438–449. [Google Scholar] [CrossRef]
- Menna, P.; Salvatorelli, E.; Gianni, L.; Minotti, G. Anthracycline cardiotoxicity. Top. Curr. Chem. 2008, 283, 21–44. [Google Scholar] [CrossRef]
- Fu, L.X.; Waagstein, F.; Hjalmarson, Ã. A new insight into adriamycin-induced cardiotoxicity. Int. J. Cardiol. 1990, 29, 15–20. [Google Scholar] [CrossRef]
- Sag, C.M.; Kohler, A.C.; Anderson, M.E.; Backs, J.; Maier, L.S. CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 51, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Berthiaume, J.M.; Oliveira, P.J.; Fariss, M.W.; Wallace, K.B. Dietary vitamin E decreases doxorubicin-induced oxidative stress without preventing mitochondrial dysfunction. Cardiovasc. Toxicol. 2005, 5, 257–267. [Google Scholar] [CrossRef]
- Abdoul-Azize, S.; Buquet, C.; Li, H.; Picquenot, J.M.; Vannier, J.P. Integration of Ca2+ signaling regulates the breast tumor cell response to simvastatin and doxorubicin. Oncogene 2018, 37, 4979–4993. [Google Scholar] [CrossRef] [PubMed]
- Solem, L.E.; Heller, L.J.; Wallace, K.B. Dose-dependent Increase in Sensitivity to Calcium-induced Mitochondrial Dysfunction and Cardiomyocyte Cell Injury by Doxorubicin. J. Mol. Cell. Cardiol. 1996, 28, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.S.; Meng, L.J.; Ding, P.G.; Sang, M.X. Epigenetic regulation of MAGE family in human cancer progression-DNA methylation, histone modification, and non-coding RNAs. Clin. Epigenetics 2018, 10, 115. [Google Scholar] [CrossRef]
- Kimball, T.H.; Vondriska, T.M. Metabolism, Epigenetics, and Causal Inference in Heart Failure. Trends Endocrinol. Metab. 2020, 31, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Hanf, A.; Oelze, M.; Manea, A.; Li, H.G.; Munzel, T.; Daiber, A. The anti-cancer drug doxorubicin induces substantial epigenetic changes in cultured cardiomyocytes. Chem. Biol. Interact. 2019, 313, 108834. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Cunha-Oliveira, T.; Simoes, R.F.; Carvalho, F.S.; Burgeiro, A.; Nordgren, K.; Wallace, K.B.; Oliveira, P.J. Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology 2017, 390, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Kumari, H.; Huang, W.H.; Chan, M.W.Y. Review on the Role of Epigenetic Modifications in Doxorubicin-Induced Cardiotoxicity. Front. Cardiovasc. Med. 2020, 7, 56. [Google Scholar] [CrossRef]
- Cui, L.; Guo, J.B.; Zhang, Q.; Yin, J.; Li, J.; Zhou, W.; Zhang, T.F.; Yuan, H.T.; Zhao, J.; Zhang, L.; et al. Erythropoietin activates SIRT1 to protect human cardiomyocytes against doxorubicin-induced mitochondrial dysfunction and toxicity. Toxicol. Lett. 2017, 275, 28–38. [Google Scholar] [CrossRef]
- Song, R.; Yang, Y.; Lei, H.; Wang, G.; Huang, Y.; Xue, W.; Wang, Y.; Yao, L.; Zhu, Y. HDAC6 inhibition protects cardiomyocytes against doxorubicin-induced acute damage by improving α-tubulin acetylation. J. Mol. Cell. Cardiol. 2018, 124, 58–69. [Google Scholar] [CrossRef]
- Tony, H.; Yu, K.W.; Qiutang, Z. MicroRNA-208a Silencing Attenuates Doxorubicin Induced Myocyte Apoptosis and Cardiac Dysfunction. Oxid. Med. Cell. Longev. 2015, 2015, 597032. [Google Scholar] [CrossRef]
- Han, D.; Wang, Y.J.; Wang, Y.B.; Dai, X.C.; Zhou, T.W.; Chen, J.W.; Tao, B.; Zhang, J.B.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges miR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity Through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, E108–E125. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Garg, A.; Avramopoulos, P.; Engelhardt, S.; Streckfuss-Bomeke, K.; Batkai, S.; Thum, T. miR-212/132 Cluster Modulation Prevents Doxorubicin-Mediated Atrophy and Cardiotoxicity. Mol. Ther. 2019, 27, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, C.; Deidda, M.; Piras, A.; Cadeddu, C.; Demurtas, L.; Puzzoni, M.; Piscopo, G.; Scartozzi, M.; Mercuro, G. Pathophysiology of cardiotoxicity induced by nonanthracycline chemotherapy. J. Cardiovasc. Med. 2016, 17, e12–e18. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Naziroglu, M.; Karaoglu, A.; Aksoy, A.O. Selenium and high dose vitamin E administration protects cisplatin-induced oxidative damage to renal, liver and lens tissues in rats. Toxicology 2004, 195, 221–230. [Google Scholar] [CrossRef]
- Spagnoli, L.G.; Orlandi, A.; Marino, B.; Mauriello, A.; De Angelis, C.; Ramacci, M.T. Propionyl-L-carnitine prevents the progression of atherosclerotic lesions in aged hyperlipemic rabbits. Atherosclerosis 1995, 114, 29–44. [Google Scholar] [CrossRef]
- Yeh, E.T.; Bickford, C.L. Cardiovascular complications of cancer therapy: Incidence, pathogenesis, diagnosis, and management. J. Am. Coll. Cardiol. 2009, 53, 2231–2247. [Google Scholar] [CrossRef]
- El-Awady, E.-S.E.; Moustafa, Y.M.; Abo-Elmatty, D.M.; Radwan, A. Cisplatin-induced cardiotoxicity: Mechanisms and cardioprotective strategies. Eur. J. Pharmacol. 2011, 650, 335–341. [Google Scholar] [CrossRef]
- Ma, H.; Jones, K.R.; Guo, R.; Xu, P.S.; Shen, Y.Q.; Ren, J. Cisplatin compromises myocardial contractile function and mitochondrial ultrastructure: Role of endoplasmic reticulum stress. Clin. Exp. Pharmacol. Physiol. 2010, 37, 460–465. [Google Scholar] [CrossRef]
- Lenneman, C.G.; Sawyer, D.B. Cardio-Oncology An Update on Cardiotoxicity of Cancer-Related Treatment. Circ. Res. 2016, 118, 1008–1020. [Google Scholar] [CrossRef]
- Feldman, D.R.; Jacobsen, E.P.; Woo, K.; Steingart, R.; Fleisher, M.; Patil, S.; Carousso, M.; Laguerre, S.; Morales, T.D.; Sheinfeld, J.; et al. Acute changes in endothelial function with cisplatin among germ cell tumor (GCT) patients (Pts). J. Clin. Oncol. 2014, 32, 9587. [Google Scholar] [CrossRef]
- Nuver, J.; Smit, A.J.; Sleijfer, D.T.; van Gessel, A.I.; van Roon, A.M.; van der Meer, J.; van den Berg, M.P.; Burgerhof, J.G.M.; Hoekstra, H.J.; Sluiter, W.J.; et al. Micro albuminuria, decreased fibrinolysis, and inflammation as early signs of atherosclerosis in long-term survivors of disseminated testicular cancer. Eur. J. Cancer 2004, 40, 701–706. [Google Scholar] [CrossRef]
- Qi, L.Y.; Luo, Q.; Zhang, Y.Y.; Jia, F.F.; Zhao, Y.; Wang, F.Y. Advances in Toxicological Research of the Anticancer Drug Cisplatin. Chem. Res. Toxicol. 2019, 32, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Podgurskaya, A.D.; Slotvitsky, M.M.; Tsvelaya, V.A.; Frolova, S.R.; Romanova, S.G.; Balashov, V.A.; Agladze, K.I. Cyclophosphamide arrhythmogenicitytesting using human-induced pluripotent stem cell-derived cardiomyocytes. Sci. Rep. 2021, 11, 2336. [Google Scholar] [CrossRef] [PubMed]
- Katayama, M.; Imai, Y.; Hashimoto, H.; Kurata, M.; Nagai, K.; Tamita, K.; Morioka, S.; Furukawa, Y. Fulminant fatal cardiotoxicity following cyclophosphamide therapy. J. Cardiol. 2009, 54, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Iqubal, A.; Iqubal, M.K.; Sharma, S.; Ansari, M.A.; Najmi, A.K.; Ali, S.M.; Ali, J.; Haque, S.E. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: Old drug with a new vision. Life Sci. 2019, 218, 112–131. [Google Scholar] [CrossRef]
- Dionisio, F.; Araujo, A.M.; Duarte-Araujo, M.; Bastos, M.D.; de Pinho, P.G.; Carvalho, F.; Costa, V.M. Cardiotoxicity of cyclophosphamide’s metabolites: An in vitro metabolomics approach in AC16 human cardiomyocytes. Arch. Toxicol. 2022, 96, 653–671. [Google Scholar] [CrossRef]
- Swamy, A.H.M.V.; Patel, U.M.; Koti, B.C.; Gadad, P.C.; Patel, N.L.; Thippeswamy, A.H.M. Cardioprotective effect of Saraca indica against cyclophosphamide induced cardiotoxicity in rats: A biochemical, electrocardiographic and histopathological study. Indian J. Pharmacol. 2013, 45, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, E.H.M.; Abd El-Ghafar, O.A.M.; Ahmed, M.A.; Sayed, A.M.; Gad-Elrab, W.M.; Ajarem, J.S.; Allam, A.A.; Mahmoud, A.M. Edaravone and Acetovanillone Upregulate Nrf2 and PI3K/Akt/mTOR Signaling and Prevent Cyclophosphamide Cardiotoxicity in Rats. Drug Des. Dev. Ther. 2020, 14, 5275–5288. [Google Scholar] [CrossRef] [PubMed]
- Ismahil, M.A.; Hamid, T.; Haberzettl, P.; Gu, Y.; Chandrasekar, B.; Srivastava, S.; Bhatnagar, A.; Prabhu, S.D. Chronic oral exposure to the aldehyde pollutant acrolein induces dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2050–H2060. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Hill, B.G.; Gu, Y.; Cai, J.; Srivastava, S.; Bhatnagar, A.; Prabhu, S.D. Mechanisms of acrolein-induced myocardial dysfunction: Implications for environmental and end ogenous aldehyde exposure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3673–H3684. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Peters, G.J. Novel Developments in the Use of Antimetabolites. Nucleosides Nucleotides Nucleic Acids 2014, 33, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.B. Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef]
- Jain, D.; Ahmad, T.; Cairo, M.; Aronow, W. Cardiotoxicity of cancer chemotherapy: Identification, prevention and treatment. Ann. Transl. Med. 2017, 5, 348. [Google Scholar] [CrossRef]
- Ridker, P.M. Testing the inflammatory hypothesis of atherothrombosis: Scientific rationale for the cardiovascular inflammation reduction trial (CIRT). J. Thromb. Haemost. 2009, 7, 332–339. [Google Scholar] [CrossRef]
- Marks, J.L.; Edwards, C.J. Protective effect of methotrexate in patients with rheumatoid arthritis and cardiovascular comorbidity. Ther. Adv. Musculoskelet. Dis. 2012, 4, 149–157. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Zhao, P.; Li, A.H.; Lv, X.L.; Gao, Y.; Sun, H.G.; Ding, Y.L.; Liu, J. Effects of Methotrexate on Plasma Cytokines and Cardiac Remodeling and Function in Postmyocarditis Rats. Mediat. Inflamm. 2009, 2009, 389720. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gui, Y.; Wang, Y.; Qian, L.; Liu, X.; Jiang, Q.; Song, H. Effects of methotrexate on the developments of heart and vessel in zebrafish. Acta Biochim. Biophys. Sin. 2009, 41, 86–96. [Google Scholar] [CrossRef]
- Tousson, E.; Hafez, E.; Zaki, S.; Gad, A. The cardioprotective effects of L-carnitine on rat cardiac injury, apoptosis, and oxidative stress caused by amethopterin. Environ. Sci. Pollut. Res. 2016, 23, 20600–20608. [Google Scholar] [CrossRef]
- Al-Taher, A.Y.; Morsy, M.A.; Rifaai, R.A.; Zenhom, N.M.; Abdel-Gaber, S.A. Paeonol Attenuates Methotrexate-Induced Cardiac Toxicity in Rats by Inhibiting Oxidative Stress and Suppressing TLR4-Induced NF-kappa B Inflammatory Pathway. Mediat. Inflamm. 2020, 2020, 8641026. [Google Scholar] [CrossRef]
- Focaccetti, C.; Bruno, A.; Magnani, E.; Bartolini, D.; Principi, E.; Dallaglio, K.; Bucci, E.O.; Finzi, G.; Sessa, F.; Noonan, D.M.; et al. Effects of 5-Fluorouracil on Morphology, Cell Cycle, Proliferation, Apoptosis, Autophagy and ROS Production in Endothelial Cells and Cardiomyocytes. PLoS ONE 2015, 10, e0115686. [Google Scholar] [CrossRef] [PubMed]
- Brell, J.M. 5-Fluorouracil Cardiotoxicity Known But Unknown. JACC Cardiooncol. 2021, 3, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Pinedo, H.M.; Peters, G.F. Fluorouracil: Biochemistry and pharmacology. J. Clin. Oncol. 1988, 6, 1653–1664. [Google Scholar] [CrossRef]
- Muneoka, K.; Shirai, Y.; Yokoyama, N.; Wakai, T.; Hatakeyama, K. 5-Fluorouracil cardiotoxicity induced by alpha-fluoro-beta-alanine. Int. J. Clin. Oncol. 2005, 10, 441–443. [Google Scholar] [CrossRef]
- Gmeiner, W.H. Chemistry of Fluorinated Pyrimidines in the Era of Personalized Medicine. Molecules 2020, 25, 3438. [Google Scholar] [CrossRef]
- Matsubara, I.; Kamiya, J.; Imai, S. Cardiotoxic effects of 5-fluorouracil in the guinea pig. Jpn. J. Pharmacol. 1980, 30, 871–879. [Google Scholar] [CrossRef]
- Li, Y.Y.; Zhang, Y.F.; Zhou, X.Z.; Lei, X.H.; Li, X.H.; Wei, L.P. Dynamic observation of 5-fluorouracil-induced myocardial injury and mitochondrial autophagy in aging rats. Exp. Ther. Med. 2021, 22, 1451. [Google Scholar] [CrossRef] [PubMed]
- Durak, I.; Karaayvaz, M.; Kavutcu, M.; Cimen, M.Y.B.; Kacmaz, M.; Buyukkocak, S.; Ozturk, H.S. Reduced antioxidant defense capacity in myocardial tissue from guinea pigs treated with 5-fluorouracil. J. Toxicol. Environ. Health Part A 2000, 59, 585–589. [Google Scholar]
- Spasojevic, I.; Jelic, S.; Zakrzewska, J.; Bacic, G. Decreased Oxygen Transfer Capacity of Erythrocytes as a Cause of 5-Fluorouracil Related Ischemia. Molecules 2009, 14, 53–67. [Google Scholar] [CrossRef]
- Thyss, A.; Gaspard, M.H.; Marsault, R.; Milano, G.; Frelin, C.; Schneider, M. Very high endothelin plasma levels in patients with 5-FU cardiotoxicity. Ann. Oncol. 1992, 3, 88. [Google Scholar] [CrossRef]
- Alter, P.; Herzum, M.; Soufi, M.; Schaefer, J.R.; Maisch, B. Cardiotoxicity of 5-fluorouracil. Cardiovasc. Hematol. Agents Med. Chem. 2006, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Polk, A.; Vistisen, K.; Vaage-Nilsen, M.; Nielsen, D.L. A systematic review of the pathophysiology of 5-fluorouracil-induced cardiotoxicity. BMC Pharmacol. Toxicol. 2014, 15, 47. [Google Scholar] [CrossRef]
- Newbery, G.; Lima, N.A.; Gurgel, L.A.; Driscoll, R.; Lima, C.C.V. Persistent heart failure following melphalan and fludarabine conditioning. J. Cardiol. Cases 2019, 20, 88–91. [Google Scholar] [CrossRef]
- Yang, X.; Liu, W.; Lyons, A.; Song, Z.; Zhai, S.; Hu, K. Pericarditis associated with cytarabine therapy for acute myelocytic leukemia: A case report. Eur. J. Clin. Pharmacol. 2018, 74, 181–182. [Google Scholar] [CrossRef]
- Reykdal, S.; Sham, R.; Kouides, P. Cytarabine-induced pericarditis: A case report and review of the literature of the cardio-pulmonary complications of cytarabine therapy. Leuk. Res. 1995, 19, 141–144. [Google Scholar] [CrossRef]
- Bradley, J.A. The current status of intraoperative radiation therapy in breast cancer: Challenges and promises. Breast J. 2018, 24, 713–714. [Google Scholar] [CrossRef] [PubMed]
- Megwalu, U.C.; Orloff, L.A.; Ma, Y. Adjuvant external beam radiotherapy for locally invasive papillary thyroid cancer. Head Neck 2019, 41, 1719–1724. [Google Scholar] [CrossRef] [PubMed]
- Alterio, D.; Marvaso, G.; Ferrari, A.; Volpe, S.; Orecchia, R.; Jereczek-Fossa, B.A. Modern radiotherapy for head and neck cancer. Semin. Oncol. 2019, 46, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Minniti, G.; Goldsmith, C.; Brada, M. Radiotherapy. Handb. Clin. Neurol. 2012, 104, 215–228. [Google Scholar] [CrossRef]
- Nabialek-Trojanowska, I.; Lewicka, E.; Wrona, A.; Kaleta, A.M.; Lewicka-Potocka, Z.; Raczak, G.; Dziadziuszko, R. Cardiovascular complications after radiotherapy. Cardiol. J. 2020, 27, 836–847. [Google Scholar] [CrossRef]
- Ping, Z.; Peng, Y.; Lang, H.; Xinyong, C.; Zhiyi, Z.; Xiaocheng, W.; Hong, Z.; Liang, S. Oxidative Stress in Radiation-Induced Cardiotoxicity. Oxid. Med. Cell. Longev. 2020, 2020, 3579143. [Google Scholar] [CrossRef]
- Wang, H.; Wei, J.; Zheng, Q.; Meng, L.; Xin, Y.; Yin, X.; Jiang, X. Radiation-induced heart disease: A review of classification, mechanism and prevention. Int. J. Biol. Sci. 2019, 15, 2128–2138. [Google Scholar] [CrossRef]
- Kuribayashi, K.; Finnberg, N.; Jeffers, J.R.; Zambetti, G.P.; El-Deiry, W.S. The relative contribution of pro-apoptotic p53-target genes in the triggering of apoptosis following DNA damage in vitro and in vivo. Cell Cycle 2011, 10, 2380–2389. [Google Scholar] [CrossRef]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Seo, D.; Kim, D.; Kim, K.; Kim, E.; Kang, J.; Seong, K.M.; Youn, H.; Youn, B. Cellular Stress Responses in Radiotherapy. Cells 2019, 8, 1105. [Google Scholar] [CrossRef] [Green Version]
- Taunk, N.K.; Haffty, B.G.; Kostis, J.B.; Goyal, S. Radiation-induced heart disease: Pathologic abnormalities and putative mechanisms. Front. Oncol. 2015, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Verheij, M.; Dewit, L.G.; Boomgaard, M.N.; Brinkman, H.J.; van Mourik, J.A. Ionizing radiation enhances platelet adhesion to the extracellular matrix of human endothelial cells by an increase in the release of von Willebrand factor. Radiat. Res. 1994, 137, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Biej, M.; Smolarczyk, R.; Cichon, T.; Kulach, N. Tumor Microenvironment as A “Game Changer” in Cancer Radiotherapy. Int. J. Mol. Sci. 2019, 20, 3212. [Google Scholar] [CrossRef]
- Boerma, M.; Roberto, K.A.; Hauer-Jensen, M. Prevention and treatment of functional and structural radiation injury in the rat heart by pentoxifylline and alpha-tocopherol. Int. J. Radiat. Oncol. 2008, 72, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, J.; Laurence, J. Role of Platelet-Derived Transforming Growth Factor-beta1 and Reactive Oxygen Species in Radiation-Induced Organ Fibrosis. Antioxid. Redox Signal. 2017, 27, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.G.; Kovacs, Z.Z.A.; Varga, Z.; Szucs, G.; Freiwan, M.; Farkas, K.; Kovari, B.; Cserni, G.; Kriston, A.; Kovacs, F.; et al. Investigation of the Antihypertrophic and Antifibrotic Effects of Losartan in a Rat Model of Radiation-Induced Heart Disease. Int. J. Mol. Sci. 2021, 22, 12963. [Google Scholar] [CrossRef]
- Yang, R.; Tan, C.; Najafi, M. Cardiac inflammation and fibrosis following chemo/radiation therapy: Mechanisms and therapeutic agents. Inflammopharmacology 2022, 30, 73–89. [Google Scholar] [CrossRef]
- Dokmanovic, M.; Wu, W.J. Monitoring Trastuzumab Resistance and Cardiotoxicity: A Tale of Personalized Medicine. Adv. Clin. Chem. 2015, 70, 95–130. [Google Scholar] [CrossRef]
- Joo, W.D.; Visintin, I.; Mor, G. Targeted cancer therapy—Are the days of systemic chemotherapy numbered? Maturitas 2013, 76, 308–314. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef]
- Nowsheen, S.; Aziz, K.; Park, J.Y.; Lerman, A.; Villarraga, H.R.; Ruddy, K.J.; Herrmann, J. Trastuzumab in Female Breast Cancer Patients With Reduced Left Ventricular Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e008637. [Google Scholar] [CrossRef] [PubMed]
- Viani, G.A.; Afonso, S.L.; Stefano, E.J.; De Fendi, L.I.; Soares, F.V. Adjuvant trastuzumab in the treatment of her-2-positive early breast cancer: A meta-analysis of published randomized trials. BMC Cancer 2007, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.I.; Burke, M.A.; Singh, A.T.K.; Prachand, S.; Lieberman, E.D.; Sun, L.; Naik, T.J.; Prasad, S.V.N.; Ardehali, H. Blockade of the erbB2 Receptor Induces Cardiomyocyte Death through Mitochondrial and Reactive Oxygen Species-dependent Pathways. J. Biol. Chem. 2009, 284, 2080–2087. [Google Scholar] [CrossRef] [PubMed]
- ElZarrad, M.K.; Mukhopadhyay, P.; Mohan, N.; Hao, E.; Dokmanovic, M.; Hirsch, D.S.; Shen, Y.; Pacher, P.; Wu, W.J. Trastuzumab alters the expression of genes essential for cardiac function and induces ultrastructural changes of cardiomyocytes in mice. PLoS ONE 2013, 8, e79543. [Google Scholar] [CrossRef]
- Touyz, R.M.; Herrmann, J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis. Oncol. 2018, 2, 13. [Google Scholar] [CrossRef]
- Bottoni, C.; Scambia, G.; Fagotti, A.; Petrillo, M. The safety of bevazicumab for the treatment of ovarian cancer. Expert Opin. Drug Saf. 2018, 17, 1107–1113. [Google Scholar] [CrossRef]
- Du, F.; Li, P.; Chen, J.; Gong, Z.; Chi, C.; Hu, B.; Chu, H. Adjuvant chemotherapy with bevacizumab (i.p.) can prolong survival time of patients with advanced ovarian cancer after cytoreduction. Neoplasma 2017, 64, 108–113. [Google Scholar] [CrossRef]
- Li, Y.; Tian, W.; Yue, D.; Chen, C.; Li, C.; Zhang, Z.; Wang, C. Bevacizumab-Induced Mitochondrial Dysfunction, Endoplasmic Reticulum Stress, and ERK Inactivation Contribute to Cardiotoxicity. Oxid. Med. Cell. Longev. 2021, 2021, 5548130. [Google Scholar] [CrossRef]
- Sabet, N.S.; Atashbar, S.; Khanlou, E.M.; Kahrizi, F.; Salimi, A. Curcumin attenuates bevacizumab-induced toxicity via suppressing oxidative stress and preventing mitochondrial dysfunction in heart mitochondria. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 1447–1457. [Google Scholar] [CrossRef]
- Anand, K.; Ensor, J.; Trachtenberg, B.; Bernicker, E.H. Osimertinib-Induced Cardiotoxicity A Retrospective Review of the FDA Adverse Events Reporting System (FAERS). JACC Cardiooncol. 2019, 1, 172–178. [Google Scholar] [CrossRef]
- Oyakawa, T.; Nakashima, K.; Naito, T. Cardiac Dysfunction Caused by Osimertinib. J. Thorac. Oncol. 2017, 12, e159–e160. [Google Scholar] [CrossRef] [PubMed]
- Hsiue, E.H.; Lee, J.H.; Lin, C.C.; Yang, J.C. Safety of gefitinib in non-small cell lung cancer treatment. Expert Opin. Drug Saf. 2016, 15, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R., Jr.; Kickler, T.S.; Rade, J.J. Recurrent myocardial infarction associated with gefitinib therapy. J. Thromb. Thrombolysis 2011, 32, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Gronich, N.; Lavi, I.; Barnett-Griness, O.; Saliba, W.; Abernethy, D.R.; Rennert, G. Tyrosine kinase-targeting drugs-associated heart failure. Br. J. Cancer 2017, 116, 1366–1373. [Google Scholar] [CrossRef]
- Alhoshani, A.; Alanazi, F.E.; Alotaibi, M.R.; Attwa, M.W.; Kadi, A.A.; Aldhfyan, A.; Akhtar, S.; Hourani, S.; Agouni, A.; Zeidan, A.; et al. EGFR Inhibitor Gefitinib Induces Cardiotoxicity through the Modulation of Cardiac PTEN/Akt/FoxO3a Pathway and Reactive Metabolites Formation: In Vivo and in Vitro Rat Studies. Chem. Res. Toxicol. 2020, 33, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- AlAsmari, A.F.; Ali, N.; AlAsmari, F.; AlAnazi, W.A.; AlShammari, M.A.; Al-Harbi, N.O.; Alhoshani, A.; As Sobeai, H.M.; AlSwayyed, M.; AlAnazi, M.M.; et al. Liraglutide attenuates gefitinib-induced cardiotoxicity and promotes cardioprotection through the regulation of MAPK/NF-kappaB signaling pathways. Saudi Pharm. J. 2020, 28, 509–518. [Google Scholar] [CrossRef]
- Choi, H.D.; Chang, M.J. Cardiac toxicities of lapatinib in patients with breast cancer and other HER2-positive cancers: A meta-analysis. Breast Cancer Res. Treat. 2017, 166, 927–936. [Google Scholar] [CrossRef]
- Tao, Z.; Li, S.X.; Shen, K.; Zhao, Y.; Zeng, H.; Ma, X. Safety and Efficacy Profile of Neratinib: A Systematic Review and Meta-Analysis of 23 Prospective Clinical Trials. Clin. Drug Investig. 2019, 39, 27–43. [Google Scholar] [CrossRef]
- Ewer, M.S.; Patel, K.; O’Brien, D.; Lorence, R.M. Cardiac safety of afatinib: A review of data from clinical trials. Cardiooncology 2015, 1, 3. [Google Scholar] [CrossRef]
- Chitturi, K.R.; Burns, E.A.; Muhsen, I.N.; Anand, K.; Trachtenberg, B.H. Cardiovascular Risks with Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors and Monoclonal Antibody Therapy. Curr. Oncol. Rep. 2022, 24, 475–491. [Google Scholar] [CrossRef]
- Aparicio-Gallego, G.; Blanco, M.; Figueroa, A.; Garcia-Campelo, R.; Valladares-Ayerbes, M.; Grande-Pulido, E.; Anton-Aparicio, L. New Insights into Molecular Mechanisms of Sunitinib-Associated Side Effects. Mol. Cancer Ther. 2011, 10, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.F.; Rupnick, M.A.; Kerkela, R.; Dallabrida, S.M.; Zurakowski, D.; Nguyen, L.; Woulfe, K.; Pravda, E.; Cassiola, F.; Desai, J.; et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 2007, 370, 2011–2019. [Google Scholar] [CrossRef]
- Richards, C.J.; Je, Y.; Schutz, F.A.; Heng, D.Y.; Dallabrida, S.M.; Moslehi, J.J.; Choueiri, T.K. Incidence and risk of congestive heart failure in patients with renal and nonrenal cell carcinoma treated with sunitinib. J. Clin. Oncol. 2011, 29, 3450–3456. [Google Scholar] [CrossRef] [PubMed]
- Truitt, R.; Mu, A.; Corbin, E.A.; Vite, A.; Brandimarto, J.; Ky, B.; Margulies, K.B. Increased Afterload Augments Sunitinib-Induced Cardiotoxicity in an Engineered Cardiac Microtissue Model. JACC Basic Transl. Sci. 2018, 3, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Bouitbir, J.; Alshaikhali, A.; Panajatovic, M.V.; Abegg, V.F.; Paech, F.; Krahenbuhl, S. Mitochondrial oxidative stress plays a critical role in the cardiotoxicity of sunitinib: Running title: Sunitinib and oxidative stress in hearts. Toxicology 2019, 426, 152281. [Google Scholar] [CrossRef]
- Blanca, A.J.; Ruiz-Armenta, M.V.; Zambrano, S.; Miguel-Carrasco, J.L.; Arias, J.L.; Arevalo, M.; Mate, A.; Aramburu, O.; Vazquez, C.M. Inflammatory and fibrotic processes are involved in the cardiotoxic effect of sunitinib: Protective role of L-carnitine. Toxicol. Lett. 2016, 241, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xue, T.; Yang, X.; Zhu, H.; Ding, X.; Lou, L.; Lu, W.; Yang, B.; He, Q. Autophagy plays an important role in sunitinib-mediated cell death in H9c2 cardiac muscle cells. Toxicol. Appl. Pharmacol. 2010, 248, 20–27. [Google Scholar] [CrossRef]
- Yang, Y.; Li, N.; Chen, T.; Zhang, C.; Liu, L.; Qi, Y.; Bu, P. Trimetazidine ameliorates sunitinib-induced cardiotoxicity in mice via the AMPK/mTOR/autophagy pathway. Pharm. Biol. 2019, 57, 625–631. [Google Scholar] [CrossRef]
- Yang, Y.; Li, N.; Chen, T.; Zhang, C.; Li, J.; Liu, L.; Qi, Y.; Zheng, X.; Zhang, C.; Bu, P. Sirt3 promotes sensitivity to sunitinib-induced cardiotoxicity via inhibition of GTSP1/JNK/autophagy pathway in vivo and in vitro. Arch. Toxicol. 2019, 93, 3249–3260. [Google Scholar] [CrossRef]
- Abdel-Rahman, O.; Fouad, M. Risk of cardiovascular toxicities in patients with solid tumors treated with sorafenib: An updated systematic review and meta-analysis. Future Oncol. 2014, 10, 1981–1992. [Google Scholar] [CrossRef]
- Cheng, H.; Kari, G.; Dicker, A.P.; Rodeck, U.; Koch, W.W.J.; Force, T. A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity. Circ. Res. 2011, 109, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, M.; Umemoto, N.; Shimada, Y.; Nishimura, Y.; Zhang, B.B.; Kuroyanagi, J.; Miyabe, M.; Tanaka, T. Downregulation of Stanniocalcin 1 Is Responsible for Sorafenib-Induced Cardiotoxicity. Toxicol. Sci. 2015, 143, 374–384. [Google Scholar] [CrossRef]
- Ma, W.Z.; Liu, M.; Liang, F.F.; Zhao, L.L.; Gao, C.Y.; Jiang, X.X.; Zhang, X.; Zhan, H.Q.; Hu, H.; Zhao, Z.H. Cardiotoxicity of sorafenib is mediated through elevation of ROS level and CaMKII activity and dysregulation of calcium homoeostasis. Basic Clin. Pharmacol. Toxicol. 2020, 126, 166–180. [Google Scholar] [CrossRef]
- Nave, O.; Elbaz, M.; Bunimovich-Mendrazitsky, S. Analysis of a breast cancer mathematical model by a new method to find an optimal protocol for HER2-positive cancer. Biosystems 2020, 197, 104191. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Zhang, Z.M. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Hayase, E.; Jenq, R.R. Role of the intestinal microbiome and microbial-derived metabolites in immune checkpoint blockade immunotherapy of cancer. Genome Med. 2021, 13, 107. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Hu, J.R.; Florido, R.; Lipson, E.J.; Naidoo, J.; Ardehali, R.; Tocchetti, C.G.; Lyon, A.R.; Padera, R.F.; Johnson, D.B.; Moslehi, J. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc. Res. 2019, 115, 854–868. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Salem, J.E.; Manouchehri, A.; Moey, M.; Lebrun-Vignes, B.; Bastarache, L.; Pariente, A.; Gobert, A.; Spano, J.P.; Balko, J.M.; Bonaca, M.P.; et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: An observational, retrospective, pharmacovigilance study. Lancet Oncol. 2018, 19, 1579–1589. [Google Scholar] [CrossRef]
- Lucas, J.A.; Menke, J.; Rabacal, W.A.; Schoen, F.J.; Sharpe, A.H.; Kelley, V.R. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J. Immunol. 2008, 181, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Okazaki, I.M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Delgobo, M.; Frantz, S. Heart failure in cancer: Role of checkpoint inhibitors. J. Thorac. Dis. 2018, 10, S4323–S4334. [Google Scholar] [CrossRef]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef]
- Sun, J.Y.; Qu, Q.; Lou, Y.X.; Hua, Y.; Sun, G.Z.; Sun, W.; Kong, X.Q. Cardiotoxicity in cancer immune-checkpoint therapy: Mechanisms, clinical evidence, and management strategies. Int. J. Cardiol. 2021, 344, 170–178. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Chen, D.H.; Guha, A.; Mackenzie, S.; Walker, J.M.; Roddie, C. CAR T Cell Therapy-Related Cardiovascular Outcomes and Management: Systemic Disease or Direct Cardiotoxicity? JACC CardioOncol. 2020, 2, 97–109. [Google Scholar] [CrossRef]
- Patel, N.P.; Doukas, P.G.; Gordon, L.I.; Akhter, N. Cardiovascular Toxicities of CAR T-cell Therapy. Curr. Oncol. Rep. 2021, 23, 78. [Google Scholar] [CrossRef]
- Stein-Merlob, A.F.; Rothberg, M.V.; Ribas, A.; Yang, E.H. Cardiotoxicities of novel cancer immunotherapies. Heart 2021, 107, 1694–1703. [Google Scholar] [CrossRef]
- Morra, F.; Merolla, F.; Picardi, I.; Russo, D.; Ilardi, G.; Varricchio, S.; Liotti, F.; Pacelli, R.; Palazzo, L.; Mascolo, M.; et al. CAF-1 Subunits Levels Suggest Combined Treatments with PARP-Inhibitors and Ionizing Radiation in Advanced HNSCC. Cancers 2019, 11, 1582. [Google Scholar] [CrossRef]
- Cerrato, A.; Visconti, R.; Celetti, A. The rationale for druggability of CCDC6-tyrosine kinase fusions in lung cancer. Mol. Cancer 2018, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Jerusalem, G.; Lancellotti, P.; Kim, S.B. HER2+ breast cancer treatment and cardiotoxicity: Monitoring and management. Breast Cancer Res. Treat. 2019, 177, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, D.J.; Parikh, R.V.; Kamath, M.; Calfon-Press, M.; Moriarty, J.M.; Aksoy, O.; Lopez-Mattei, J.; Palaskas, N.; Iliescu, C.A.; Yang, E.H. Structural Transcatheter Cardiac Interventions in the Cardio-Oncology Population. Curr. Treat. Options Cardiovasc. Med. 2021, 23, 20. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X. Future perspectives of personalized medicine in traditional Chinese medicine: A systems biology approach. Complement. Ther. Med. 2012, 20, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Cardinale, D.; Suter, T.; Plataniotis, G.; de Azambuja, E.; Sandri, M.T.; Criscitiello, C.; Goldhirsch, A.; Cipolla, C.; Roila, F.; et al. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO Clinical Practice Guidelines. Ann. Oncol. 2012, 23 (Suppl. S7), vii155–vii166. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Aronow, W. Cardiotoxicity of cancer chemotherapy in clinical practice. Hosp. Pract. 2019, 47, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Ozer, H.; Armitage, J.O.; Bennett, C.L.; Crawford, J.; Demetri, G.D.; Pizzo, P.A.; Schiffer, C.A.; Smith, T.J.; Somlo, G.; Wade, J.C.; et al. 2000 update of recommendations for the use of hematopoietic colony-stimulating factors: Evidence-based, clinical practice guidelines. J. Clin. Oncol. 2000, 18, 3558–3585. [Google Scholar] [CrossRef]
- Gharib, M.I.; Burnett, A.K. Chemotherapy-induced cardiotoxicity: Current practice and prospects of prophylaxis. Eur. J. Heart Fail. 2002, 4, 235–242. [Google Scholar] [CrossRef]
- Galetta, F.; Franzoni, F.; Cervetti, G.; Regoli, F.; Fallahi, P.; Tocchini, L.; Carpi, A.; Antonelli, A.; Petrini, M.; Santoro, G. In vitro and in vivo study on the antioxidant activity of dexrazoxane. Biomed. Pharmacother. 2010, 64, 259–263. [Google Scholar] [CrossRef]
- Langer, S.W. Dexrazoxane for the treatment of chemotherapy-related side effects. Cancer Manag. Res. 2014, 6, 357–363. [Google Scholar] [CrossRef]
- Yu, X.; Ruan, Y.; Huang, X.; Dou, L.; Lan, M.; Cui, J.; Chen, B.; Gong, H.; Wang, Q.; Yan, M.; et al. Dexrazoxane ameliorates doxorubicin-induced cardiotoxicity by inhibiting both apoptosis and necroptosis in cardiomyocytes. Biochem. Biophys. Res. Commun. 2020, 523, 140–146. [Google Scholar] [CrossRef]
- Venturini, M.; Michelotti, A.; Del Mastro, L.; Gallo, L.; Carnino, F.; Garrone, O.; Tibaldi, C.; Molea, N.; Bellina, R.C.; Pronzato, P.; et al. Multicenter randomized controlled clinical trial to evaluate cardioprotection of dexrazoxane versus no cardioprotection in women receiving epirubicin chemotherapy for advanced breast cancer. J. Clin. Oncol. 1996, 14, 3112–3120. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.; Espie, M.; Llombart, A.; Monnier, A.; Rapoport, B.L.; Stahalova, V.; Dexrazoxane Study Group. Multicenter randomized phase III study of the cardioprotective effect of dexrazoxane (Cardioxane®) in advanced/metastatic breast cancer patients treated with anthracycline-based chemotherapy. Ann. Oncol. 2006, 17, 614–622. [Google Scholar] [CrossRef]
- Safdar, Z.; Cho, E. Effect of spironolactone use in pulmonary arterial hypertension–analysis from pivotal trial databases. Pulm. Circ. 2021, 11, 20458940211045618. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Liu, G.Z.; Liu, Y.W.; Wang, R.J.; Hou, T.T.; Chen, C.Y.; Zheng, S.J.; Dong, Z.X. Spironolactone Attenuates Doxorubicin-induced Cardiotoxicity in Rats. Cardiovasc. Ther. 2016, 34, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Akpek, M.; Ozdogru, I.; Sahin, O.; Inanc, M.; Dogan, A.; Yazici, C.; Berk, V.; Karaca, H.; Kalay, N.; Oguzhan, A.; et al. Protective effects of spironolactone against anthracycline-induced cardiomyopathy. Eur. J. Heart Fail. 2015, 17, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Yavas, G.; Elsurer, R.; Yavas, C.; Elsurer, C.; Ata, O. Does spironolactone ameliorate trastuzumab-induced cardiac toxicity? Med. Hypotheses 2013, 81, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Kalay, N.; Basar, E.; Ozdogru, I.; Er, O.; Cetinkaya, Y.; Dogan, A.; Inanc, T.; Oguzhan, A.; Eryol, N.K.; Topsakal, R.; et al. Protective effects of carvedilol against anthracycline-induced cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 2258–2262. [Google Scholar] [CrossRef] [Green Version]
- Flesch, M.; Maack, C.; Cremers, B.; Baumer, A.T.; Sudkamp, M.; Bohm, M. Effect of beta-blockers on free radical-induced cardiac contractile dysfunction. Circulation 1999, 100, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Spallarossa, P.; Garibaldi, S.; Altieri, P.; Fabbi, P.; Manca, V.; Nasti, S.; Rossettin, P.; Ghigliotti, G.; Ballestrero, A.; Patrone, F.; et al. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J. Mol. Cell. Cardiol. 2004, 37, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.J.; Bjork, J.A.; Santos, M.S.; Leino, R.L.; Froberg, M.K.; Moreno, A.J.; Wallace, K.B. Carvedilol-mediated antioxidant protection against doxorubicin-induced cardiac mitochondrial toxicity. Toxicol. Appl. Pharmacol. 2004, 200, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.S.; Ayub-Ferreira, S.M.; de Barros Wanderley, M.R., Jr.; das Dores Cruz, F.; Goncalves Brandao, S.M.; Rigaud, V.O.C.; Higuchi-Dos-Santos, M.H.; Hajjar, L.A.; Kalil Filho, R.; Hoff, P.M.; et al. Carvedilol for Prevention of Chemotherapy-Related Cardiotoxicity: The CECCY Trial. J. Am. Coll. Cardiol. 2018, 71, 2281–2290. [Google Scholar] [CrossRef]
- Hunt, S.A.; Abraham, W.T.; Chin, M.H.; Feldman, A.M.; Francis, G.S.; Ganiats, T.G.; Jessup, M.; Konstam, M.A.; Mancini, D.M.; Michl, K.; et al. Focused Update Incorporated Into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines (vol 119, p. e391, 2009). Circulation 2009, 121, E258. [Google Scholar] [CrossRef]
- Swedberg, K.; Kjekshus, J. Effects of enalapril on mortality in severe congestive heart failure: Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). Am. J. Cardiol. 1988, 62, 60A–66A. [Google Scholar] [CrossRef]
- El-Aziz, M.A.A.; Othman, A.I.; Amer, M.; El-Missiry, M.A. Potential protective role of angiotensin-converting enzyme inhibitors captopril and enalapril against adriamycin-induced acute cardiac and hepatic toxicity in rats. J. Appl. Toxicol. 2001, 21, 469–473. [Google Scholar] [CrossRef]
- Janbabai, G.; Nabati, M.; Faghihinia, M.; Azizi, S.; Borhani, S.; Yazdani, J. Effect of Enalapril on Preventing Anthracycline-Induced Cardiomyopathy. Cardiovasc. Toxicol. 2017, 17, 130–139. [Google Scholar] [CrossRef]
- Jafari, M.; Mousavi, S.M.; Asgharzadeh, A.; Yazdani, N. Coenzyme Q10 in the treatment of heart failure: A systematic review of systematic reviews. Indian Heart J. 2018, 70 (Suppl. S1), S111–S117. [Google Scholar] [CrossRef]
- Conklin, K.A. Coenzyme q10 for prevention of anthracycline-induced cardiotoxicity. Integr. Cancer Ther. 2005, 4, 110–130. [Google Scholar] [CrossRef]
- Crestanello, J.A.; Doliba, N.M.; Doliba, N.M.; Babsky, A.M.; Niborii, K.; Osbakken, M.D.; Whitman, G.J. Effect of coenzyme Q10 supplementation on mitochondrial function after myocardial ischemia reperfusion. J. Surg. Res. 2002, 102, 221–228. [Google Scholar] [CrossRef]
- Botelho, A.F.M.; Lempek, M.R.; Branco, S.E.M.T.; Nogueira, M.M.; de Almeida, M.E.; Costa, A.G.; Freitas, T.G.; Rocha, M.C.R.C.; Moreira, M.V.L.; Barreto, T.O.; et al. Coenzyme Q10 Cardioprotective Effects Against Doxorubicin-Induced Cardiotoxicity in Wistar Rat. Cardiovasc. Toxicol. 2020, 20, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Hou, C.W.; Shibu, M.A.; Day, C.H.; Pai, P.Y.; Liu, Z.R.; Lin, T.Y.; Viswanadha, V.P.; Kuo, C.H.; Huang, C.Y. Protective effect of Co-enzyme Q10 On doxorubicin-induced cardiomyopathy of rat hearts. Environ. Toxicol. 2017, 32, 679–689. [Google Scholar] [CrossRef]
- Iarussi, D.; Auricchio, U.; Agretto, A.; Murano, A.; Giuliano, M.; Casale, F.; Indolfi, P.; Iacono, A. Protective effect of coenzyme Q10 on anthracyclines cardiotoxicity: Control study in children with acute lymphoblastic leukemia and non-Hodgkin lymphoma. Mol. Asp. Med. 1994, 15, s207–s212. [Google Scholar] [CrossRef]
- Shah, S.M.A.; Akram, M.; Riaz, M.; Munir, N.; Rasool, G. Cardioprotective Potential of Plant-Derived Molecules: A Scientific and Medicinal Approach. Dose Response 2019, 17, 1559325819852243. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Saibabu, V.; Fatima, Z.; Khan, L.A.; Hameed, S. Therapeutic Potential of Dietary Phenolic Acids. Adv. Pharmacol. Sci. 2015, 2015, 823539. [Google Scholar] [CrossRef] [PubMed]
- Hollman, P.C.H. Evidence for health benefits of plant phenols: Local or systemic effects? J. Sci. Food Agric. 2001, 81, 842–852. [Google Scholar] [CrossRef]
- Ho, C.C.; Huang, A.C.; Yu, C.S.; Lien, J.C.; Wu, S.H.; Huang, Y.P.; Huang, H.Y.; Kuo, J.H.; Liao, W.Y.; Yang, J.S.; et al. Ellagic acid induces apoptosis in TSGH8301 human bladder cancer cells through the endoplasmic reticulum stress- and mitochondria-dependent signaling pathways. Environ. Toxicol. 2014, 29, 1262–1274. [Google Scholar] [CrossRef]
- Kong, C.S.; Jeong, C.H.; Choi, J.S.; Kim, K.J.; Jeong, J.W. Antiangiogenic effects of p-coumaric acid in human endothelial cells. Phytother. Res. 2013, 27, 317–323. [Google Scholar] [CrossRef]
- Wang, L.; Li, W.F.; Lin, M.Q.; Garcia, M.; Mulholland, D.; Lilly, M.; Martins-Green, M. Luteolin, ellagic acid and punicic acid are natural products that inhibit prostate cancer metastasis. Carcinogenesis 2014, 35, 2321–2330. [Google Scholar] [CrossRef]
- Tangney, C.C.; Rasmussen, H.E. Polyphenols, Inflammation, and Cardiovascular Disease. Curr. Atheroscler. Rep. 2013, 15, 324. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhang, L.; Li, M.; Wu, W.; Yang, M.; Wang, J.; Guo, D.A. Salvianolic acids prevent acute doxorubicin cardiotoxicity in mice through suppression of oxidative stress. Food Chem. Toxicol. 2008, 46, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.C.; Yang, L.; Liu, Y.S.; He, J.X.; Yang, L.; Zhang, Q.; Liu, F.; Li, J.; Liu, J.; Sumi, S.; et al. Resveratrol attenuates doxorubicin-induced cardiotoxicity in rats by up-regulation of vascular endothelial growth factor B. J. Nutr. Biochem. 2020, 79, 108132. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Hu, W.; Zhang, D.D. Resveratrol, a polyphenol phytoalexin, protects against doxorubicin-induced cardiotoxicity. J. Cell. Mol. Med. 2015, 19, 2324–2328. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Xu, J.Z.; Dirain, M.L.S.; Leeuwenburgh, C. Calorie restriction combined with resveratrol induces autophagy and protects 26-month-old rat hearts from doxorubicin-induced toxicity (vol 74, p. 252, 2014). Free. Radic. Biol. Med. 2015, 81, 183. [Google Scholar] [CrossRef]
- Zong, A.Z.; Cao, H.Z.; Wang, F.S. Anticancer polysaccharides from natural resources: A review of recent research. Carbohydr. Polym. 2012, 90, 1395–1410. [Google Scholar] [CrossRef]
- Meng, X.; Liang, H.; Luo, L. Antitumor polysaccharides from mushrooms: A review on the structural characteristics, antitumor mechanisms and immunomodulating activities. Carbohydr. Res. 2016, 424, 30–41. [Google Scholar] [CrossRef]
- Dong, Q.; Lin, X.; Shen, L.; Feng, Y. The protective effect of herbal polysaccharides on ischemia-reperfusion injury. Int. J. Biol. Macromol. 2016, 92, 431–440. [Google Scholar] [CrossRef]
- Xu, F.; Li, X.; Xiao, X.; Liu, L.F.; Zhang, L.; Lin, P.P.; Zhang, S.L.; Li, Q.S. Effects of Ganoderma lucidum polysaccharides against doxorubicin-induced cardiotoxicity. Biomed. Pharmacother. 2017, 95, 504–512. [Google Scholar] [CrossRef]
- Cao, Y.; Ruan, Y.; Shen, T.; Huang, X.; Li, M.; Yu, W.; Zhu, Y.; Man, Y.; Wang, S.; Li, J. Astragalus polysaccharide suppresses doxorubicin-induced cardiotoxicity by regulating the PI3k/Akt and p38MAPK pathways. Oxid. Med. Cell. Longev. 2014, 2014, 674219. [Google Scholar] [CrossRef] [PubMed]
- Li, W.J.; Zhang, X.Y.; Wu, R.T.; Song, Y.H.; Xie, M.Y. Ganoderma atrum polysaccharide improves doxorubicin-induced cardiotoxicity in mice by regulation of apoptotic pathway in mitochondria. Carbohydr. Polym. 2018, 202, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.Y.; Shang, P.P.; Li, D.Y. Luteolin: A Flavonoid that Has Multiple Cardio-Protective Effects and Its Molecular Mechanisms. Front. Pharmacol. 2017, 8, 692. [Google Scholar] [CrossRef] [PubMed]
- Bisol, A.; de Campos, P.S.; Lamers, M.L. Flavonoids as anticancer therapies: A systematic review of clinical trials. Phytother. Res. 2020, 34, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Batra, P.; Sharma, A.K. Anti-cancer potential of flavonoids: Recent trends and future perspectives. 3 Biotech 2013, 3, 439–459. [Google Scholar] [CrossRef]
- Bondonno, C.P.; Yang, X.; Croft, K.D.; Considine, M.J.; Ward, N.C.; Rich, L.; Puddey, I.B.; Swinny, E.; Mubarak, A.; Hodgson, J.M. Flavonoid-rich apples and nitrate-rich spinach augment nitric oxide status and improve endothelial function in healthy men and women: A randomized controlled trial. Free Radic. Biol. Med. 2012, 52, 95–102. [Google Scholar] [CrossRef]
- Wang, C.Z.; Mehendale, S.R.; Calway, T.; Yuan, C.S. Botanical flavonoids on coronary heart disease. Am. J. Chin. Med. 2011, 39, 661–671. [Google Scholar] [CrossRef]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Busselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef]
- Mantawy, E.M.; Esmat, A.; El-Bakly, W.M.; Salah ElDin, R.A.; El-Demerdash, E. Mechanistic clues to the protective effect of chrysin against doxorubicin-induced cardiomyopathy: Plausible roles of p53, MAPK and AKT pathways. Sci. Rep. 2017, 7, 4795. [Google Scholar] [CrossRef]
- Qi, W.; Boliang, W.; Xiaoxi, T.; Guoqiang, F.; Jianbo, X.; Gang, W. Cardamonin protects against doxorubicin-induced cardiotoxicity in mice by restraining oxidative stress and inflammation associated with Nrf2 signaling. Biomed. Pharmacother. 2020, 122, 109547. [Google Scholar] [CrossRef]
- Zhai, J.; Tao, L.; Zhang, S.; Gao, H.; Zhang, Y.; Sun, J.; Song, Y.; Qu, X. Calycosin ameliorates doxorubicin-induced cardiotoxicity by suppressing oxidative stress and inflammation via the sirtuin 1-NOD-like receptor protein 3 pathway. Phytother. Res. 2020, 34, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Elekofehinti, O.O.; Iwaloye, O.; Olawale, F.; Ariyo, E.O. Saponins in Cancer Treatment: Current Progress and Future Prospects. Pathophysiology 2021, 28, 250–272. [Google Scholar] [CrossRef] [PubMed]
- Ragupathi, G.; Gardner, J.R.; Livingston, P.O.; Gin, D.Y. Natural and synthetic saponin adjuvant QS-21 for vaccines against cancer. Expert Rev. Vaccines 2011, 10, 463–470. [Google Scholar] [CrossRef]
- Sun, H.X.; Xie, Y.; Ye, Y.P. Advances in saponin-based adjuvants. Vaccine 2009, 27, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.J.; Duan, J.L.; Wu, X.X.; Guo, C.; Yin, Y.; Zhu, Y.R.; Hu, T.X.; Wei, G.; Wen, A.D.; Xi, M.M. Total saponins from Aralia taibaiensis protect against myocardial ischemia/reperfusion injury through AMPK pathway. Int. J. Mol. Med. 2015, 36, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Ning, K.; Jiang, L.; Hu, T.; Wang, X.; Liu, A.; Bao, Y. ATP-Sensitive Potassium Channels Mediate the Cardioprotective Effect of Panax notoginseng Saponins against Myocardial Ischaemia-Reperfusion Injury and Inflammatory Reaction. BioMed Res. Int. 2020, 2020, 3039184. [Google Scholar] [CrossRef]
- Dong, Y.; Duan, L.; Chen, H.W.; Liu, Y.M.; Zhang, Y.; Wang, J. Network Pharmacology-Based Prediction and Verification of the Targets and Mechanism for Panax Notoginseng Saponins against Coronary Heart Disease. Evid. Based Complement. Altern. Med. 2019, 2019, 6503752. [Google Scholar] [CrossRef]
- Wang, X.; Chen, L.; Wang, T.; Jiang, X.; Zhang, H.; Li, P.; Lv, B.; Gao, X. Ginsenoside Rg3 antagonizes adriamycin-induced cardiotoxicity by improving endothelial dysfunction from oxidative stress via upregulating the Nrf2-ARE pathway through the activation of akt. Phytomedicine 2015, 22, 875–884. [Google Scholar] [CrossRef]
- Lin, J.; Fang, L.; Li, H.; Li, Z.; Lyu, L.; Wang, H.; Xiao, J. Astragaloside IV alleviates doxorubicin induced cardiomyopathy by inhibiting NADPH oxidase derived oxidative stress. Eur. J. Pharmacol. 2019, 859, 172490. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Wu, S.S.; Chen, X.M.; Pi, J.K.; Cheng, Y.F.; Zhang, Y.; Wang, X.J.; Luo, D.; Zhou, J.H.; Xu, J.Y.; et al. Saikosaponin D Alleviates DOX-induced Cardiac Injury In Vivo and In Vitro. J. Cardiovasc. Pharmacol. 2022, 79, 558–567. [Google Scholar] [CrossRef]
- Yang, W.; Ma, L.; Li, S.; Cui, K.; Lei, L.; Ye, Z. Evaluation of the Cardiotoxicity of Evodiamine In Vitro and In Vivo. Molecules 2017, 22, 943. [Google Scholar] [CrossRef]
- Ye, Q.; Liu, H.; Fang, C.; Liu, Y.; Liu, X.; Liu, J.; Zhang, C.; Zhang, T.; Peng, C.; Guo, L. Cardiotoxicity evaluation and comparison of diterpene alkaloids on zebrafish. Drug Chem. Toxicol. 2021, 44, 294–301. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Yu, Y.Y.; Wang, S.F.; Zhang, Q.; Wu, H.W.; Wei, J.Y.; Yang, W.; Li, S.Y.; Yang, H.J. Cardiotoxicity evaluation of nine alkaloids from Rhizoma Coptis. Hum. Exp. Toxicol. 2018, 37, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, S.; Wang, K.; Bao, X.; Liu, Y.; Zhou, S.; Liu, H.; Qiu, Y.; Wang, T.; Yu, H. Alkaloids from Traditional Chinese Medicine against hepatocellular carcinoma. Biomed. Pharmacother. 2019, 120, 109543. [Google Scholar] [CrossRef] [PubMed]
- Zou, K.; Li, Z.; Zhang, Y.; Zhang, H.Y.; Li, B.; Zhu, W.L.; Shi, J.Y.; Jia, Q.; Li, Y.M. Advances in the study of berberine and its derivatives: A focus on anti-inflammatory and anti-tumor effects in the digestive system. Acta Pharmacol. Sin. 2017, 38, 157–167. [Google Scholar] [CrossRef]
- Wang, S.; Liu, H.; Zhang, Y.; Ren, L. Efficacy of Alkaloids in Alleviating Myocardial Ischemia-Reperfusion Injury in Rats: A Meta-Analysis of Animal Studies. BioMed Res. Int. 2021, 2021, 6661526. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Kar, A.; Sharma, P.; Sharma, A. Cardioprotective potential of N,alpha-L-rhamnopyranosyl vincosamide, an indole alkaloid, isolated from the leaves of Moringa oleifera in isoproterenol induced cardiotoxic rats: In vivo and in vitro studies. Bioorg. Med. Chem. Lett. 2013, 23, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Sastre Torano, J.; Ramautar, R.; de Jong, G. Advances in capillary electrophoresis for the life sciences. J. Chromatogr. B 2019, 1118–1119, 116–136. [Google Scholar] [CrossRef]
- Samadi, P.; Sarvarian, P.; Gholipour, E.; Asenjan, K.S.; Aghebati-Maleki, L.; Motavalli, R.; Hojjat-Farsangi, M.; Yousefi, M. Berberine: A novel therapeutic strategy for cancer. IUBMB Life 2020, 72, 2065–2079. [Google Scholar] [CrossRef]
- Cai, Y.; Xin, Q.; Lu, J.; Miao, Y.; Lin, Q.; Cong, W.; Chen, K. A New Therapeutic Candidate for Cardiovascular Diseases: Berberine. Front. Pharmacol. 2021, 12, 631100. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, J.; Tong, N.; Liao, X.; Wang, E.; Li, Z.; Luo, Y.; Zuo, H. Berberine attenuates doxorubicin-induced cardiotoxicity in mice. J. Int. Med. Res. 2011, 39, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Bharathi Priya, L.; Baskaran, R.; Huang, C.Y.; Vijaya Padma, V. Neferine modulates IGF-1R/Nrf2 signaling in doxorubicin treated H9c2 cardiomyoblasts. J. Cell. Biochem. 2018, 119, 1441–1452. [Google Scholar] [CrossRef]
- Wu, Y.Z.; Zhang, L.; Wu, Z.X.; Shan, T.T.; Xiong, C. Berberine Ameliorates Doxorubicin-Induced Cardiotoxicity via a SIRT1/p66Shc-Mediated Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 2150394. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Qiu, H.; Li, C.S.; Cai, P.P.; Qi, F.H. The positive role of traditional Chinese medicine as an adjunctive therapy for cancer. Biosci. Trends 2021, 15, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Hao, P.; Jiang, F.; Cheng, J.; Ma, L.; Zhang, Y.; Zhao, Y. Traditional Chinese Medicine for Cardiovascular Disease: Evidence and Potential Mechanisms. J. Am. Coll. Cardiol. 2017, 69, 2952–2966. [Google Scholar] [CrossRef]
- Cui, Y.; Li, C.; Zeng, C.; Li, J.; Zhu, Z.; Chen, W.; Huang, A.; Qi, X. Tongmai Yangxin pills anti-oxidative stress alleviates cisplatin-induced cardiotoxicity: Network pharmacology analysis and experimental evidence. Biomed. Pharmacother. 2018, 108, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Liu, S.; Qin, Y.; Sun, C.; Chen, L.; Wu, C.; Bao, Y. Cardioprotective effect of Platycodon grandiflorum in patients with early breast cancer receiving anthracycline-based chemotherapy: Study protocol for a randomized controlled trial. Trials 2017, 18, 386. [Google Scholar] [CrossRef]
- Shah, S.; Mohan, M.; Kasture, S.; Ballero, M.; Maxia, A.; Sanna, C. Protective effect of Hypericum hircinum on doxorubicin-induced cardiotoxicity in rats. Nat. Prod. Res. 2013, 27, 1502–1507. [Google Scholar] [CrossRef]
- Xin, Y.F.; Zhou, G.L.; Deng, Z.Y.; Chen, Y.X.; Wu, Y.G.; Xu, P.S.; Xuan, Y.X. Protective effect of Lycium barbarum on doxorubicin-induced cardiotoxicity. Phytother. Res. 2007, 21, 1020–1024. [Google Scholar] [CrossRef]
- Wu, B.Y.; Liu, C.T.; Chen, S.Y.; Tsai, M.Y. A case of chemotherapy-induced congestive heart failure successfully treated with Chinese herbal medicine. Complement. Ther. Med. 2015, 23, 251–256. [Google Scholar] [CrossRef]
- Jagetia, G.C.; Reddy, T.K.; Malagi, K.J.; Nayak, B.S.; Naidu, M.B.; Ravikiran, P.B.; Kamath, S.U.; Shetty, P.C.; Reddy, D.S. Antarth, a polyherbal preparation protects against the doxorubicin-induced toxicity without compromising its Antineoplastic activity. Phytother. Res. 2005, 19, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Guo, J.; Yang, R.; Peng, H.; Zhao, J.; Li, L.; Peng, S. Cyclovirobuxine D Attenuates Doxorubicin-Induced Cardiomyopathy by Suppression of Oxidative Damage and Mitochondrial Biogenesis Impairment. Oxid. Med. Cell. Longev. 2015, 2015, 151972. [Google Scholar] [CrossRef] [PubMed]
- Hasin, T.; Gerber, Y.; McNallan, S.M.; Weston, S.A.; Kushwaha, S.S.; Nelson, T.J.; Cerhan, J.R.; Roger, V.L. Patients with heart failure have an increased risk of incident cancer. J. Am. Coll. Cardiol. 2013, 62, 881–886. [Google Scholar] [CrossRef] [PubMed]
- de Wit, S.; de Boer, R.A. From Studying Heart Disease and Cancer Simultaneously to Reverse Cardio-Oncology. Circulation 2021, 144, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Newman, A.A.C.; Afonso, M.S.; van Solingen, C.; Corr, E.M.; Brown, E.J.; Albers, K.B.; Yamaguchi, N.; Narke, D.; Schlegel, M.; et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat. Med. 2020, 26, 1452–1458. [Google Scholar] [CrossRef]
- Avraham, S.; Abu-Sharki, S.; Shofti, R.; Haas, T.; Korin, B.; Kalfon, R.; Friedman, T.; Shiran, A.; Saliba, W.; Shaked, Y.; et al. Early Cardiac Remodeling Promotes Tumor Growth and Metastasis. Circulation 2020, 142, 670–683. [Google Scholar] [CrossRef]
- Awwad, L.; Goldenberg, T.; Langier-Goncalves, I.; Aronheim, A. Cardiac Remodeling in the Absence of Cardiac Contractile Dysfunction Is Sufficient to Promote Cancer Progression. Cells 2022, 11, 1108. [Google Scholar] [CrossRef]
- Awwad, L.; Aronheim, A. Cardiac Dysfunction Promotes Cancer Progression via Multiple Secreted Factors. Cancer Res. 2022, 82, 1753–1761. [Google Scholar] [CrossRef]
- Shi, C.; Aboumsallem, J.P.; de Wit, S.; Schouten, E.M.; Bracun, V.; Meijers, W.C.; Sillje, H.H.W.; de Boer, R.A. Evaluation of renal cancer progression in a mouse model of heart failure. Cancer Commun. 2021, 41, 796–799. [Google Scholar] [CrossRef]
- Katoh, M. Cardio-miRNAs and onco-miRNAs: Circulating miRNA-based diagnostics for non-cancerous and cancerous diseases. Front. Cell Dev. Biol. 2014, 2, 61. [Google Scholar] [CrossRef] [Green Version]
- van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Liao, X.; Ye, C.; Yin, X.; Liu, M.; Hong, Y.; Yu, M.; Liu, Y.; Liang, H.; Zhang, C.Y.; et al. ING5 suppresses breast cancer progression and is regulated by miR-24. Mol. Cancer 2017, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.; Xu, H.; Zhou, S.; Zheng, Y.; Keller, B.B.; Cai, L. Emerging roles of microRNA-208a in cardiology and reverse cardio-oncology. Med. Res. Rev. 2021, 41, 2172–2194. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Z.; He, Y.; Hu, X. Cardio-Oncology: Mechanisms, Drug Combinations, and Reverse Cardio-Oncology. Int. J. Mol. Sci. 2022, 23, 10617. https://doi.org/10.3390/ijms231810617
Liang Z, He Y, Hu X. Cardio-Oncology: Mechanisms, Drug Combinations, and Reverse Cardio-Oncology. International Journal of Molecular Sciences. 2022; 23(18):10617. https://doi.org/10.3390/ijms231810617
Chicago/Turabian StyleLiang, Zehua, Yuquan He, and Xin Hu. 2022. "Cardio-Oncology: Mechanisms, Drug Combinations, and Reverse Cardio-Oncology" International Journal of Molecular Sciences 23, no. 18: 10617. https://doi.org/10.3390/ijms231810617