
RNA Polymerase II “Pause” Prepares Promoters for Upcoming Transcription during Drosophila Development

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Tissue Collection and Developmental Stage Verification Using Marker Genes Transcription
2.2. Genes Induced at 6–8 h of Embryogenesis Moderately Use the “Paused” RNA Polymerase II to Prepare for the Upcoming Transcription
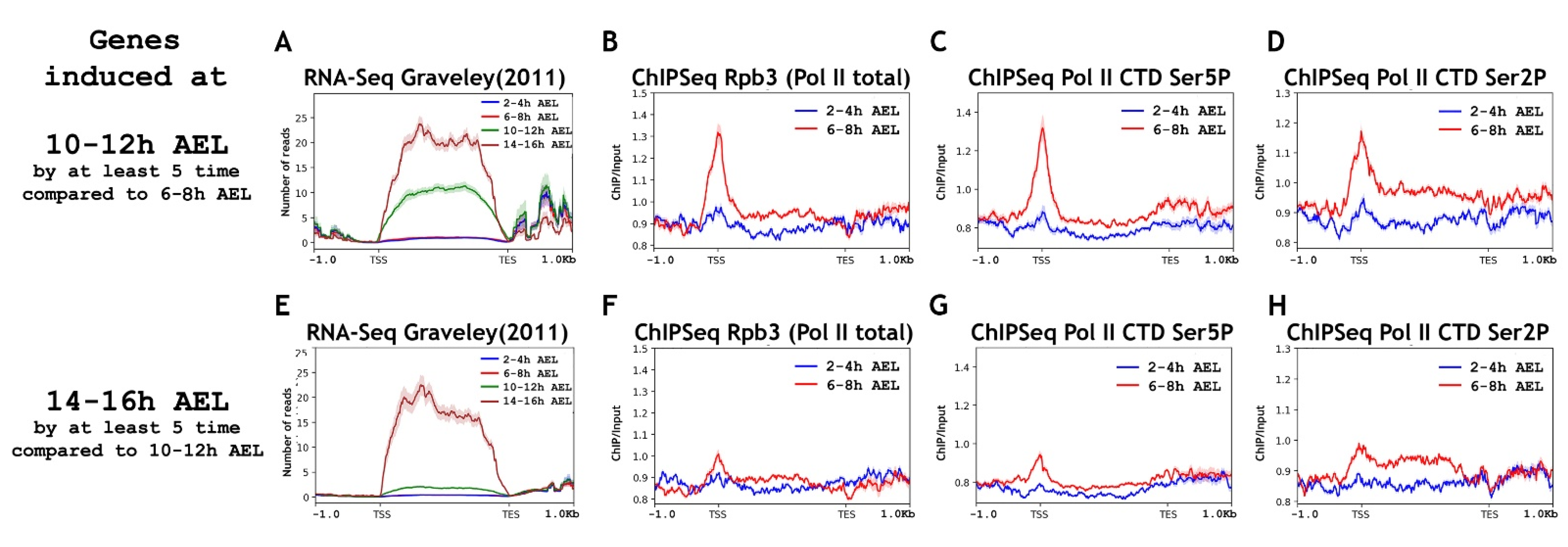
2.3. Genes Induced at 10–12 h AEL, but Not Genes Induced at 14–16 h AEL, Possess “Paused” Pol II on the Promoters at 6–8 h AEL
2.4. Brd4/Fs(1)h Protein Is Maternity Loaded and Its Expression Is Delayed until the Mid-Embryogenesis
2.5. Genes Induced during Metamorphosis Use Promoter-Proximal Pausing to Prepare Promoters for Upcoming Transcription
2.6. The Pool of Ecdysone-Dependent Genes That Is Iteratively Induced during Development Is Controlled by Promoter-Proximal Pausing
3. Discussion
3.1. Genes That become Active in Mid-Embryogenesis and in the Early Stages of Metamorphosis Use Elongation Control to Prepare Promoters for Transcription
3.2. Elongation Regulators Are Recruited to Promoters during Drosophila Development Only When RNA Polymerase II Is Recruited
3.3. Release of Developmental Genes from Elongation Control
4. Conclusions
5. Materials and Methods
5.1. Collection of the Material Corresponding to Different Drosophila Developmental Stages
5.2. ChIP-Seq Analysis
5.3. Antibodies and Western Blotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Darbo, E.; Herrmann, C.; Lecuit, T.; Thieffry, D.; van Helden, J. Transcriptional and Epigenetic Signatures of Zygotic Genome Activation during Early Drosophila Embryogenesis. BMC Genom. 2013, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Papatsenko, I.; Levine, M.; Papatsenko, D. Temporal Waves of Coherent Gene Expression during Drosophila Embryogenesis. Bioinformatics 2010, 26, 2731–2736. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural Basis of Transcription Initiation by RNA Polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Jishage, M.; Yu, X.; Shi, Y.; Ganesan, S.J.; Chen, W.-Y.; Sali, A.; Chait, B.T.; Asturias, F.J.; Roeder, R.G. Architecture of Pol II(G) and Molecular Mechanism of Transcription Regulation by Gdown1. Nat. Struct. Mol. Biol. 2018, 25, 859–867. [Google Scholar] [CrossRef]
- Eick, D.; Geyer, M. The RNA Polymerase II Carboxy-Terminal Domain (CTD) Code. Chem. Rev. 2013, 113, 8456–8490. [Google Scholar] [CrossRef]
- Mayfield, J.E.; Robinson, M.R.; Cotham, V.C.; Irani, S.; Matthews, W.L.; Ram, A.; Gilmour, D.S.; Cannon, J.R.; Zhang, Y.J.; Brodbelt, J.S. Mapping the Phosphorylation Pattern of Drosophila Melanogaster RNA Polymerase II Carboxyl-Terminal Domain Using Ultraviolet Photodissociation Mass Spectrometry. ACS Chem. Biol. 2017, 12, 153–162. [Google Scholar] [CrossRef]
- Petrenko, N.; Jin, Y.; Wong, K.H.; Struhl, K. Mediator Undergoes a Compositional Change during Transcriptional Activation. Mol. Cell 2016, 64, 443–454. [Google Scholar] [CrossRef]
- Nilson, K.A.; Guo, J.; Turek, M.E.; Brogie, J.E.; Delaney, E.; Luse, D.S.; Price, D.H. THZ1 Reveals Roles for Cdk7 in Co-Transcriptional Capping and Pausing. Mol. Cell 2015, 59, 576–587. [Google Scholar] [CrossRef]
- Wong, K.H.; Jin, Y.; Struhl, K. TFIIH Phosphorylation of the Pol II CTD Stimulates Mediator Dissociation from the Preinitiation Complex and Promoter Escape. Mol. Cell 2014, 54, 601–612. [Google Scholar] [CrossRef]
- Buratowski, S. Progression through the RNA Polymerase II CTD Cycle. Mol. Cell 2009, 36, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Ebmeier, C.C.; Erickson, B.; Allen, B.L.; Allen, M.A.; Kim, H.; Fong, N.; Jacobsen, J.R.; Liang, K.; Shilatifard, A.; Dowell, R.D.; et al. Human TFIIH Kinase CDK7 Regulates Transcription-Associated Chromatin Modifications. Cell Rep. 2017, 20, 1173–1186. [Google Scholar] [CrossRef]
- Guo, Y.E.; Manteiga, J.C.; Henninger, J.E.; Sabari, B.R.; Dall’Agnese, A.; Hannett, N.M.; Spille, J.-H.; Afeyan, L.K.; Zamudio, A.V.; Shrinivas, K.; et al. Pol II Phosphorylation Regulates a Switch between Transcriptional and Splicing Condensates. Nature 2019, 572, 543–548. [Google Scholar] [CrossRef]
- Adelman, K.; Lis, J.T. Promoter-Proximal Pausing of RNA Polymerase II: Emerging Roles in Metazoans. Nat. Rev. Genet. 2012, 13, 720–731. [Google Scholar] [CrossRef]
- Core, L.; Adelman, K. Promoter-Proximal Pausing of RNA Polymerase II: A Nexus of Gene Regulation. Genes Dev. 2019, 33, 960–982. [Google Scholar] [CrossRef]
- Duarte, F.M.; Fuda, N.J.; Mahat, D.B.; Core, L.J.; Guertin, M.J.; Lis, J.T. Transcription Factors GAF and HSF Act at Distinct Regulatory Steps to Modulate Stress-Induced Gene Activation. Genes Dev. 2016, 30, 1731–1746. [Google Scholar] [CrossRef]
- Aoi, Y.; Takahashi, Y.; Shah, A.P.; Iwanaszko, M.; Rendleman, E.J.; Khan, N.H.; Cho, B.-K.; Goo, Y.A.; Ganesan, S.; Kelleher, N.L.; et al. SPT5 Stabilization of Promoter-Proximal RNA Polymerase II. Mol. Cell 2021, 81, 4413–4424.e5. [Google Scholar] [CrossRef]
- Gilchrist, D.A.; Fromm, G.; dos Santos, G.; Pham, L.N.; McDaniel, I.E.; Burkholder, A.; Fargo, D.C.; Adelman, K. Regulating the Regulators: The Pervasive Effects of Pol II Pausing on Stimulus-Responsive Gene Networks. Genes Dev. 2012, 26, 933–944. [Google Scholar] [CrossRef]
- Levine, M. Paused RNA Polymerase II as a Developmental Checkpoint. Cell 2011, 145, 502–511. [Google Scholar] [CrossRef]
- Vorobyeva, N.E.; Mazina, M.Y. The Elongation Regulators and Architectural Proteins as New Participants of Eukaryotic Gene Transcription. Russ. J. Genet. 2021, 57, 751–763. [Google Scholar] [CrossRef]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a Novel Transcription Elongation Factor That Regulates RNA Polymerase II Processivity, Is Composed of Human Spt4 and Spt5 Homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sansó, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin-Dependent Kinase Control of the Initiation-to-Elongation Switch of RNA Polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef]
- Schulz, S.; Gietl, A.; Smollett, K.; Tinnefeld, P.; Werner, F.; Grohmann, D. TFE and Spt4/5 Open and Close the RNA Polymerase Clamp during the Transcription Cycle. Proc. Natl. Acad. Sci. USA 2016, 113, E1816–E1825. [Google Scholar] [CrossRef]
- Vos, S.M.; Farnung, L.; Urlaub, H.; Cramer, P. Structure of Paused Transcription Complex Pol II–DSIF–NELF. Nature 2018, 560, 601–606. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Inukai, N.; Narita, T.; Wada, T.; Handa, H. Evidence That Negative Elongation Factor Represses Transcription Elongation through Binding to a DRB Sensitivity-Inducing Factor/RNA Polymerase II Complex and RNA. Mol. Cell. Biol. 2002, 22, 2918–2927. [Google Scholar] [CrossRef]
- Vos, S.M.; Pöllmann, D.; Caizzi, L.; Hofmann, K.B.; Rombaut, P.; Zimniak, T.; Herzog, F.; Cramer, P. Architecture and RNA Binding of the Human Negative Elongation Factor. eLife 2016, 5, e14981. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a Multisubunit Complex Containing RD, Cooperates with DSIF to Repress RNA Polymerase II Elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Vos, S.M.; Farnung, L.; Boehning, M.; Wigge, C.; Linden, A.; Urlaub, H.; Cramer, P. Structure of Activated Transcription Complex Pol II–DSIF–PAF–SPT6. Nature 2018, 560, 607–612. [Google Scholar] [CrossRef]
- Chen, F.; Gao, X.; Shilatifard, A. Stably Paused Genes Revealed through Inhibition of Transcription Initiation by the TFIIH Inhibitor Triptolide. Genes Dev. 2015, 29, 39–47. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shibata, H.; Handa, H. Transcription Elongation Factors DSIF and NELF: Promoter-Proximal Pausing and Beyond. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2013, 1829, 98–104. [Google Scholar] [CrossRef]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to Run: Control of Transcription Elongation by RNA Polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef]
- Gaub, A.; Sheikh, B.N.; Basilicata, M.F.; Vincent, M.; Nizon, M.; Colson, C.; Bird, M.J.; Bradner, J.E.; Thevenon, J.; Boutros, M.; et al. Evolutionary Conserved NSL Complex/BRD4 Axis Controls Transcription Activation via Histone Acetylation. Nat. Commun. 2020, 11, 2243. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Kockmann, T.; Gerstung, M.; Schlumpf, T.; Xhinzhou, Z.; Hess, D.; Beerenwinkel, N.; Beisel, C.; Paro, R. The BET Protein FSH Functionally Interacts with ASH1 to Orchestrate Global Gene Activity in Drosophila. Genome Biol. 2013, 14, R18. [Google Scholar] [CrossRef] [PubMed]
- Chetverina, D.; Vorobyeva, N.E.; Mazina, M.Y.; Fab, L.V.; Lomaev, D.; Golovnina, A.; Mogila, V.; Georgiev, P.; Ziganshin, R.H.; Erokhin, M. Comparative Interactome Analysis of the PRE DNA-Binding Factors: Purification of the Combgap-, Zeste-, Psq-, and Adf1-Associated Proteins. Cell. Mol. Life Sci. 2022, 79, 353. [Google Scholar] [CrossRef]
- Cubeñas-Potts, C.; Rowley, M.J.; Lyu, X.; Li, G.; Lei, E.P.; Corces, V.G. Different Enhancer Classes in Drosophila Bind Distinct Architectural Proteins and Mediate Unique Chromatin Interactions and 3D Architecture. Nucleic Acids Res. 2017, 45, 1714–1730. [Google Scholar] [CrossRef]
- Kuroda, M.I.; Kang, H.; De, S.; Kassis, J.A. Dynamic Competition of Polycomb and Trithorax in Transcriptional Programming. Annu. Rev. Biochem. 2020, 89, 235–253. [Google Scholar] [CrossRef]
- Kininis, M.; Isaacs, G.D.; Core, L.J.; Hah, N.; Kraus, W.L. Postrecruitment Regulation of RNA Polymerase II Directs Rapid Signaling Responses at the Promoters of Estrogen Target Genes. Mol. Cell. Biol. 2009, 29, 1123–1133. [Google Scholar] [CrossRef]
- Phatnani, H.P.; Greenleaf, A.L. Phosphorylation and Functions of the RNA Polymerase II CTD. Genes Dev. 2006, 20, 2922–2936. [Google Scholar] [CrossRef]
- Aoi, Y.; Smith, E.R.; Shah, A.P.; Rendleman, E.J.; Marshall, S.A.; Woodfin, A.R.; Chen, F.X.; Shiekhattar, R.; Shilatifard, A. NELF Regulates a Promoter-Proximal Step Distinct from RNA Pol II Pause-Release. Mol. Cell 2020, 78, 261–274.e5. [Google Scholar] [CrossRef]
- Smolle, M.; Workman, J.L. Transcription-Associated Histone Modifications and Cryptic Transcription. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2013, 1829, 84–97. [Google Scholar] [CrossRef]
- Wang, Z.; Song, A.; Xu, H.; Hu, S.; Tao, B.; Peng, L.; Wang, J.; Li, J.; Yu, J.; Wang, L.; et al. Coordinated Regulation of RNA Polymerase II Pausing and Elongation Progression by PAF1. Sci. Adv. 2022, 8, eabm5504. [Google Scholar] [CrossRef]
- Aoi, Y.; Shah, A.P.; Ganesan, S.; Soliman, S.H.A.; Cho, B.-K.; Goo, Y.A.; Kelleher, N.L.; Shilatifard, A. SPT6 Functions in Transcriptional Pause/Release via PAF1C Recruitment. Mol. Cell 2022, in press. [CrossRef]
- Hendrix, D.A.; Hong, J.-W.; Zeitlinger, J.; Rokhsar, D.S.; Levine, M.S. Promoter Elements Associated with RNA Pol II Stalling in the Drosophila Embryo. Proc. Natl. Acad. Sci. USA 2008, 105, 7762–7767. [Google Scholar] [CrossRef]
- Zeitlinger, J.; Stark, A.; Kellis, M.; Hong, J.-W.; Nechaev, S.; Adelman, K.; Levine, M.; Young, R.A. RNA Polymerase Stalling at Developmental Control Genes in the Drosophila Melanogaster Embryo. Nat. Genet. 2007, 39, 1512–1516. [Google Scholar] [CrossRef]
- Bothma, J.P.; Garcia, H.G.; Esposito, E.; Schlissel, G.; Gregor, T.; Levine, M. Dynamic Regulation of Eve Stripe 2 Expression Reveals Transcriptional Bursts in Living Drosophila Embryos. Proc. Natl. Acad. Sci. USA 2014, 111, 10598–10603. [Google Scholar] [CrossRef]
- Lagha, M.; Bothma, J.P.; Esposito, E.; Ng, S.; Stefanik, L.; Tsui, C.; Johnston, J.; Chen, K.; Gilmour, D.S.; Zeitlinger, J.; et al. Paused Pol II Coordinates Tissue Morphogenesis in the Drosophila Embryo. Cell 2013, 153, 976–987. [Google Scholar] [CrossRef]
- Gaertner, B.; Johnston, J.; Chen, K.; Wallaschek, N.; Paulson, A.; Garruss, A.S.; Gaudenz, K.; De Kumar, B.; Krumlauf, R.; Zeitlinger, J. Poised RNA Polymerase II Changes over Developmental Time and Prepares Genes for Future Expression. Cell Rep. 2012, 2, 1670–1683. [Google Scholar] [CrossRef]
- Mazina, M.Y.; Kovalenko, E.V.; Vorobyeva, N.E. The Negative Elongation Factor NELF Promotes Induced Transcriptional Response of Drosophila Ecdysone-Dependent Genes. Sci. Rep. 2021, 11, 172. [Google Scholar] [CrossRef]
- Ghavi-Helm, Y.; Klein, F.A.; Pakozdi, T.; Ciglar, L.; Noordermeer, D.; Huber, W.; Furlong, E.E.M. Enhancer Loops Appear Stable during Development and Are Associated with Paused Polymerase. Nature 2014, 512, 96–100. [Google Scholar] [CrossRef]
- McKay, D.J.; Lieb, J.D. A Common Set of DNA Regulatory Elements Shapes Drosophila Appendages. Dev. Cell 2013, 27, 306–318. [Google Scholar] [CrossRef] [Green Version]
- Graveley, B.R.; Brooks, A.N.; Carlson, J.W.; Duff, M.O.; Landolin, J.M.; Yang, L.; Artieri, C.G.; van Baren, M.J.; Boley, N.; Booth, B.W.; et al. The Developmental Transcriptome of Drosophila Melanogaster. Nature 2011, 471, 473–479. [Google Scholar] [CrossRef]
- Fitz, J.; Neumann, T.; Pavri, R. Regulation of RNA Polymerase II Processivity by Spt5 Is Restricted to a Narrow Window during Elongation. EMBO J. 2018, 37, e97965. [Google Scholar] [CrossRef]
- Mazina, M.Y.; Derevyanko, P.K.; Kocheryzhkina, E.V.; Nikolenko, Y.V.; Krasnov, A.N.; Vorobyeva, N.E. Coactivator Complexes Participate in Different Stages of the Drosophila Melanogaster Hsp70 Gene Transcription. Russ. J. Genet. 2017, 2, 178–186. [Google Scholar] [CrossRef]
- Shetty, A.; Kallgren, S.P.; Demel, C.; Maier, K.C.; Spatt, D.; Alver, B.H.; Cramer, P.; Park, P.J.; Winston, F. Spt5 Plays Vital Roles in the Control of Sense and Antisense Transcription Elongation. Mol. Cell 2017, 66, 77–88.e5. [Google Scholar] [CrossRef]
- Stoiber, M.; Celniker, S.; Cherbas, L.; Brown, B.; Cherbas, P. Diverse Hormone Response Networks in 41 Independent Drosophila Cell Lines. G3 2016, 6, 683–694. [Google Scholar] [CrossRef]
- He, Q.; Johnston, J.; Zeitlinger, J. ChIP-Nexus Enables Improved Detection of in Vivo Transcription Factor Binding Footprints. Nat. Biotechnol. 2015, 33, 395–401. [Google Scholar] [CrossRef]
- Bacon, C.W.; D’Orso, I. CDK9: A Signaling Hub for Transcriptional Control. Transcription 2019, 10, 57–75. [Google Scholar] [CrossRef]
- Merkel, P.; Khoury, N.; Bertolotto, C.; Perfetti, R. Insulin and Glucose Regulate the Expression of the DNA Repair Enzyme XPD. Mol. Cell. Endocrinol. 2003, 201, 75–85. [Google Scholar] [CrossRef]
- Donnio, L.-M.; Miquel, C.; Vermeulen, W.; Giglia-Mari, G.; Mari, P.-O. Cell-Type Specific Concentration Regulation of the Basal Transcription Factor TFIIH in XPBy/y Mice Model. Cancer Cell Int. 2019, 19, 237. [Google Scholar] [CrossRef] [PubMed]
- Vorobyeva, N.E.; Erokhin, M.; Chetverina, D.; Krasnov, A.N.; Mazina, M.Y. Su(Hw) Primes 66D and 7F Drosophila Chorion Genes Loci for Amplification through Chromatin Decondensation. Sci. Rep. 2021, 11, 16963. [Google Scholar] [CrossRef] [PubMed]
- Vorobyeva, N.E.; Mazina, M.U.; Golovnin, A.K.; Kopytova, D.V.; Gurskiy, D.Y.; Nabirochkina, E.N.; Georgieva, S.G.; Georgiev, P.G.; Krasnov, A.N. Insulator Protein Su(Hw) Recruits SAGA and Brahma Complexes and Constitutes Part of Origin Recognition Complex-Binding Sites in the Drosophila Genome. Nucleic Acids Res. 2013, 41, 5717–5730. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Celniker, S.E.; Dillon, L.A.L.; Gerstein, M.B.; Gunsalus, K.C.; Henikoff, S.; Karpen, G.H.; Kellis, M.; Lai, E.C.; Lieb, J.D.; MacAlpine, D.M.; et al. Unlocking the Secrets of the Genome. Nature 2009, 459, 927–930. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Mazina, M.Y.; Kovalenko, E.V.; Derevyanko, P.K.; Nikolenko, J.V.; Krasnov, A.N.; Vorobyeva, N.E. One Signal Stimulates Different Transcriptional Activation Mechanisms. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2018, 1861, 178–189. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazina, M.Y.; Kovalenko, E.V.; Evdokimova, A.A.; Erokhin, M.; Chetverina, D.; Vorobyeva, N.E. RNA Polymerase II “Pause” Prepares Promoters for Upcoming Transcription during Drosophila Development. Int. J. Mol. Sci. 2022, 23, 10662. https://doi.org/10.3390/ijms231810662
Mazina MY, Kovalenko EV, Evdokimova AA, Erokhin M, Chetverina D, Vorobyeva NE. RNA Polymerase II “Pause” Prepares Promoters for Upcoming Transcription during Drosophila Development. International Journal of Molecular Sciences. 2022; 23(18):10662. https://doi.org/10.3390/ijms231810662
Chicago/Turabian StyleMazina, Marina Yu., Elena V. Kovalenko, Aleksandra A. Evdokimova, Maksim Erokhin, Darya Chetverina, and Nadezhda E. Vorobyeva. 2022. "RNA Polymerase II “Pause” Prepares Promoters for Upcoming Transcription during Drosophila Development" International Journal of Molecular Sciences 23, no. 18: 10662. https://doi.org/10.3390/ijms231810662
APA StyleMazina, M. Y., Kovalenko, E. V., Evdokimova, A. A., Erokhin, M., Chetverina, D., & Vorobyeva, N. E. (2022). RNA Polymerase II “Pause” Prepares Promoters for Upcoming Transcription during Drosophila Development. International Journal of Molecular Sciences, 23(18), 10662. https://doi.org/10.3390/ijms231810662