

Chidamide plus Tyrosine Kinase Inhibitor Remodel the Tumor Immune Microenvironment and Reduce Tumor Progression When Combined with Immune Checkpoint Inhibitor in Naïve and Anti-PD-1 Resistant CT26-Bearing Mice
 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Anticancer Effect of VEGFR-TKIs Combined with Anti-PD-1 Ab in CT26-Bearing Mice
2.2. Anticancer Activity of Anti-PD-1 Antibody Combined with Cabozantinib or Regorafenib Plus Chidamide-k30 in CT26-Bearing Mice
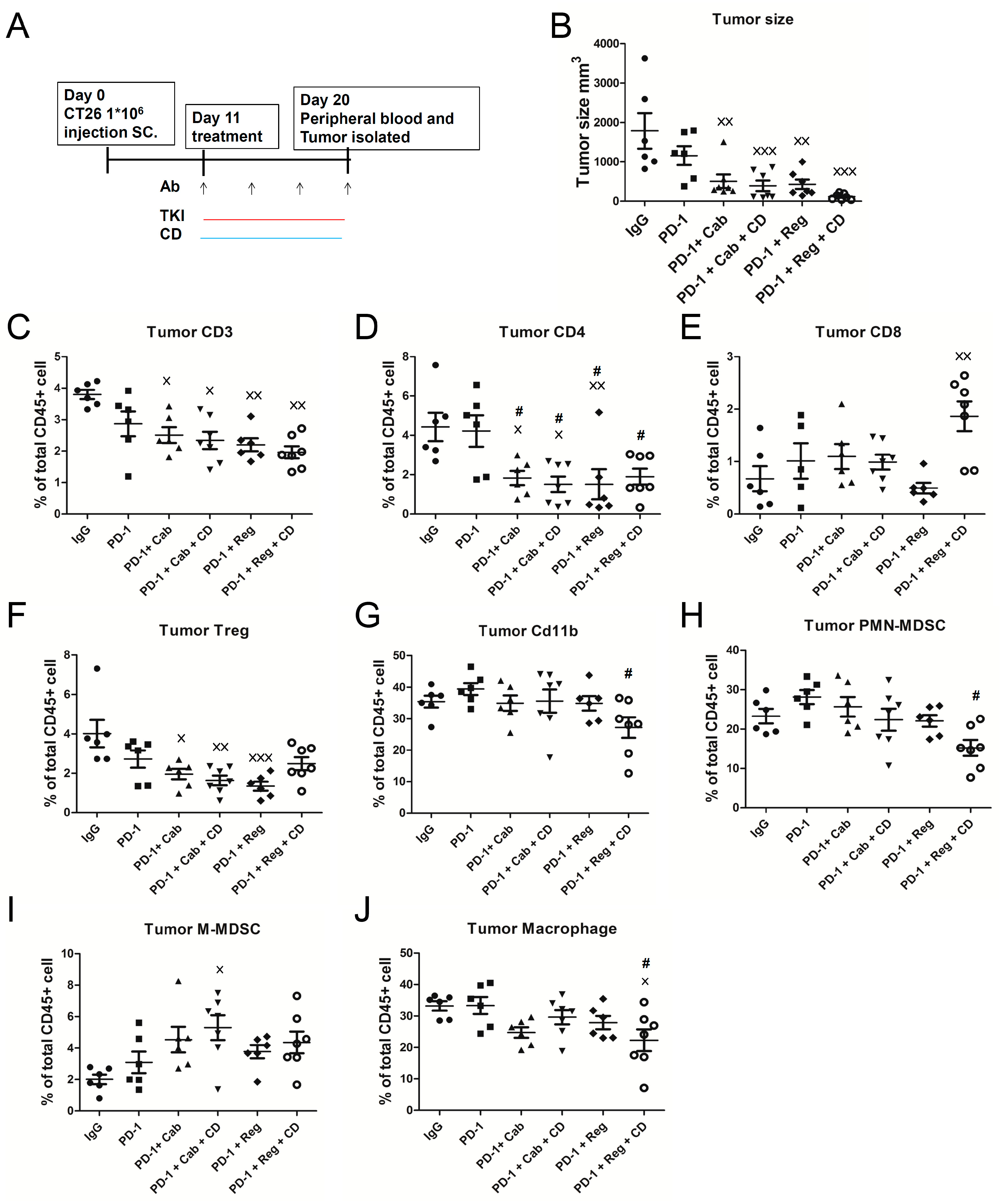
2.3. Chidamide Is a Key Component in Triplet Combination Regimens for Significantly Regulating Immune Cell Population and Gene Expression in the TME of CT26 Tumor-Bearing Mice
2.4. Anticancer Activity of Several ICIs Combined with Cabozantinib/Regorafenib Plus Chidamide in CT26-Bearing Mice
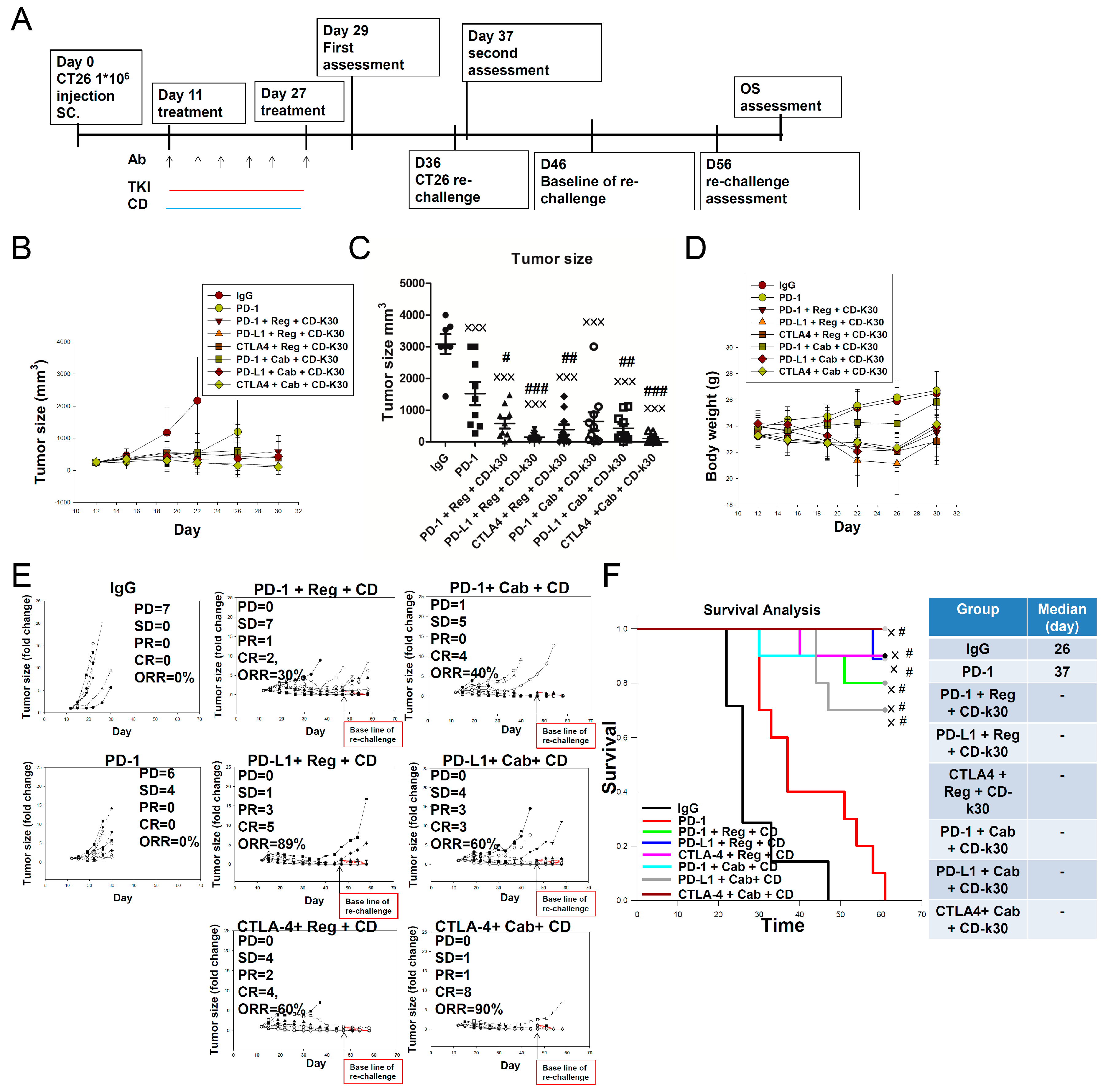
2.5. Overcoming First-Line Anti-PD-1 Antibody Treatment-Induced Drug Resistance Using Cabozantinib/Regorafenib plus Chidamide Combined with Anti-CTLA-4 Antibody in CT26-Bearing Mice
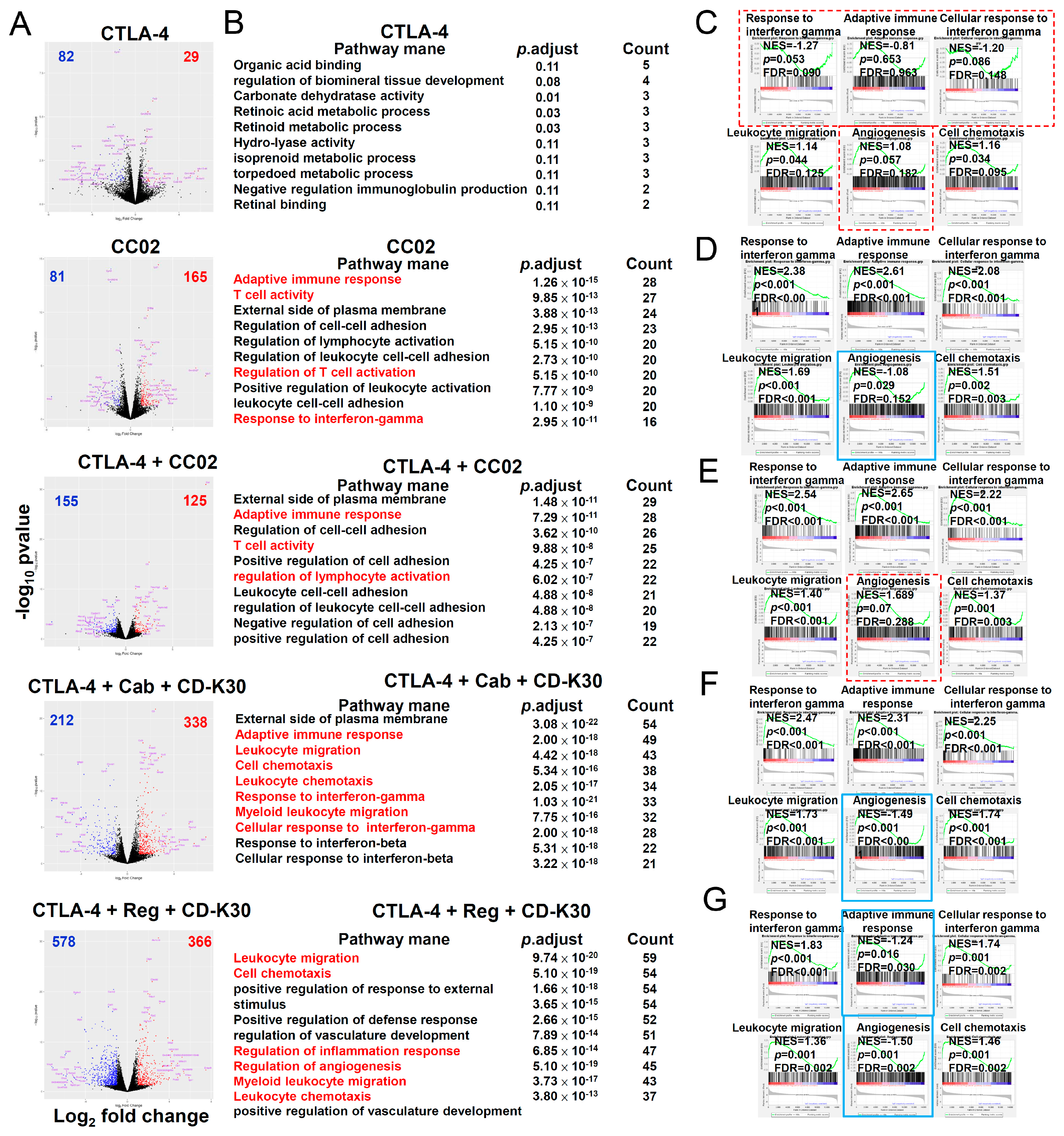
2.6. IntraTumor Gene Expression in First-Line Anti-PD-1 Antibody-Resistant CT-26 Tumor-Bearing Mice after Second-Line Therapy with Chidamide-k30 Combined with Cabozantinib/Regorafenib Plus Anti-CTLA-4 Antibody
3. Discussions
4. Material and Methods
4.1. Anti-Colorectal Cancer Activity in Animal Models
4.2. Tumor Rechallenge in Animal Models
4.3. Survival Rate in Animal Models
4.4. Flow Cytometry
4.5. Overcoming Primary Resistance and HPD Induced by First-Line PD-1 Checkpoint Blockade Therapy
4.6. RNA Quantification and Qualification
4.7. Library Preparation for Transcriptome Sequencing
4.8. Bioinformatics
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, D. CAR-T “the living drugs”, immune checkpoint inhibitors, and precision medicine: A new era of cancer therapy. J. Hematol. Oncol. 2019, 12, 113. [Google Scholar] [CrossRef] [PubMed]
- Chyuan, I.T.; Chu, C.L.; Hsu, P.N. Targeting the Tumor Microenvironment for Improving Therapeutic Effectiveness in Cancer Immunotherapy: Focusing on Immune Checkpoint Inhibitors and Combination Therapies. Cancers 2021, 13, 1188. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Hamid, O.; Daud, A.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.J.; Weber, J.S.; Gangadhar, T.C.; Joseph, R.W.; et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients With Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1668–1674. [Google Scholar] [CrossRef]
- Adam, T.; Becker, T.M.; Chua, W.; Bray, V.; Roberts, T.L. The Multiple Potential Biomarkers for Predicting Immunotherapy Response-Finding the Needle in the Haystack. Cancers 2021, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef]
- Vukadin, S.; Khaznadar, F.; Kizivat, T.; Vcev, A.; Smolic, M. Molecular Mechanisms of Resistance to Immune Checkpoint Inhibitors in Melanoma Treatment: An Update. Biomedicines 2021, 9, 835. [Google Scholar] [CrossRef]
- Wang, S.; He, Z.; Wang, X.; Li, H.; Liu, X.S. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. eLife 2019, 8, e49020. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed]
- Narita, Y.; Kitamura, H.; Wakita, D.; Sumida, K.; Masuko, K.; Terada, S.; Nakano, K.; Nishimura, T. The key role of IL-6-arginase cascade for inducing dendritic cell-dependent CD4(+) T cell dysfunction in tumor-bearing mice. J. Immunol. 2013, 190, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, J.B.; Ha, S.J.; Kim, H.R. Clinical Perspectives to Overcome Acquired Resistance to Anti-Programmed Death-1 and Anti-Programmed Death Ligand-1 Therapy in Non-Small Cell Lung Cancer. Mol. Cells 2021, 44, 363–373. [Google Scholar] [CrossRef]
- Wang, X.; Wang, F.; Zhong, M.; Yarden, Y.; Fu, L. The biomarkers of hyperprogressive disease in PD-1/PD-L1 blockage therapy. Mol. Cancer 2020, 19, 81. [Google Scholar] [CrossRef]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef]
- Tada, Y.; Togashi, Y.; Kotani, D.; Kuwata, T.; Sato, E.; Kawazoe, A.; Doi, T.; Wada, H.; Nishikawa, H.; Shitara, K. Targeting VEGFR2 with Ramucirumab strongly impacts effector/ activated regulatory T cells and CD8(+) T cells in the tumor microenvironment. J. Immunother. Cancer 2018, 6, 106. [Google Scholar] [CrossRef]
- Meder, L.; Schuldt, P.; Thelen, M.; Schmitt, A.; Dietlein, F.; Klein, S.; Borchmann, S.; Wennhold, K.; Vlasic, I.; Oberbeck, S.; et al. Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res. 2018, 78, 4270–4281. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Gao, J.; Di, W.; Wu, X. Anti-angiogenesis therapy overcomes the innate resistance to PD-1/PD-L1 blockade in VEGFA-overexpressed mouse tumor models. Cancer Immunol. Immunother. CII 2020, 69, 1781–1799. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Heishi, T.; Khan, O.F.; Kowalski, P.S.; Incio, J.; Rahbari, N.N.; Chung, E.; Clark, J.W.; Willett, C.G.; Luster, A.D.; et al. Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J. Clin. Investig. 2017, 127, 3039–3051. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.D.; Hoang, A.; Zhou, L.; Kalra, S.; Yetil, A.; Sun, M.; Ding, Z.; Zhang, X.; Bai, S.; German, P.; et al. Resistance to Antiangiogenic Therapy Is Associated with an Immunosuppressive Tumor Microenvironment in Metastatic Renal Cell Carcinoma. Cancer Immunol. Res. 2015, 3, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Kheshtchin, N.; Arab, S.; Ajami, M.; Mirzaei, R.; Ashourpour, M.; Mousavi, N.; Khosravianfar, N.; Jadidi-Niaragh, F.; Namdar, A.; Noorbakhsh, F.; et al. Inhibition of HIF-1alpha enhances anti-tumor effects of dendritic cell-based vaccination in a mouse model of breast cancer. Cancer Immunol. Immunother. CII 2016, 65, 1159–1167. [Google Scholar] [CrossRef]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006, 66, 8814–8821. [Google Scholar] [CrossRef]
- Miyake, K.; Yoshizumi, T.; Imura, S.; Sugimoto, K.; Batmunkh, E.; Kanemura, H.; Morine, Y.; Shimada, M. Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: Correlation with poor prognosis with possible regulation. Pancreas 2008, 36, e1–e9. [Google Scholar] [CrossRef]
- Chen, J.S.; Chou, C.H.; Wu, Y.H.; Yang, M.H.; Chu, S.H.; Chao, Y.S.; Chen, C.N. CC-01 (chidamide plus celecoxib) modifies the tumor immune microenvironment and reduces tumor progression combined with immune checkpoint inhibitor. Sci. Rep. 2022, 12, 1100. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-gamma in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; Kim, R.; Kim, T.W.; O’Connor, J.M.; Strickler, J.H.; Malka, D.; Sartore-Bianchi, A.; Bi, F.; Yamaguchi, K.; Yoshino, T.; et al. Third- or Later-line Therapy for Metastatic Colorectal Cancer: Reviewing Best Practice. Clin. Colorectal Cancer 2019, 18, e117–e129. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schutz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.K.; Wilhelm, A.; Antoniades, C.G. TAM receptor tyrosine kinase function and the immunopathology of liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G899–G905. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Efremova, M.; Rieder, D.; Klepsch, V.; Charoentong, P.; Finotello, F.; Hackl, H.; Hermann-Kleiter, N.; Lower, M.; Baier, G.; Krogsdam, A.; et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat. Commun. 2018, 9, 32. [Google Scholar] [CrossRef]
- Zhong, W.; Myers, J.S.; Wang, F.; Wang, K.; Lucas, J.; Rosfjord, E.; Lucas, J.; Hooper, A.T.; Yang, S.; Lemon, L.A.; et al. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genom. 2020, 21, 2. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Doleschel, D.; Hoff, S.; Koletnik, S.; Rix, A.; Zopf, D.; Kiessling, F.; Lederle, W. Regorafenib enhances anti-PD1 immunotherapy efficacy in murine colorectal cancers and their combination prevents tumor regrowth. J. Exp. Clin. Cancer Res. CR 2021, 40, 288. [Google Scholar] [CrossRef]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Hansen, W.; Hutzler, M.; Abel, S.; Alter, C.; Stockmann, C.; Kliche, S.; Albert, J.; Sparwasser, T.; Sakaguchi, S.; Westendorf, A.M.; et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med. 2012, 209, 2001–2016. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Hsu, C.; Chan, S.L.; Choo, S.P.; Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 2020, 72, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, K.; Matsui, A.; Kikuchi, H.; Klein, S.; Mamessier, E.; Chen, I.X.; Aoki, S.; Kitahara, S.; Inoue, K.; Shigeta, A.; et al. Regorafenib combined with PD1 blockade increases CD8 T-cell infiltration by inducing CXCL10 expression in hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e001435. [Google Scholar] [CrossRef]
- Zopf, D.; Fichtner, I.; Bhargava, A.; Steinke, W.; Thierauch, K.H.; Diefenbach, K.; Wilhelm, S.; Hafner, F.T.; Gerisch, M. Pharmacologic activity and pharmacokinetics of metabolites of regorafenib in preclinical models. Cancer Med. 2016, 5, 3176–3185. [Google Scholar] [CrossRef] [PubMed]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Coukos, G. Deciphering and reversing tumor immune suppression. Immunity 2013, 39, 61–73. [Google Scholar] [CrossRef]
- Liu, C.; He, H.; Li, X.; Su, M.A.; Cao, Y. Correction: Dynamic metrics-based biomarkers to predict responders to anti-PD-1 immunotherapy. Br. J. Cancer 2019, 120, 772. [Google Scholar] [CrossRef]
- Lund, A.W.; Duraes, F.V.; Hirosue, S.; Raghavan, V.R.; Nembrini, C.; Thomas, S.N.; Issa, A.; Hugues, S.; Swartz, M.A. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012, 1, 191–199. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef]
- Omar, N.E.; El-Fass, K.A.; Abushouk, A.I.; Elbaghdady, N.; Barakat, A.E.M.; Noreldin, A.E.; Johar, D.; Yassin, M.; Hamad, A.; Elazzazy, S.; et al. Diagnosis and Management of Hematological Adverse Events Induced by Immune Checkpoint Inhibitors: A Systematic Review. Front. Immunol. 2020, 11, 1354. [Google Scholar] [CrossRef]
- Schwartz, G.; Darling, J.O.; Mindo, M.; Damicis, L. Management of Adverse Events Associated with Cabozantinib Treatment in Patients with Advanced Hepatocellular Carcinoma. Target. Oncol. 2020, 15, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regimens | Initial Tumor Volume (mm3) | ORR (%) | PD | SD | PR | CR | Survival Rate (%) | Relapse * (Recurrence) (%) | Immunity # (Rechallenge) (%) |
|---|---|---|---|---|---|---|---|---|---|
| anti-PD-1 Ab | 227 | 0 | 8 | 0 | 0 | 0 | 0 | - | - |
| anti-PD-1 Ab + reg | 13 | 1 | 6 | 0 | 1 | 13 | 0 | 100 | |
| anti-PD-1 Ab + cab | 0 | 1 | 5 | 0 | 0 | 0 | - | - | |
| anti-PD-1 Ab + cab+ CD-k30 | 50 | 0 | 4 | 1 | 3 | 50 | 50 | 100 | |
| anti-PD-1 Ab + reg + CD-k30 | 43 | 0 | 4 | 0 | 3 | 86 | 0 | 100 |
| Regimens | Initial Tumor Volume (mm3) | ORR (%) | PD | SD | PR | CR | ORR (%) & | PD & | SD & | PR & | CR & | Survival Rate (%) | Relapse * (Recurrence) (%) | Immunity # (Rechallenge) (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| anti-PD-1 Ab | 243 | 0 | 6 | 4 | 0 | 0 | 0 | 8 | 2 | 0 | 0 | 0 | - | - |
| anti-PD-1 Ab + reg + CD-k30 | 30 | 0 | 7 | 1 | 2 | 60 | 1 | 3 | 2 | 4 | 80 | 0 | 100 | |
| anti-PD-L1 Ab + reg + CD-k30 | 89 | 0 | 1 | 3 | 5 | 89 | 0 | 1 | 0 | 8 | 89 | 13 | 100 | |
| anti-CTLA-4 Ab + reg + CD-k30 | 60 | 0 | 4 | 2 | 4 | 80 | 1 | 1 | 1 | 7 | 90 | 0 | 100 | |
| anti-PD-1 Ab + cab+ CD-k30 | 40 | 1 | 5 | 0 | 4 | 60 | 3 | 1 | 1 | 5 | 70 | 0 | 100 | |
| Anti-PD-L1 Ab + cab + CD-k30 | 60 | 0 | 4 | 3 | 3 | 60 | 2 | 2 | 1 | 5 | 70 | 17 | 100 | |
| anti-CTLA-4 Ab + cab + CD-k30 | 90 | 0 | 1 | 1 | 8 | 90 | 0 | 1 | 0 | 9 | 100 | 0 | 100 |
| Number of Mice | Response to First-Line Anti-PD-1 Antibody Therapy | Types of Drug Resistance to First-Line Anti-PD-1 Antibody Therapy |
|---|---|---|
| 10 | Treatment with anti-IgG antibody (as negative control) | N/A |
| 17 | Yes | Response * |
| 91 | NO | Primary resistance ** |
| 11 | NO | HPD *** |
| Therapeutic Resistance | Regimens | Initial Tumor Volume (mm3) | ORR (%) | PD | SD | PR | CR | ORR (%) & | PD & | SD & | PR & | CR & | Survival Rate (%) | Relapse * (%) | Immunity # (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Primary resistance | anti-PD-1 | 396 | 0 | 4 | 1 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | - | - |
| Anti-CTLA-4 | 0 | 0 | 7 | 0 | 0 | 0 | 2 | 5 | 0 | 0 | 0 | - | - | ||
| Anti-CTLA-4 + CC-02 | 37.5 | 1 | 4 | 0 | 3 | 37.5 | 2 | 3 | 0 | 3 | 37.5 | 0 | 100 | ||
| Anti-CTLA-4 + reg+ CD-k30 | 62.5 | 0 | 2 | 1 | 5 | 87.5 | 0 | 1 | 0 | 7 | 87.5 | 0 | 100 | ||
| Anti-CTLA-4 + cab + CD-k30 | 57.1 | 0 | 3 | 1 | 3 | 100 | 0 | 0 | 3 | 4 | 71.4 | 0 | 100 | ||
| HPD | Anti-CTLA-4 + cab + CDHCl | 669 | 18.1 | 0 | 9 | 0 | 2 | 45.4% | 3 | 3 | 3 | 2 | 45.4 | 0 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-S.; Hsieh, Y.-C.; Chou, C.-H.; Wu, Y.-H.; Yang, M.-H.; Chu, S.-H.; Chao, Y.-S.; Chen, C.-N. Chidamide plus Tyrosine Kinase Inhibitor Remodel the Tumor Immune Microenvironment and Reduce Tumor Progression When Combined with Immune Checkpoint Inhibitor in Naïve and Anti-PD-1 Resistant CT26-Bearing Mice. Int. J. Mol. Sci. 2022, 23, 10677. https://doi.org/10.3390/ijms231810677
Chen J-S, Hsieh Y-C, Chou C-H, Wu Y-H, Yang M-H, Chu S-H, Chao Y-S, Chen C-N. Chidamide plus Tyrosine Kinase Inhibitor Remodel the Tumor Immune Microenvironment and Reduce Tumor Progression When Combined with Immune Checkpoint Inhibitor in Naïve and Anti-PD-1 Resistant CT26-Bearing Mice. International Journal of Molecular Sciences. 2022; 23(18):10677. https://doi.org/10.3390/ijms231810677
Chicago/Turabian StyleChen, Jia-Shiong, Yi-Chien Hsieh, Cheng-Han Chou, Yi-Hong Wu, Mu-Hsuan Yang, Sz-Hao Chu, Ye-Su Chao, and Chia-Nan Chen. 2022. "Chidamide plus Tyrosine Kinase Inhibitor Remodel the Tumor Immune Microenvironment and Reduce Tumor Progression When Combined with Immune Checkpoint Inhibitor in Naïve and Anti-PD-1 Resistant CT26-Bearing Mice" International Journal of Molecular Sciences 23, no. 18: 10677. https://doi.org/10.3390/ijms231810677