
Transcriptome Sequencing and Metabolome Analysis Reveals the Molecular Mechanism of Drought Stress in Millet

Abstract
1. Introduction
2. Results
2.1. Transcriptome Characteristics of HQ and YS10 under Drought Stress
2.2. Differential Gene Identification and Functional Analysis under Drought Stress
2.3. The Effects of Drought Stress on Energy Metabolism
2.4. Dynamic Changes in the Transcriptome of Plant Hormone Signal Transduction
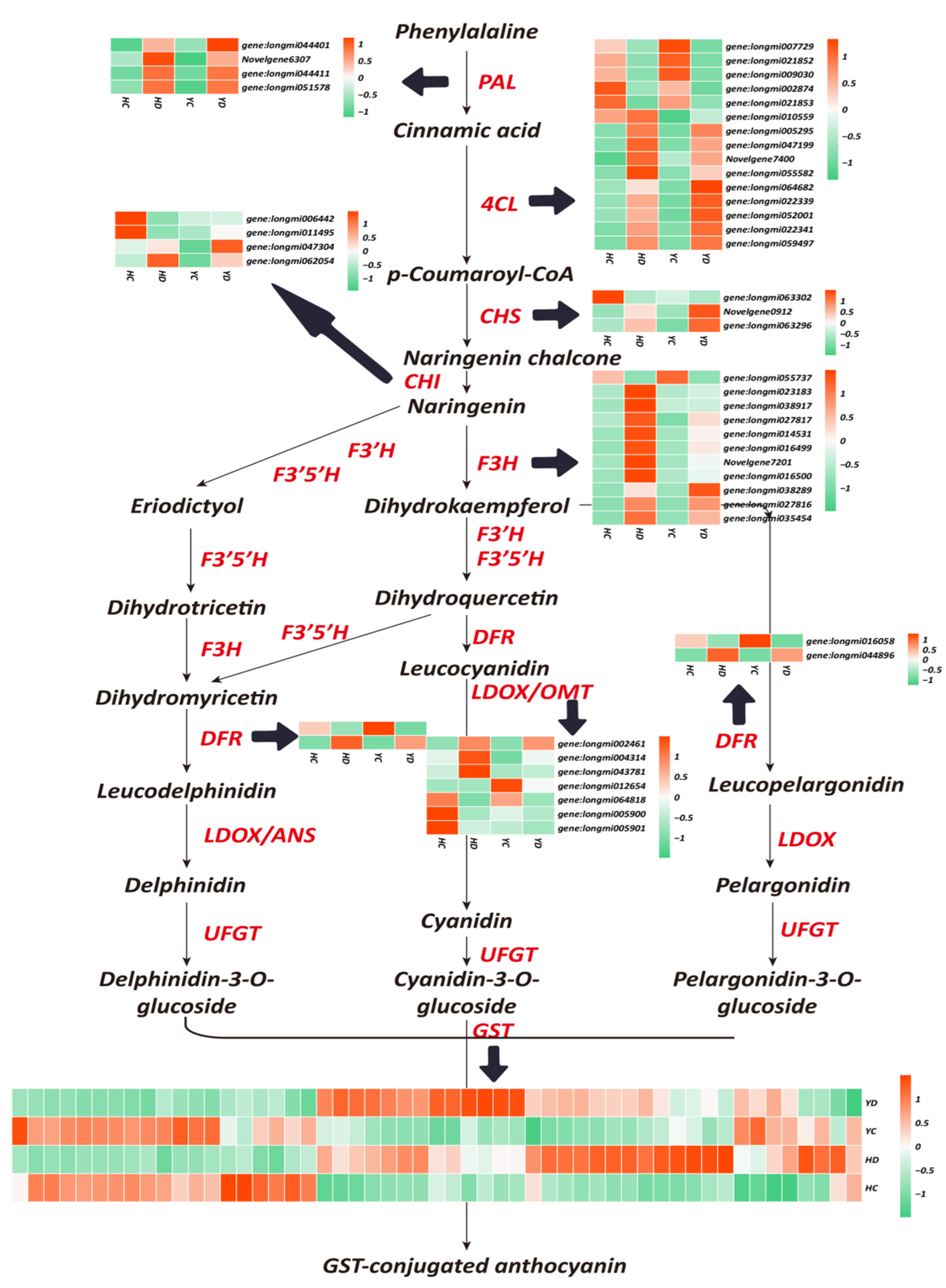
2.5. Increased Anthocyanin Metabolism Improves the Drought Resistance of HQ and YS10
2.6. Expression Profiling of Photosynthesis-Related Genes
2.7. Identification and Expression Analysis of Transcription Factors Related to Drought Stress
2.8. Overview of the Metabolomes of the HQ and YS10 Strains
2.9. qRT-PCR Verification of the RNA-Seq Data
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Cultivation and Treatment
4.2. Transcriptome Sequencing and Metabolome Detection
4.3. Bioinformatics Analysis
4.4. qRT-PCR
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DEGs | Differentially expressed genes |
| DEMs | differential metabolites |
| HRMS | high-resolution mass spectrometry |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genome |
| bHLH | basic helix-loop-helix |
| C3H | Cys3His zinc finger proteins |
| MYB | MYB domain proteins |
| WRKY | WRKY proteins |
| TCPs | TCP proteins |
| bZIPs | basic region-leucine zipper |
| C2H2s | C2H2 zinc-finger proteins |
| JA | jasmonic acid |
| SA | salicylic acid |
| ABA | abscisic acid |
| CK | cytokinin |
| GA | gibberellic acid |
| HK | hexokinase |
| MDH | malate dehydrogenase |
| SDH | succinate dehydrogenase |
| PAL | phenylalanine ammonia lyase |
| C4H | cinnamate 4-hydroxylase |
| 4CL | 4-coumaroyl-CoA ligase |
| CHS | chalcone synthase |
| CHI | chalcone isomerase |
| F3H | flavanone 3-hydroxylase |
| DFR | dihydroflavonol-4-reductase |
| OMT | O-methyltransferase |
| GST | glutathione S-transferase |
| PSB | photosystem II reaction center subunits |
| PSA | photosystem I reaction center subunits |
| LHC | light harvesting complex |
| ATPS | ATP synthase |
Appendix A



References
- Lu, H.; Zhang, J.; Liu, K.; Wu, N.; Li, Y.; Zhou, K.; Ye, M.; Zhang, T.; Zhang, H.; Yang, X.; et al. Earliest domestication of common millet (Panicum miliaceum) in East Asia extended to 10,000 years ago. Proc. Natl. Acad. Sci. USA 2009, 106, 7367–7372. [Google Scholar] [CrossRef] [PubMed]
- Habiyaremye, C.; Matanguihan, J.; D’Alpoim Guedes, J.; Ganjyal, G.; Whiteman, M.; Kidwell, K.; Murphy, K. Proso Millet (Panicum miliaceum L.) and Its Potential for Cultivation in the Pacific Northwest, U.S.: A Review. Front. Plant. Sci. 2016, 7, 1961. [Google Scholar] [CrossRef] [PubMed]
- Diao, X.M. Production and genetic improvement of minor cereals in China. Crop J. 2017, 5, 103–104. [Google Scholar] [CrossRef]
- Diao, X.M. Progresses in Stress Tolerance and Field Cultivation Studies of Orphan Cereals in China. Sci. Agric. Sinica. 2019, 52, 3943–3949. [Google Scholar] [CrossRef]
- Hussain, H.; Hussain, S.; Khaliq, A.; Ashraf, U.; Anjum, S.; Men, S.; Wang, L. Chilling and Drought Stresses in Crop Plants: Implications, Cross Talk, and Potential Management Opportunities. Front. Plant. Sci. 2018, 9, 393. [Google Scholar] [CrossRef]
- Zou, C.S.; Li, L.T.; Miki, D.; Li, D.L.; Tang, Q.M.; Xiao, L.H.; Rajput, S.; Deng, P.; Peng, L.; Jia, W.; et al. The genome of broomcorn millet. Nat. Commun. 2019, 10, 436. [Google Scholar] [CrossRef]
- Karyudi Fletcher, R.J. Osmoregulative capacity in birdseed millet under conditions of water stress. I. Variation in Setaria italica and Panicum miliaceum. Euphytica 2002, 125, 337–348. [Google Scholar] [CrossRef]
- Yuan, Y.H.; Liu, L.; Gao, Y.B.; Yang, Q.H.; Dong, K.J.; Liu, T.P.; Feng, B.L. Comparative analysis of drought-responsive physiological and transcriptome in broomcorn millet (Panicum miliaceum L.) genotypes with contrasting drought tolerance. Ind. Crops Prod. 2022, 177, 114498. [Google Scholar] [CrossRef]
- Na, X.F.; Cao, X.N.; Ma, C.X.; Ma, S.L.; Xu, P.X.; Liu, S.C.; Wang, J.J.; Wang, H.G.; Chen, L.; Qiao, Z.J. Plant stage, not drought stress, determinesthe effect of cultivars on bacterial community diversity in the rhizosphere of broomcorn millet (Panicum miliaceum L.). Front. Micro. 2019, 10, 828. [Google Scholar] [CrossRef]
- Chaves, M.M.; Flexas, J.; Pinheiro, C. Photosynthesis under drought and salt stress: Regulation mechanisms from whole plant to cell. Ann. Bot. 2009, 103, 551–560. [Google Scholar] [CrossRef]
- Shivhare, R.; Lata, C. Assessment of pearl millet genotypes for drought stress tolerance at early and late seedling stages. Acta Physiol. Plant. 2019, 41, 39. [Google Scholar] [CrossRef]
- Lawlor, D.W. Limitation to photosynthesis in water-stressed leaves: Stomata vs. metabolism and the role of ATP. Ann. Bot. 2002, 89, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.W.; Cornic, G. Photosynthetic carbon assimilation and associated metabolism in relation to water deficits in higher plants. Plant Cell Environ. 2002, 25, 275–294. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.; Davies, W. Drought, ozone, ABA and ethylene: New insights from cell to plant to community. Plant Cell Environ. 2010, 33, 510–525. [Google Scholar] [CrossRef]
- Ji, X.M.; Dong, B.D.; Shiran, B.; Talbot, M.J.; Edlington, J.E.; Hughes, T.; White, R.G.; Gubler, F.; Dolferus, R. Control of Abscisic Acid Catabolism and Abscisic Acid Homeostasis Is Important for Reproductive Stage Stress Tolerance in Cereals. Plant Physiol. 2011, 156, 647–662. [Google Scholar] [CrossRef]
- Wilkinson, S.; Kudoyarova, G.; Veselov, D.; Arkhipova, T.; Davies, W. Plant hormone interactions: Innovative targets for crop breeding and management. J. Exp. Bot. 2012, 63, 3499–3509. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; LV, B.; Li, J.; Luo, L.; Lu, S.; Zhang, X.; Ma, H.; Ming, F. The NAC family transcription factor OsNAP confers abiotic stress response through the ABA pathway. Plant Cell Physiol. 2014, 55, 604–619. [Google Scholar] [CrossRef]
- Fei, X.; Hou, L.; Shi, J.; Yang, T.; Liu, Y.; Wei, A. Patterns of Drought Response of 38 WRKY Transcription Factors of Zanthoxylum bungeanum Maxim. Int. J. Mol. Sci. 2019, 20, 68. [Google Scholar] [CrossRef]
- An, J.; Zhang, X.; Bi, S.; You, C.; Wang, X.; Hao, Y. The ERF transcription factor MdERF38 promotes drought stress-induced anthocyanin biosynthesis in apple. Plant J. 2020, 101, 573–589. [Google Scholar] [CrossRef]
- Shi, H.; Chen, L.; Ye, T.T.; Liu, X.D.; Ding, K.J.; Chan, Z.L. Modulation of auxin content in Arabidopsis confers improved drought stress resistance. Plant Physiol. Biochem. 2014, 82, 209–217. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Li, Y.P.; Hassan, M.J.; Li, Z.; Peng, Y. Indole-3-acetic acid improves drought tolerance of white clover via activating auxin, abscisic acid and jasmonic acid related genes and inhibiting senescence genes. BMC Plant Biol. 2020, 20, 150. [Google Scholar] [CrossRef] [PubMed]
- Kaya, C.; Tuna, A.L.; Alves, A.A.C. Gibberellic acid improves water defcit tolerance in maize plants. Acta Physiol. Plant. 2006, 28, 331–337. [Google Scholar] [CrossRef]
- Scarpeci, T.E.; Frea, V.S.; Zanor, M.I.; Valle, E.M. Overexpression of AtERF019 delays plant growth and senescence, and improves drought tolerance in Arabidopsis. J. Exp. Bot. 2017, 68, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Anjum, S.A.; Wang, L.; Farooq, M.; Khan, I.; Xue, L. Methyl jasmonateinduced alteration in lipid peroxidation, antioxidative defence system and yield in soybean under drought. J. Agron. Crop. Sci. 2011, 197, 296–301. [Google Scholar] [CrossRef]
- Ollas, C.D.; Dodd, I.C. Physiological impacts of ABA-JA interactions under water- limitation. Plant Mol. Biol. 2016, 91, 641–650. [Google Scholar] [CrossRef]
- Gao, J.; Shen, X.; Zhang, Z.; Peng, R.; Xiong, A.; Xu, J.; Zhu, B.; Zheng, J.; Yao, Q. The myb transcription factor MdMYB6 suppresses anthocyanin biosynthesis in transgenic Arabidopsis. Plant Cell Tiss. Organ. Cult. 2011, 106, 235–242. [Google Scholar] [CrossRef]
- Li, Q.; Chen, P.; Dai, S.; Sun, Y.; Yuan, B.; Kai, W.; Pei, Y.; He, S.; Liang, B.; Zhang, Y.; et al. PacCYP707A2 negatively regulates cherry fruit ripening while PacCYP707A1 mediates drought tolerance. J. Exp. Bot. 2015, 66, 3765–3774. [Google Scholar] [CrossRef]
- Okay, S.; Derelli, E.; Unver, T. Transcriptome-wide identification of bread wheat WRKY transcription factors in response to drought stress. Mol. Genet. Genom. 2014, 289, 765–781. [Google Scholar] [CrossRef]
- Joshi, R.; Wani, S.; Balwant, S.; Abhishek, B.; Dar, Z.A.; Lone, A.; Pareek, A.; Singla-Pareek, S. Transcription Factors and Plants Response to Drought Stress: Current Understanding and Future Directions. Front. Plant Sci. 2016, 7, 1029. [Google Scholar] [CrossRef]
- Guo, Y.; Ping, W.; Chen, J.; Zhu, L.; Zhao, Y.; Guo, J.; Huang, Y. Meta-analysis of the effects of overexpression of WRKY transcription factors on plant responses to drought stress. BMC Genet. 2019, 20, 63. [Google Scholar] [CrossRef]
- Ghodke, P.; Khandagale, K.; Thangasamy, A.; Kulkarni, A.; Narwade, N.; Shirsat, D.; Randive, P.; Roylawar, P.; Singh, I.; Gawande, S.; et al. Comparative transcriptome analyses in contrasting onion (Allium cepa L.) genotypes for drought stress. PLoS ONE 2020, 15, e0237457. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zeng, Z.; Lyu, Y.M.; Zhao, S.W. Drought-Responsive NAC Transcription Factor RcNAC72 Is Recognized by RcABF4, Interacts with RcDREB2A to Enhance Drought Tolerance in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 1755. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Liu, L.; Jian, L.L.; Xu, W.X.; Wang, J.T.; Li, Y.X.; Jiang, C.Z. Heterologous Expression of MfWRKY7 of Resurrection Plant Myrothamnus flabellifolia Enhances Salt and Drought Tolerance in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 7890. [Google Scholar] [CrossRef] [PubMed]
- Leng, P.; Zhao, J. Transcription factors as molecular switches to regulate drought adaptation in maize. Theor. Appl. Genet. 2020, 133, 1455–1456. [Google Scholar] [CrossRef]
- Hu, Q.; Ao, C.W.; Wang, X.R.; Wu, Y.F.; Du, X.Z. GhWRKY1-like, a WRKY transcription factor, mediates drought tolerance in Arabidopsis via modulating ABA biosynthesis. BMC Plant Biol. 2021, 21, 458. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.Y.; Gao, S.X.; Li, J.; Song, P.Y.; Zhang, Q.; Guo, J.F.; Wang, X.Y.; Han, X.Y.; Wang, X.J.; Zhu, Y.; et al. The bHLH transcription factor regulated gene OsWIH2 is a positive regulator of drought tolerance in rice. Plant Physiol. Bioch. 2021, 169, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.E.; Kim, T.H.; Shim, J.S.; Bang, S.W.; Yoon, H.B.; Oh, S.H.; Kim, Y.S.; Oh, S.J.; Seo, J.S.; Kim, J.K. Rice NAC17 transcription factor enhances drought tolerance by modulating lignin accumulation. Plant Sci. 2022, 323, 111404. [Google Scholar] [CrossRef]
- Zhang, G.F.; Li, G.D.; Xiang, Y.; Zhang, A.Y. The transcription factor ZmMYB-CC10 improves drought tolerance by activating ZmAPX4 expression in maize. Biochem. Biophys. Res. Commun. 2022, 604, 1–7. [Google Scholar] [CrossRef]
- Sperdouli, I.; Moustakas, M. Interaction of proline, sugars, and anthocyanins during photosynthetic acclimation of Arabidopsis thaliana to drought stress. J. Plant Physiol. 2012, 169, 577–585. [Google Scholar] [CrossRef]
- Gonzalez-Villagra, J.; Kurepin, L.; Reyes-Díaz, M. Evaluating the involvement and interaction of abscisic acid and miRNA156 in the induction of anthocyanin biosynthesis in drought-stressed plants. Planta 2017, 246, 299–312. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, W.; Wei, Y.; Shi, H. MeCIPK23 interacts with Whirly transcription factors to activate abscisic acid biosynthesis and regulate drought resistance in cassava. Plant Biotechnol. J. 2020, 18, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Jiang, Y.; Li, H.; Guo, J.; Dong, M.; Zhang, J.; Liu, G. Genome-wide analysis of the NAC transcription factor family in broomcorn millet (Panicum miliaceum L.) and expression analysis under drought stress. BMC Genom. 2020, 21, 96. [Google Scholar] [CrossRef] [PubMed]
- Dobáková, M.; Sobotka, R.; Tichy, M.; Komenda, J. Psb28 protein is involved in the biogenesis of the photosystem II inner antenna CP47 (PsbB) in the cyanobacterium Synechocystis sp. PCC 6803. Plant Physiol. 2009, 149, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.X.; Lorkovc, Z.J.; Oelm lleru, R.; Schröder, W.P. The low molecular mass PsbW protein is involved in the stabilization of the dimeric photosystem II complex in Arabidopsis thaliana. J. Biol. Chem. 2000, 275, 37945–37950. [Google Scholar] [CrossRef] [PubMed]
- García-Cerdán, J.G.; KováCS, L.; Tóth, T.; Kereïche, S.; Aseeva, E.; Boekema, E.J.; Mamedov, F.; Funk, C.; Schröder, W.P. The PsbW protein stabilizes the supramolecular organization of photosystem II in higher plants. Plant J. 2011, 65, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Fagerlunda, R.D.; Forsmana, J.A.; Biswasa, S.; Vassb, I.; Daviesa, F.K.; Summerfieldc, T.C.; Eaton-Ryea, J.J. Stabilization of Photosy-stem II by the PsbT protein impacts photodamage, repair and biogenesis. BBA-Bioenerg. 2020, 1861, 148234. [Google Scholar] [CrossRef]
- Bentley, F.K.; Hao, L.; Dilbeck, P.; Burnap, R.L.; Eaton-Rye, J.J. Effects of inactivating psbM and psbT on photodamage and assembly of photosystem II in Synechocystis sp. PCC 6803. Biochemistry 2008, 47, 11637–11646. [Google Scholar] [CrossRef] [PubMed]
- Thidholm, E.; Lindstro, V.; Tissier, C.; Robinson, C.; Schröder, W.P.; Funk, C. Novel approach reveals localisation and assembly pathway of the PsbS and PsbW proteins into the photosystem II dimer. FEBS Lett. 2002, 513, 217–222. [Google Scholar] [CrossRef]
- Wang, Y.W.; Chen, S.M.; Wang, W.J.; Huang, X.Q.; Zhou, C.F.; Zhuang, Z.; Lu, S. The DnaJ-Like Zinc Finger Domain Protein PSA2 Affects Light Acclimation and Chloroplast Development in Arabidopsis thaliana. Front. Plant Sci. 2016, 7, 360. [Google Scholar] [CrossRef]
- Shen, J.; Williams-Carrier, R.; Barkan, A. PSA3, a Protein on the Stromal Face of the Thylakoid Membrane, Promotes Photosystem I Accumulation in Cooperation with the Assembly Factor PYG7. Plant Physiol. 2017, 174, 1850–1862. [Google Scholar] [CrossRef]
- Kashino, Y.; Lauber, W.M.; Carroll, J.A.; Wang, Q.J.; Whitmarsh, J.; Satoh, K.; Pakrasi, H.B. Proteomic Analysis of a Highly Active Photosystem II Preparation from the Cyanobacterium Synechocystis sp. PCC 6803 Reveals the Presence of Novel Polypeptides. Biochemistry 2002, 41, 8004–8012. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, D.Y.; Guo, J.K.; Wu, H.; Jin, M.F.; Lu, Q.T.; Lu, C.M.; Zhang, L.X. A Psb27 homologue in Arabidopsis thaliana is required for efficient repair of photodamaged photosystem II. Plant Mol. Biol. 2006, 61, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Onokata, J. Organization of the psaH gene family of photosystem I in Nicotiana sylvestris. Plant Cell Physiol. 1994, 35, 297–302. [Google Scholar] [PubMed]
- Shanker, A.K.; Maheswari, M.; Yadav, S.K.; Desai, S.; Bhanu, D.; Attal, N.B.; Venkateswarlu, B. Drought stress responses in crops. Funct. Integr. Genom. 2014, 14, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Cao, H.L.; Lin, H.Z.; Hu, J.; Ye, Y.J.; Li, J.; Hao, Z.L.; Hao, X.Y.; Sun, Y.; Yang, Y.J.; et al. Expression patterns of alpha-amylase and beta-amylase genes provide insights into the molecular mechanisms underlying the responses of tea plants (Camellia sinensis) to stress and postharvest processing treatments. Planta 2019, 250, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khodasadi, E.; Fakheri, B.; Komatsu, S. Organ-specific proteomics of soybean seedlings under flooding and drought stresses. J. Proteom. 2017, 162, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Swanston, J.; Molina-Cano, J. Beta-amylase activity and thermostability in two mutants derived from the malting barley cv. Triumph. J. Cereal Sci. 2001, 33, 155–161. [Google Scholar] [CrossRef]
- Du, Y.L.; Zhao, Q.; Chen, L.R.; Yao, X.D.; Zhang, H.J.; Wu, J.J.; Xie, F.T. Effect of drought stress during soybean R2–R6 growth stages on sucrose metabolism in leaf and seed. Int. J. Mol. Sci. 2020, 21, 618. [Google Scholar] [CrossRef]
- Lehretz, G.G.; Sonnewald, S.; Lugassi, N.; Granot, D.; Sonnewald, U. Future-proofing potato for drought and heat tolerance by overexpression of hexokinase and SP6A. Front. Plant Sci. 2021, 11, 614534. [Google Scholar] [CrossRef]
- Xue, G.P.; McIntyre, C.L.; Donna Glassop, D.; Shorter, R. Use of expression analysis to dissect alterations in carbohydrate metabolism in wheat leaves during drought stress. Plant Mol. Biol. 2008, 67, 197–214. [Google Scholar] [CrossRef]
- Yao, K.; Wu, Y.Y. Phosphofructokinase and glucose-6-phosphate dehydrogenase in response to drought and bicarbonate stress at transcriptional and functional levels in mulberry. Russ. J. Plant Physiol. 2016, 63, 235–242. [Google Scholar] [CrossRef]
- Piero, A.R.L.; Puglisi, I.; Rapisarda, P.; Petrone, G. Anthocyanins accumulation and related gene expression in red orange fruit induced by low temperature storage. J. Agric. Food Chem. 2005, 53, 9083–9088. [Google Scholar] [CrossRef] [PubMed]
- Chalker-Scott, L. Environmental significance of anthocyanins in plant stress responses. Photochem. Photobiol. 1999, 70, 1–9. [Google Scholar] [CrossRef]
- Shao, L.; Shu, Z.; Sun, S.L.; Peng, C.L.; Wang, X.J.; Lin, Z.F. Antioxidation of anthocyanins in photosynthesis under high temperature stress. J. Int. Plant Biol. 2007, 49, 1341–1351. [Google Scholar] [CrossRef]
- Mori, K.; Goto-Yamamoto, N.; Kitayama, M.; Hashizume, K. Loss of anthocyanins in red-wine grape under high temperature. J. Exp. Bot. 2007, 58, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, V.; D’Amelia, V.; Esposito, M.; Amitrano, C.; Carillo, P.; Carputo, D.; Maggio, A. Anthocyanins are key regulators of drought stress tolerance in tobacco. Biology 2021, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, N.; Harshavardhan, V.T.; Govind, G.; Seiler, C.; Kohli, A. Contrapuntal role of ABA: Does it mediate stress tolerance or plant growth retardation under long-term drought stress? Gene 2012, 506, 265–273. [Google Scholar] [CrossRef]
- Seiler, C.; Harshavardhan, V.T.; Rajesh, K.; Reddy, P.S.; Strickert, M.; Rolletschek, H.; Scholz, U.; Wobus, U.; Sreenivasulu, N. ABA biosynthesis and degradation contributing to ABA homeostasis during barley seed development under control and terminal drought-stress conditions. J. Exp. Bot. 2011, 62, 2615–2632. [Google Scholar] [CrossRef]
- Hai, N.N.; Chuong, N.N.; Tu, N.H.C.; Kisiala, A.; Hoang, X.L.T.; Thao, N.P. Role and regulation of cytokinins in plant response to drought stress. Plants 2020, 9, 422. [Google Scholar] [CrossRef]
- Ullah, A.; Manghwar, H.; Shaban, M.; Khan, A.H.; Akbar, A.; Ali, U.; Ali, E.; Fahad, S. Phytohormones enhanced drought tolerance in plants: A coping strategy. Environ. Sci. Pollut. Res. 2018, 25, 33103–33118. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.J.; Zhang, W.J.; Yan, S.N.; Wang, R.; Zhao, J.F.; Li, Y.J.; Qi, Z.G.; Sun, Z.X.; Zhu, Z.G. The putative auxin efflux carrier OsPIN3t is involved in the drought stress response and drought tolerance. Plant J. 2012, 72, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, H.; Liu, Q.; Huang, J.; Semagn, K.; Liu, D.; Li, Y.C.; Yang, B.; He, Y.L.; Sui, C. Analysis of unigenes involved in lateral root development in Bupleurum chinense and B. scorzonerifolium. Planta 2021, 253, 1–12. [Google Scholar] [CrossRef]
- Devkar, V.; Mehterov, N.; Ali, S.; Ozgur, R.; Turkan, I.; Mueller-Roeber, B.; Balazadeh, S. NAC transcription factor JUNGBRUNNEN 1 enhances drought tolerance in tomato. Plant Biotechnol. J. 2018, 16, 354–366. [Google Scholar]
- Baldoni, E.; Genga, A.A.; Cominelli, E. Plant MYB transcription factors: Their role in drought response mechanisms. Int. J. Mol. Sci. 2015, 16, 15811–15851. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Rabara, R.C.; Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 2014, 239, 255–266. [Google Scholar] [CrossRef]
- He, G.H.; Xu, J.Y.; Wang, Y.X.; Liu, J.M.; Li, P.S.; Chen, M.; Ma, Y.Z.; Xu, Z.S. Drought-responsive WRKY transcription factor genes TaWRKY1 and TaWRKY33 from wheat confer drought and/or heat resistance in Arabidopsis. BMC Plant Biol. 2016, 16, 116. [Google Scholar] [CrossRef]
- Zhao, G.; Song, Y.; Wang, C.X.; Butt, H.I.; Wang, Q.H.; Zhang, C.J.; Yang, Z.R.; Liu, Z.; Chen, E.Y.; Zhang, X.Y.; et al. Genome-wide identifi-cation and functional analysis of the TIFY gene family in response to drought in cotton. Mol. Genet. Genom. 2016, 291, 2173–2187. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, W.; Qiao, Z.; Feng, M.; Wang, G.; Duan, Y.; Chen, L. Resistance evaluation and response of 16 millet varieties at germination stage to drought stress. Acta Agrestia Sin. 2013, 21, 302–307. [Google Scholar] [CrossRef]
- Au, K.; Underwood, J.; Lee, L.; Wong, W. Improving PacBio Long Read Accuracy by Short Read Alignment. PLoS ONE 2012, 7, e46679. [Google Scholar] [CrossRef]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K. Proteomics, Bioinformatics, PacBio Sequencing and Its Applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; PyI, P.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HQ | YS | |
|---|---|---|
| Subreads | ||
| Subreads base(G) | 26.11 | 20.77 |
| Subreads number | 20,615,482 | 17,382,103 |
| Average subreads length | 1267 | 1196 |
| FLNC | ||
| FLNC_number | 531,759 | 377,205 |
| Mean_length | 2152 | 2174 |
| Total number after correct | 288,698 | 225,872 |
| Isoforms number | 112,437 | 87,330 |
| Annotation num | ||
| NR | 4721 | 3656 |
| SwissProt | 2910 | 2269 |
| KEGG | 4446 | 3422 |
| KOG | 2081 | 1605 |
| GO | 3208 | 2545 |
| NT | 5153 | 4038 |
| PFAM | 3208 | 2545 |
| lncRNA | ||
| Sense_overlapping | 3342 | 430 |
| Sense_intronic | 241 | 2868 |
| LincRNA | 3701 | 2939 |
| Antisense | 538 | 172 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Hu, Y.; Song, J.; Feng, H.; Wang, J.; Chen, L.; Wang, L.; Diao, X.; Wan, Y.; Liu, S.; et al. Transcriptome Sequencing and Metabolome Analysis Reveals the Molecular Mechanism of Drought Stress in Millet. Int. J. Mol. Sci. 2022, 23, 10792. https://doi.org/10.3390/ijms231810792
Cao X, Hu Y, Song J, Feng H, Wang J, Chen L, Wang L, Diao X, Wan Y, Liu S, et al. Transcriptome Sequencing and Metabolome Analysis Reveals the Molecular Mechanism of Drought Stress in Millet. International Journal of Molecular Sciences. 2022; 23(18):10792. https://doi.org/10.3390/ijms231810792
Chicago/Turabian StyleCao, Xiaoning, Yulu Hu, Jian Song, Hui Feng, Junjie Wang, Ling Chen, Lun Wang, Xianmin Diao, Yan Wan, Sichen Liu, and et al. 2022. "Transcriptome Sequencing and Metabolome Analysis Reveals the Molecular Mechanism of Drought Stress in Millet" International Journal of Molecular Sciences 23, no. 18: 10792. https://doi.org/10.3390/ijms231810792
APA StyleCao, X., Hu, Y., Song, J., Feng, H., Wang, J., Chen, L., Wang, L., Diao, X., Wan, Y., Liu, S., & Qiao, Z. (2022). Transcriptome Sequencing and Metabolome Analysis Reveals the Molecular Mechanism of Drought Stress in Millet. International Journal of Molecular Sciences, 23(18), 10792. https://doi.org/10.3390/ijms231810792